A Comparative Study of the ZnO Growth on Graphene and Graphene Oxide: The Role of the Initial Oxidation State of Carbon

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
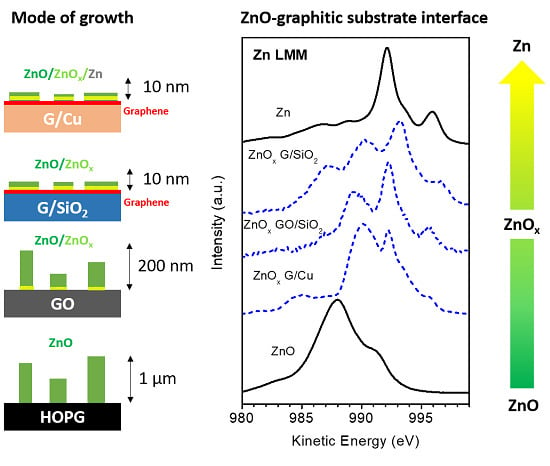
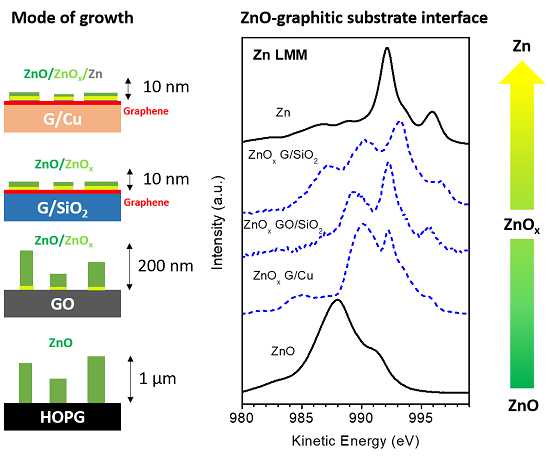
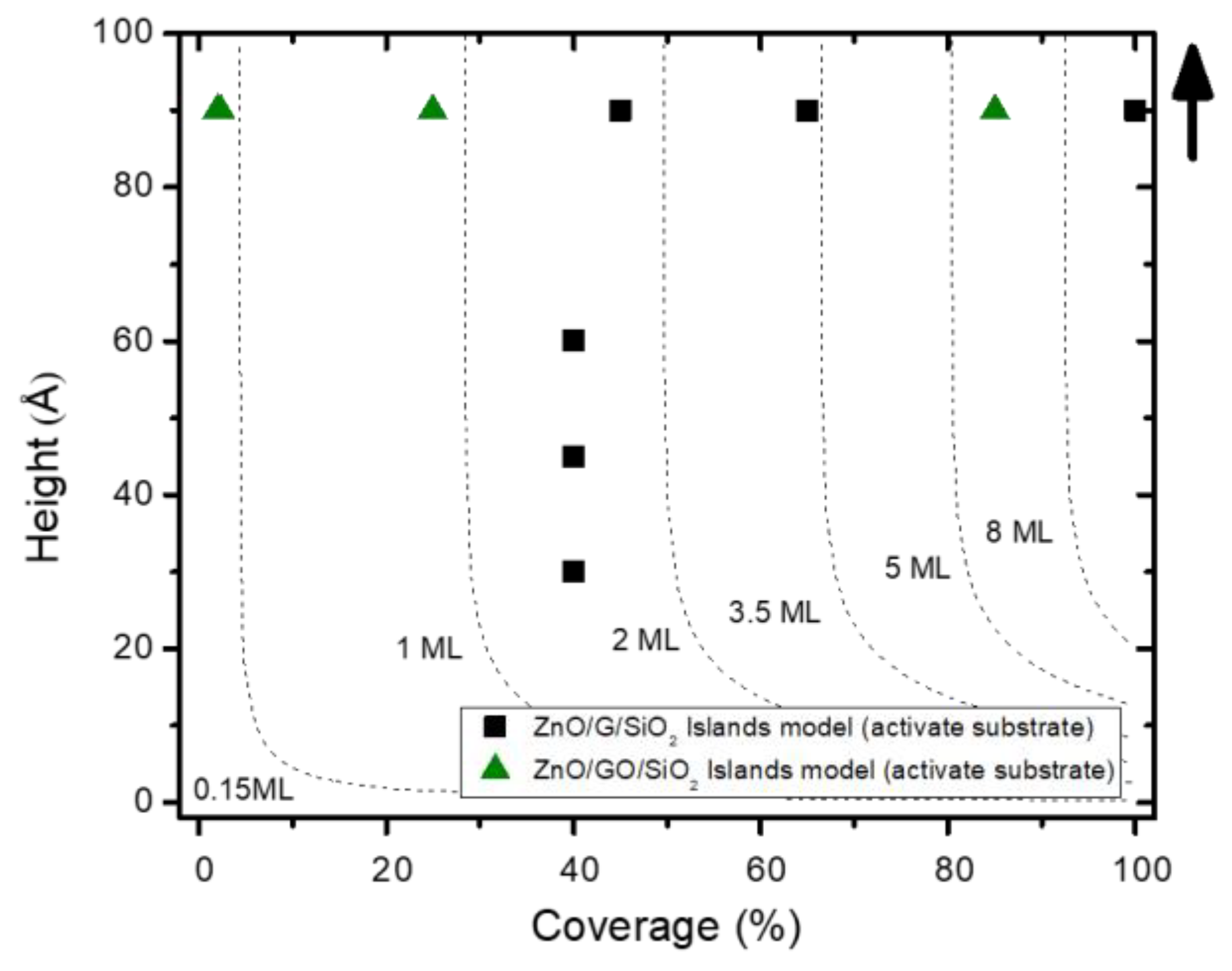
