
Exon–Intron Differential Analysis Reveals the Role of Competing Endogenous RNAs in Post-Transcriptional Regulation of Translation

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
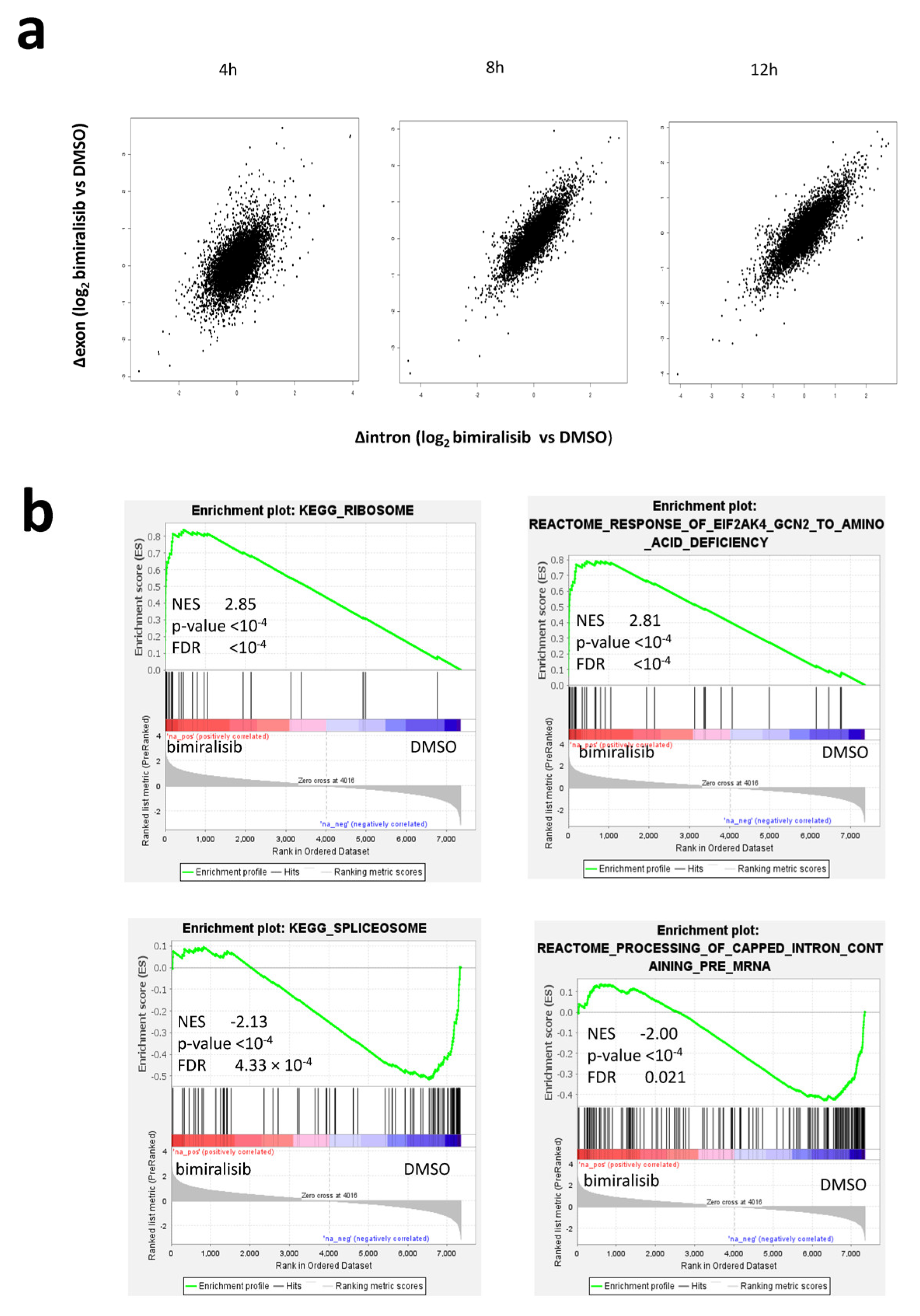
2.1. Bimiralisib Reduces the Transcription of Genes Coding for Proteasome and Ribosome Components
2.2. Post-Transcriptional Regulation of Many Transcripts Encoding for Riboproteins and Translation Regulators Is an Early Event after Dual PIK3/mTOR Inhibition
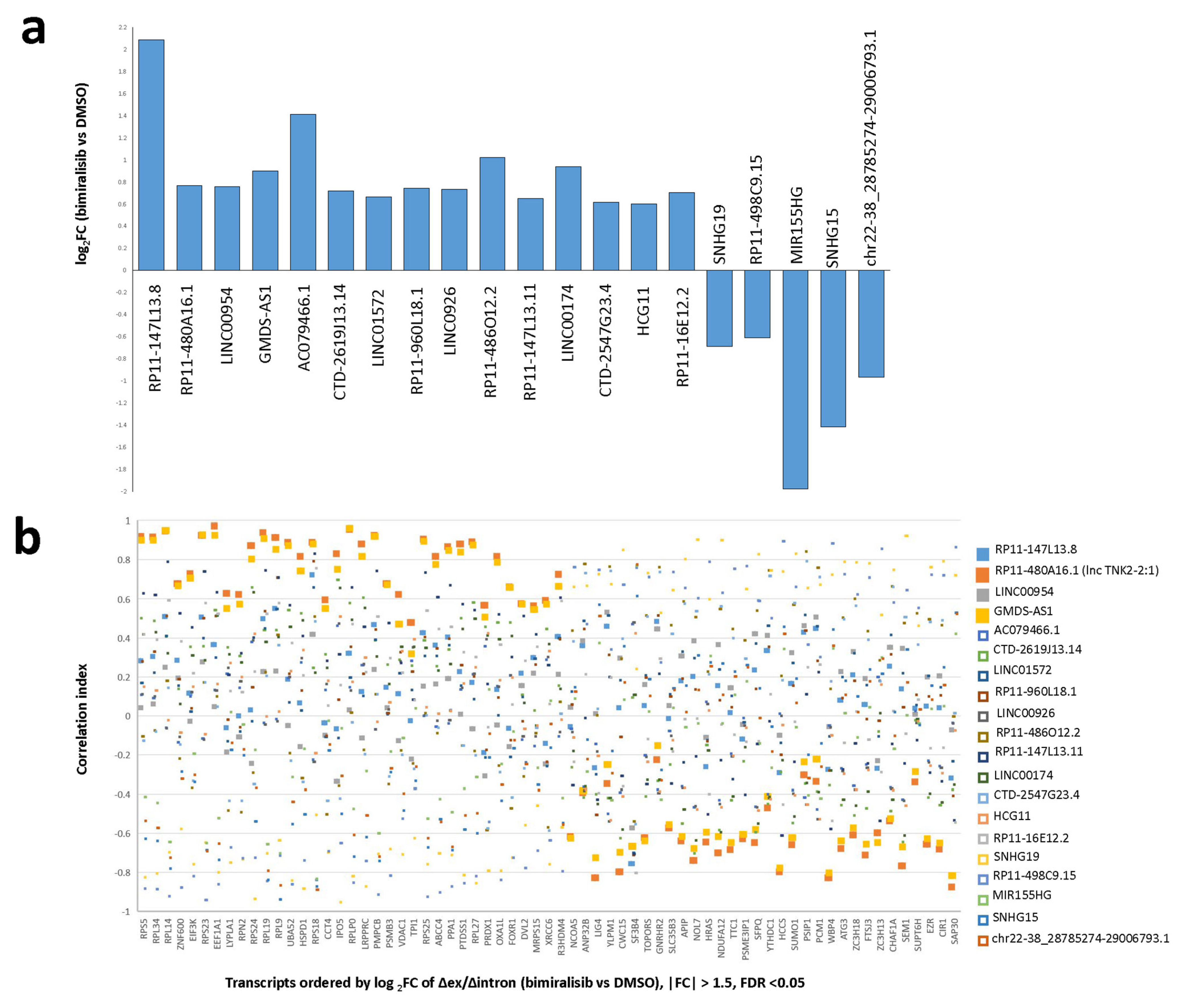
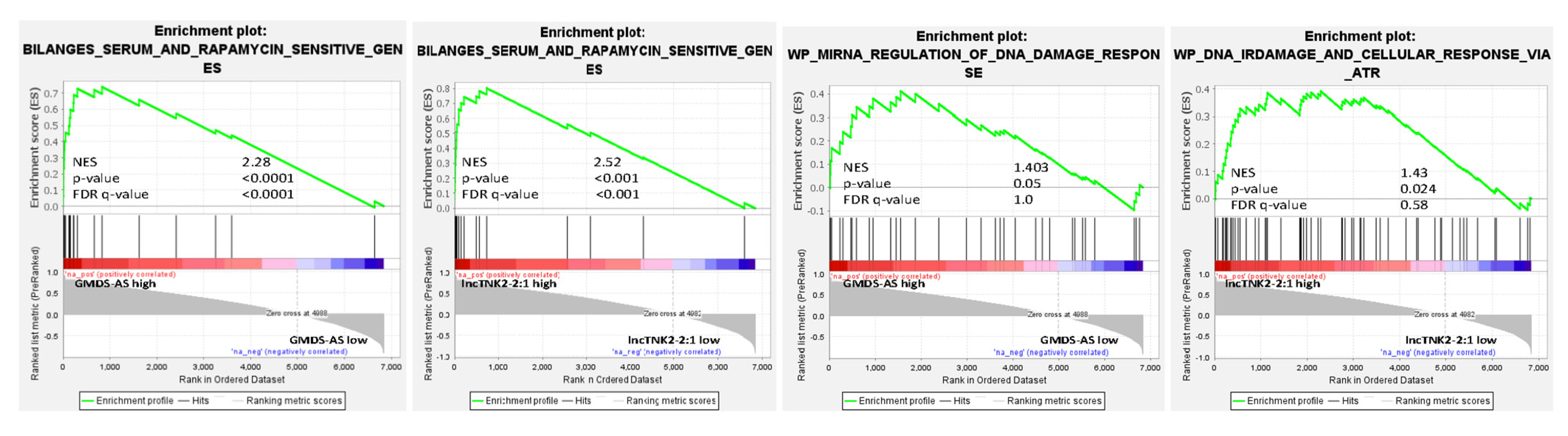
2.3. The lincRNAs RP11-480A16.1 (lncTNK2-2:1) and GMDS-AS1 Are Differentially Expressed after Dual PIK3/mTOR Inhibition and Strongly Correlated to Significantly Stabilized Transcripts
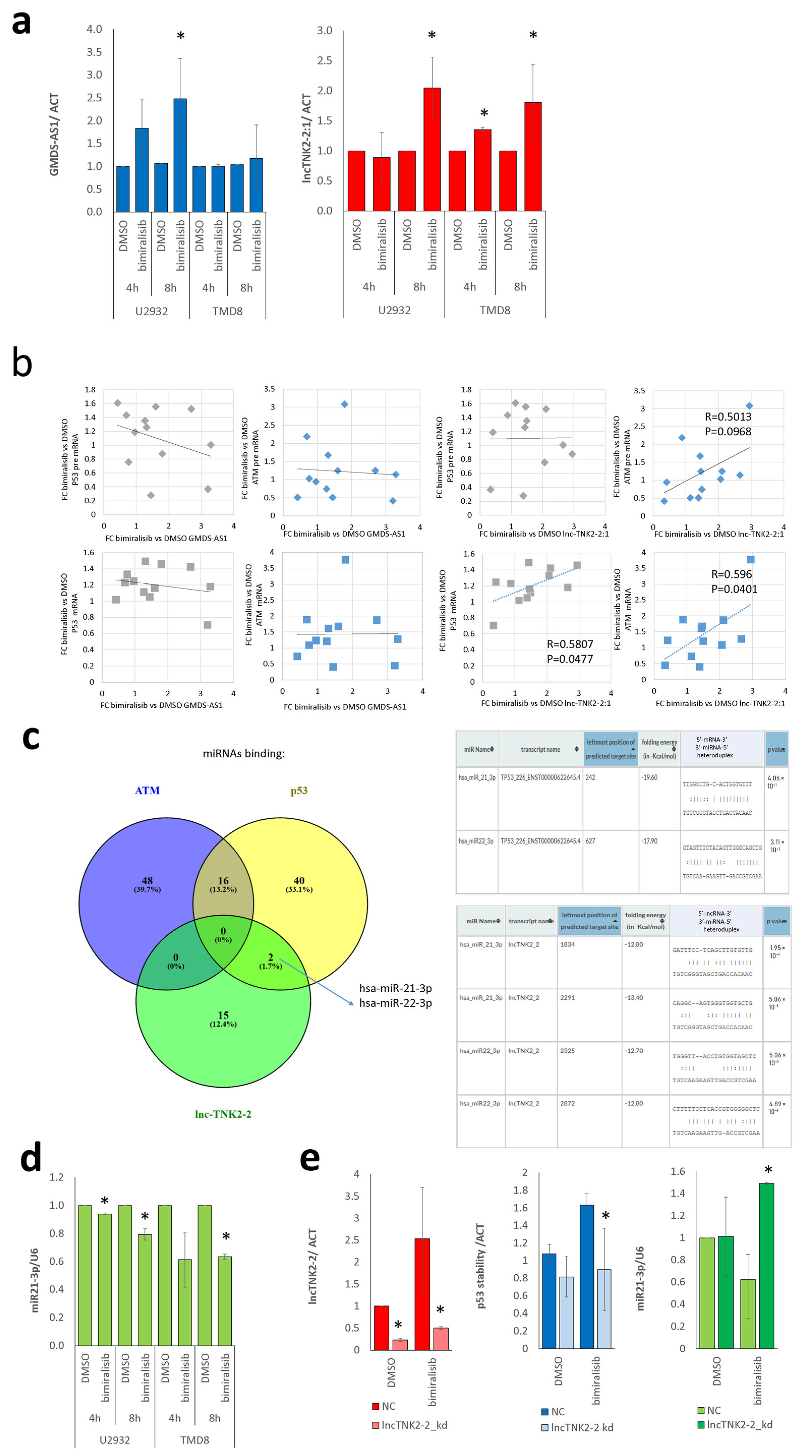
2.4. lncTNK2-2:1 Induces Stabilization of p53 and ATM by Sequestering miR21-3p
2.5. lncTNK2-2:1 Degradation Reverts Stabilization of p53 and Releases miR21-3p
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Bimiralisib Treatment
4.2. lncTNK2:2-1 Degradation
4.3. RNA-Extraction
4.4. Whole-Transcriptome Sequencing (RNA-Seq)
4.5. Data Mining
4.6. Reverse Transcription of Total RNA to cDNA
4.7. Quantitative Real-Time PCR
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.G.; Hogan, D.J.; McCullough, H.L.; Myers, J.W.; Herschlag, D.; Ferrell, J.E.; Brown, P.O. Concordant Regulation of Translation and mRNA Abundance for Hundreds of Targets of a Human microRNA. PLoS Biol. 2009, 