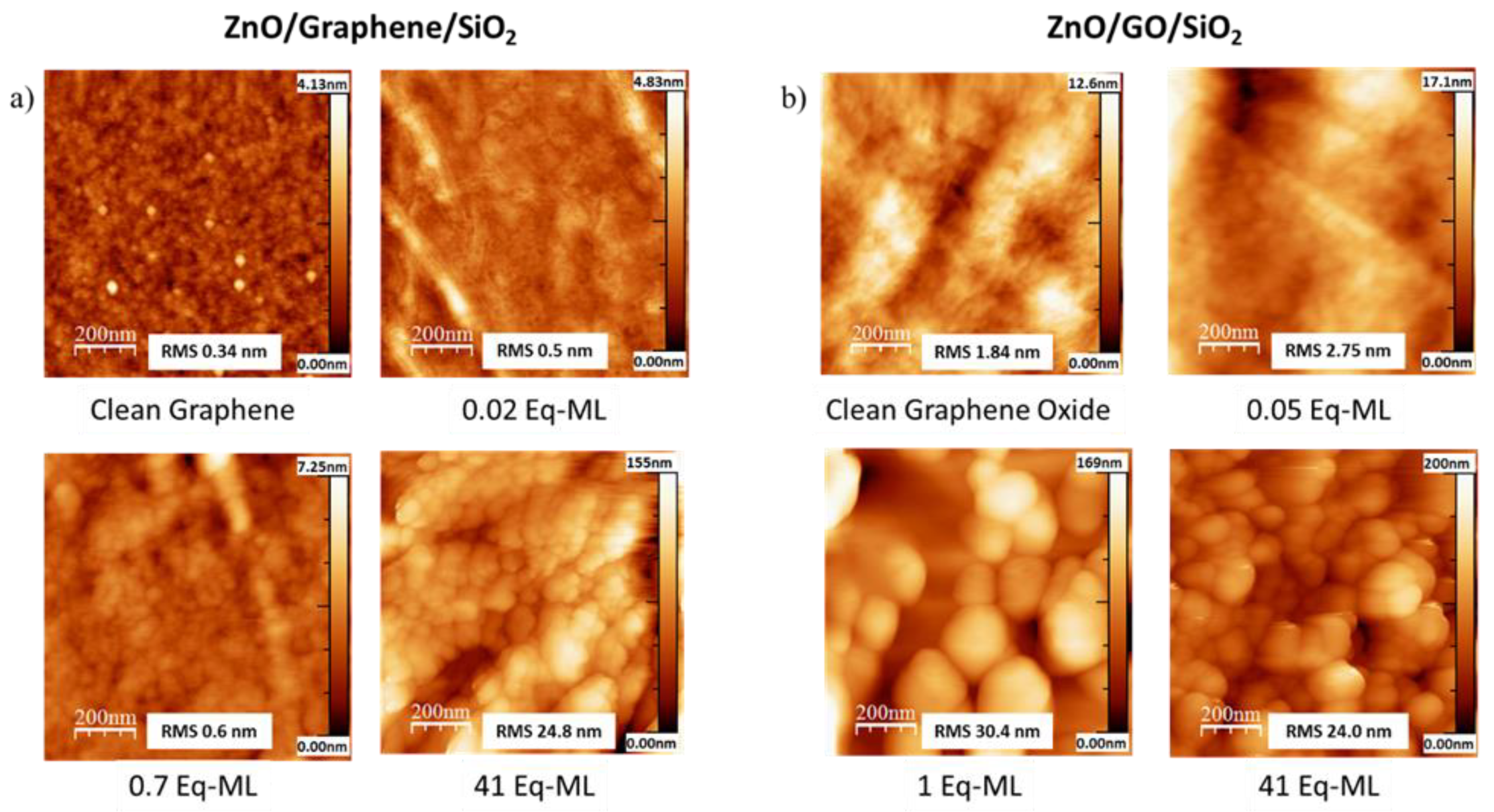
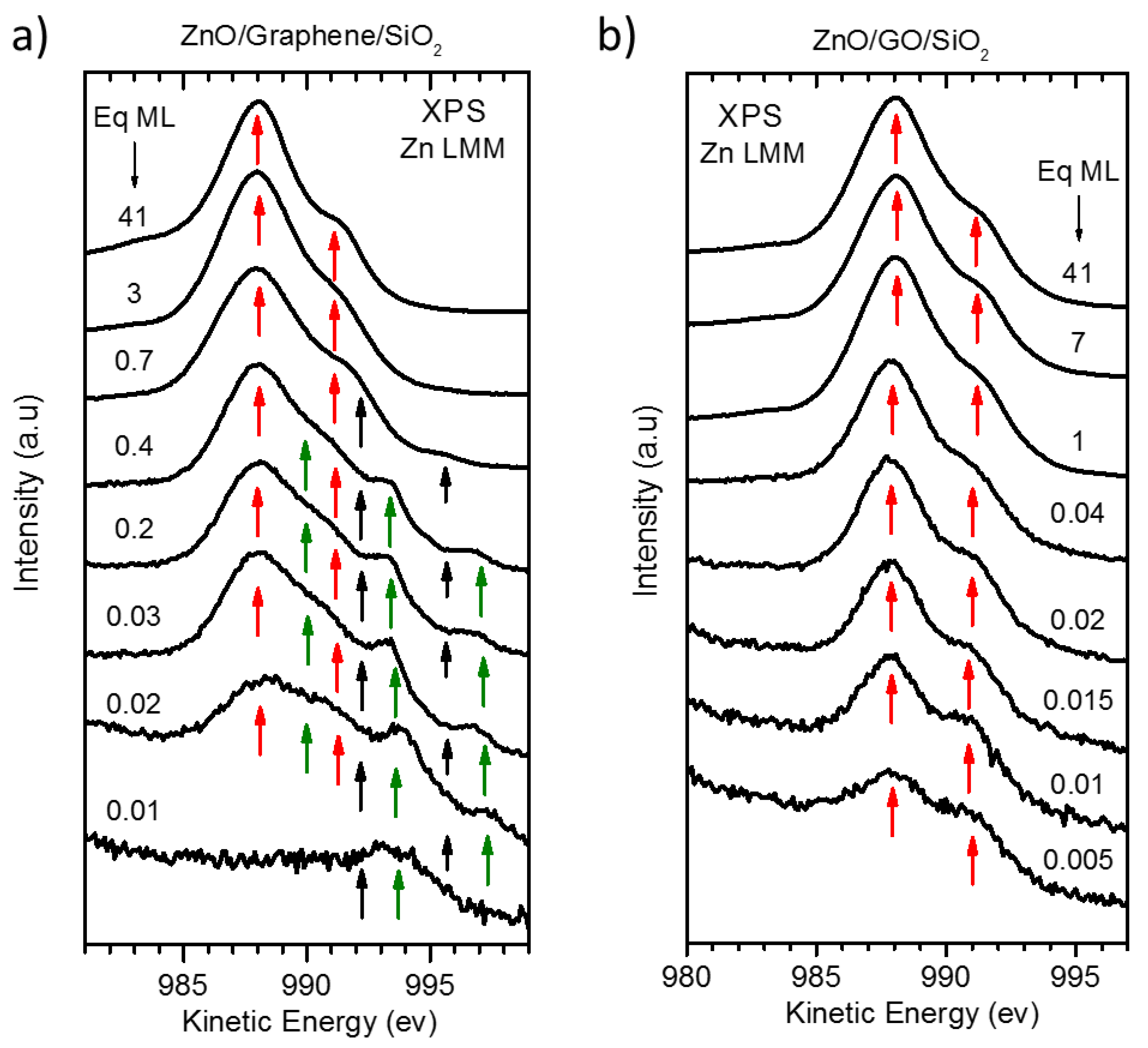
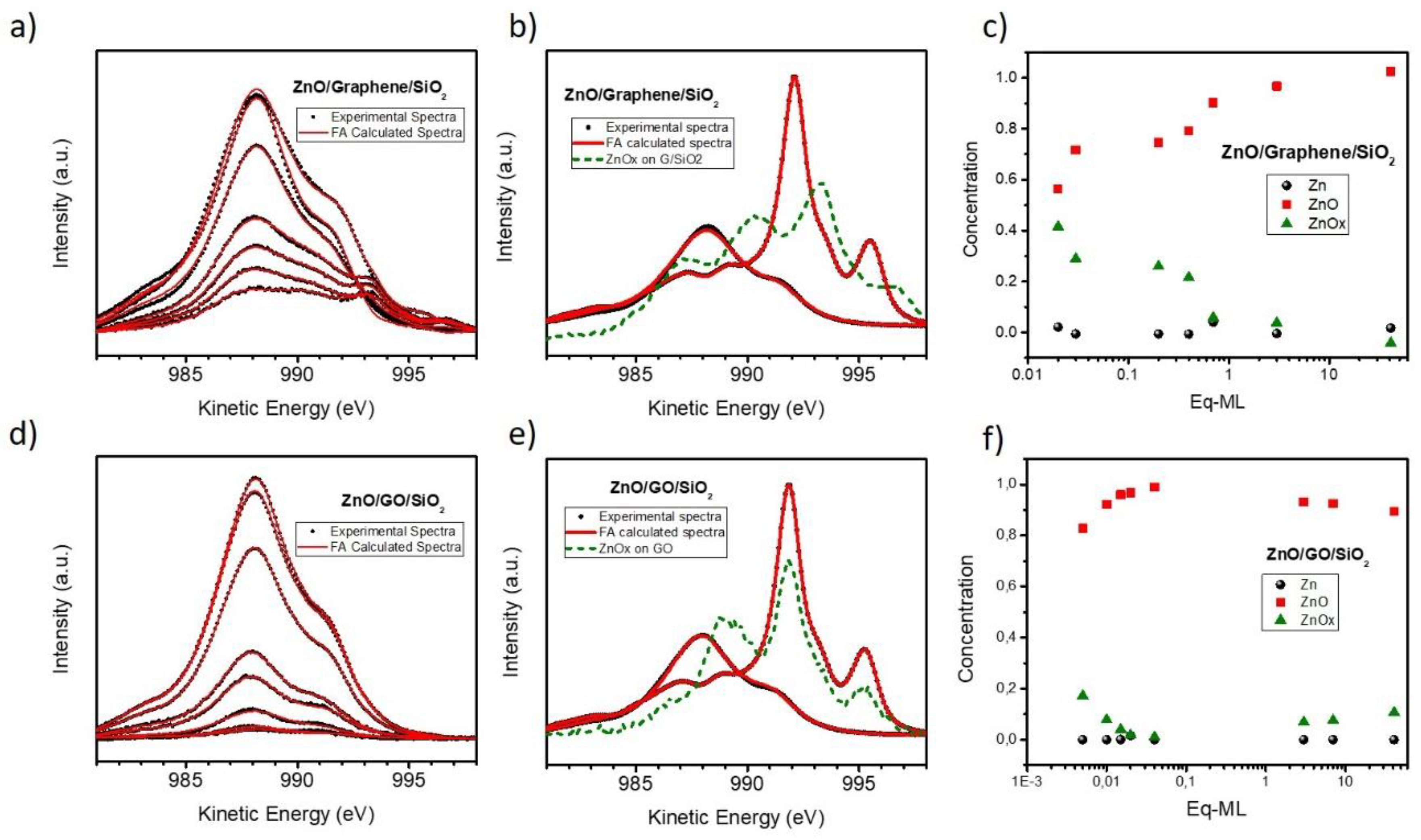