7, e1000238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagkouni, D.; Paraskevopoulou, M.D.; Tastsoglou, S.; Skoufos, G.; Karavangeli, A.; Pierros, V.; Zacharopoulou, E.; Hatzigeorgiou, A.G. DIANA-LncBase v3: Indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 2020, 48, D101–D110. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Gaidatzis, D.; Burger, L.; Florescu, M.; Stadler, M.B. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol. 2015, 33, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2017, 24, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.P.; Popat, R.; Stathis, A.; Krasniqi, F.; Eyre, T.A.; Ng, C.H.; El-Sharkawi, D.; Schmidt, C.; Wicki, A.; Ivanova, E.; et al. A Dose-Escalation (DE) Study with Expansion Evaluating Safety, Pharmacokinetics and Ef-ficacy of the Novel, Balanced PI3K/mTOR Inhibitor PQR309 in Patients with Relapsed or Refractory Lympho-ma. Blood 2016, 128, 5893. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [Green Version]
- Tarantelli, C.; Lupia, A.; Stathis, A.; Bertoni, F. Is There a Role for Dual PI3K/mTOR Inhibitors for Patients Affected with Lymphoma? Int. J. Mol. Sci. 2020, 21, 1060. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, S.; Yang, J.; Zhao, F. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Calsina, B.; Castro-Vega, L.J.; Torres-Pérez, R.; Inglada-Pérez, L.; Currás-Freixes, M.; Roldán-Romero, J.M.; Mancikova, V.; Letón, R.; Remacha, L.; Santos, M.; et al. Integrative multi-omics analysis identifies a prognostic miRNA signature and a targetable miR-21-3p/TSC2/mTOR axis in metastatic pheochromocytoma/paraganglioma. Theranostics 2019, 9, 4946. [Google Scholar] [CrossRef]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazzi, M.; Bavelloni, A.; Gallo, A.; Faenza, I.; Blalock, W.L. Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. Int. J. Mol. Sci. 2019, 20, 2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCool, M.A.; Bryant, C.J.; Baserga, S.J. MicroRNAs and long non-coding RNAs as novel regulators of ribosome biogenesis. Biochem. Soc. Trans. 2020, 48, 595–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Xin, X.F.; Zhang, J.Y.; Dai, W.; Lv, T.F.; Song, Y. LncRNA GMDS-AS1 inhibits lung adenocarcinoma development by regulating miR-96-5p/CYLD signaling. Cancer Med. 2020, 9, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, C.A. p53 is a general repressor of RNA polymerase III transcription. EMBO J. 1998, 17, 3112–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000, 20, 5930–5938. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.A.; Espinosa, J.M. The impact of post-transcriptional regulation in the p53 network. Brief. Funct. Genom. 2012, 12, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Houghton, P.J. The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 11869–11874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting ge-nome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Holik, A.Z.; Su, S.; Jansz, N.; Chen, K.; Leong, H.S.; Blewitt, M.E.; Asselin-Labat, M.-L.; Smyth, G.K.; Ritchie, M.E. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 2015, 43, e97. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.H.; Karlen, Y.; Bakker, O.; Hoff, M.J.B.V.D.; Moorman, A.F.M. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene_Name | Ensembl ID | logFC | AveExpr | t | p Value | adj. p Value | circRNA |
|---|---|---|---|---|---|---|---|
| RP11-147L13.8 | ENSG00000267731.1 | 2.08 | 4.1 | 9.74 | 7.00 × 10−7 | 6.48 × 10−5 | NO |
| RP11-480A16.1 | ENSG00000260261.1 | 0.763 | 4.62 | 9.52 | 8.89 × 10−7 | 7.32 × 10−5 | YES |
| LINC00954 | ENSG00000228784.6 | 0.76 | 3.5 | 7.74 | 7.03 × 10−6 | 0.000205749 | YES |
| GMDS-AS1 | ENSG00000250903.7 | 0.9 | 3.75 | 7.27 | 1.30 × 10−5 | 0.000290741 | YES |
| AC079466.1 | ENSG00000266976.1 | 1.41 | 4.25 | 7.06 | 1.71 × 10−5 | 0.000341307 | NO |
| CTD-2619J13.14 | ENSG00000232098.3 | 0.72 | 4.42 | 6.4 | 4.24 × 10−5 | 0.000611259 | NO |
| LINC01572 | ENSG00000261008.5 | 0.66 | 3.77 | 6.35 | 4.54 × 10−5 | 0.000636107 | YES |
| RP11-960L18.1 | ENSG00000261218.4 | 0.74 | 3.5 | 6.29 | 4.93 × 10−5 | 0.000670722 | YES |
| LINC00926 | ENSG00000247982.5 | 0.73 | 4.98 | 5.82 | 9.95 × 10−5 | 0.001049071 | YES |
| RP11-486O12.2 | ENSG00000247373.3 | 1.02 | 3.53 | 5.8 | 0.0001 | 0.001063727 | NO |
| RP11-147L13.11 | ENSG00000278730.1 | 0.65 | 4.7 | 5.5 | 0.00016 | 0.001445039 | NO |
| LINC00174 | ENSG00000179406.6 | 0.94 | 3.52 | 5.29 | 0.00022 | 0.001840434 | NO |
| CTD-2547G23.4 | ENSG00000274925.1 | 0.62 | 3.55 | 4.57 | 0.00071 | 0.004099891 | NO |
| HCG11 | ENSG00000228223.2 | 0.6 | 3.59 | 4.48 | 0.00084 | 0.004600193 | NO |
| RP11-16E12.2 | ENSG00000259772.5 | 0.7 | 3.93 | 4.3 | 0.00115 | 0.005710491 | NO |
| SNHG19 | ENSG00000260260.1 | −0.68 | 4.14 | −5.4 | 0.00018 | 0.001630628 | NO |
| RP11-498C9.15 | ENSG00000263731.1 | −0.61 | 4.96 | −5.59 | 0.00014 | 0.001328078 | NO |
| MIR155HG | ENSG00000234883.3 | −1.97 | 5.66 | −9.15 | 1.33 × 10−6 | 8.69 × 10−5 | NO |
| SNHG15 | ENSG00000232956.7 | −1.41 | 5.37 | −9.89 | 5.98 × 10−7 | 6.00 × 10−5 | NO |
| chr22-38_28785274-29006793.1 | ENSG00000279978.1 | −0.97 | 5.8 | −10.21 | 4.28 × 10−7 | 5.64 × 10−5 | NO |
| Name | Size | ES | NES | NOM p-Value | FDR q-Value | FWER p-Value |
|---|---|---|---|---|---|---|
| REACTOME_EUKARYOTIC_TRANSLATION_ELONGATION | 37.000 | 0.721 | 2.522 | 0.000 | 0.000 | 0.000 |
| REACTOME_RESPONSE_OF_EIF2AK4_GCN2_TO_AMINO_ACID_DEFICIENCY | 42.000 | 0.682 | 2.419 | 0.000 | 0.000 | 0.000 |
| REACTOME_SELENOAMINO_ACID_METABOLISM | 50.000 | 0.633 | 2.304 | 0.000 | 0.000 | 0.000 |
| REACTOME_EUKARYOTIC_TRANSLATION_INITIATION | 51.000 | 0.610 | 2.219 | 0.000 | 0.000 | 0.000 |
| REACTOME_ACTIVATION_OF_THE_MRNA_UPON_BINDING_OF_THE_CAP_BINDING_COMPLEX_AND_EIFS_AND_SUBSEQUENT_BINDING_TO_43S | 24.000 | 0.681 | 2.205 | 0.000 | 0.000 | 0.000 |
| REACTOME_NONSENSE_MEDIATED_DECAY_NMD_ | 52.000 | 0.593 | 2.187 | 0.000 | 0.000 | 0.000 |
| REACTOME_SRP_DEPENDENT_COTRANSLATIONAL_PROTEIN_TARGETING_TO_MEMBRANE | 54.000 | 0.577 | 2.133 | 0.000 | 0.000 | 0.002 |
| REACTOME_FANCONI_ANEMIA_PATHWAY | 26.000 | 0.645 | 2.112 | 0.000 | 0.000 | 0.002 |
| REACTOME_INFLUENZA_INFECTION | 74.000 | 0.499 | 1.932 | 0.000 | 0.004 | 0.032 |
| REACTOME_HDR_THROUGH_SINGLE_STRAND_ANNEALING_SSA_ | 23.000 | 0.580 | 1.873 | 0.000 | 0.011 | 0.109 |
| REACTOME_ASSOCIATION_OF_TRIC_CCT_WITH_TARGET_PROTEINS_DURING_BIOSYNTHESIS | 28.000 | 0.540 | 1.791 | 0.000 | 0.034 | 0.308 |
| REACTOME_HDR_THROUGH_HOMOLOGOUS_RECOMBINATION_HRR_ | 39.000 | 0.512 | 1.777 | 0.000 | 0.037 | 0.358 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munz, N.; Cascione, L.; Parmigiani, L.; Tarantelli, C.; Rinaldi, A.; Cmiljanovic, N.; Cmiljanovic, V.; Giugno, R.; Bertoni, F.; Napoli, S. Exon–Intron Differential Analysis Reveals the Role of Competing Endogenous RNAs in Post-Transcriptional Regulation of Translation. Non-Coding RNA 2021, 7, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7020026
Munz N, Cascione L, Parmigiani L, Tarantelli C, Rinaldi A, Cmiljanovic N, Cmiljanovic V, Giugno R, Bertoni F, Napoli S. Exon–Intron Differential Analysis Reveals the Role of Competing Endogenous RNAs in Post-Transcriptional Regulation of Translation. Non-Coding RNA. 2021; 7(2):26. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7020026
Chicago/Turabian StyleMunz, Nicolas, Luciano Cascione, Luca Parmigiani, Chiara Tarantelli, Andrea Rinaldi, Natasa Cmiljanovic, Vladimir Cmiljanovic, Rosalba Giugno, Francesco Bertoni, and Sara Napoli. 2021. "Exon–Intron Differential Analysis Reveals the Role of Competing Endogenous RNAs in Post-Transcriptional Regulation of Translation" Non-Coding RNA 7, no. 2: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7020026