3.1. Quantitative and Morphological Analysis: ZnO Growth Mode
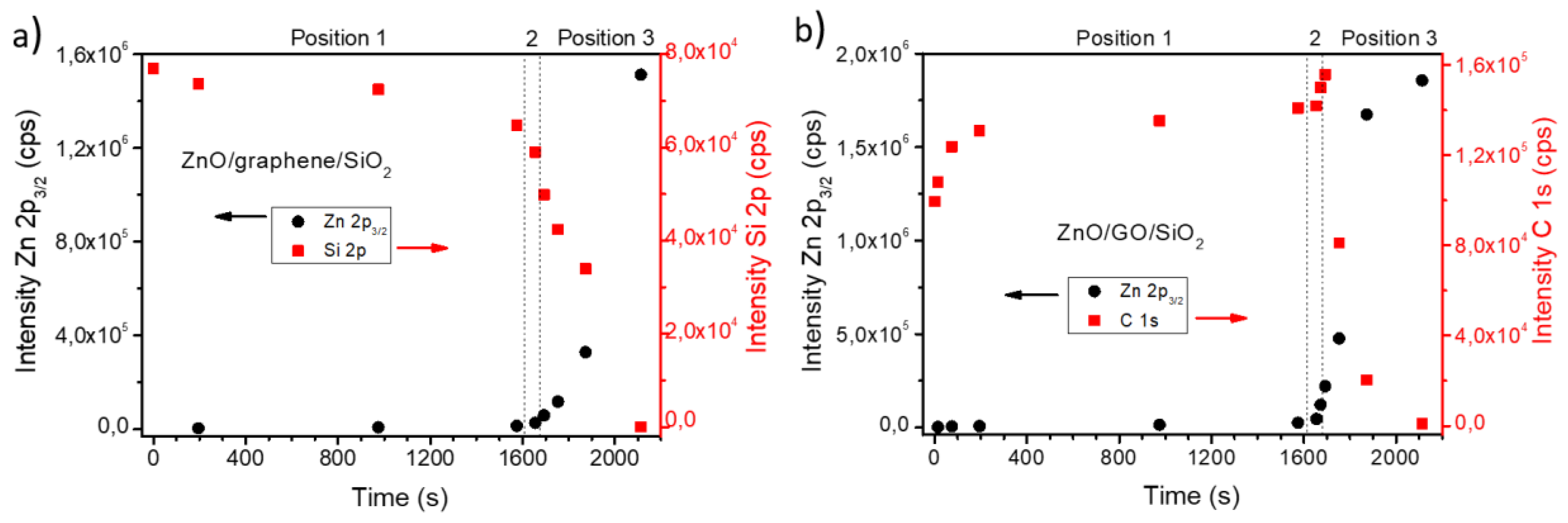
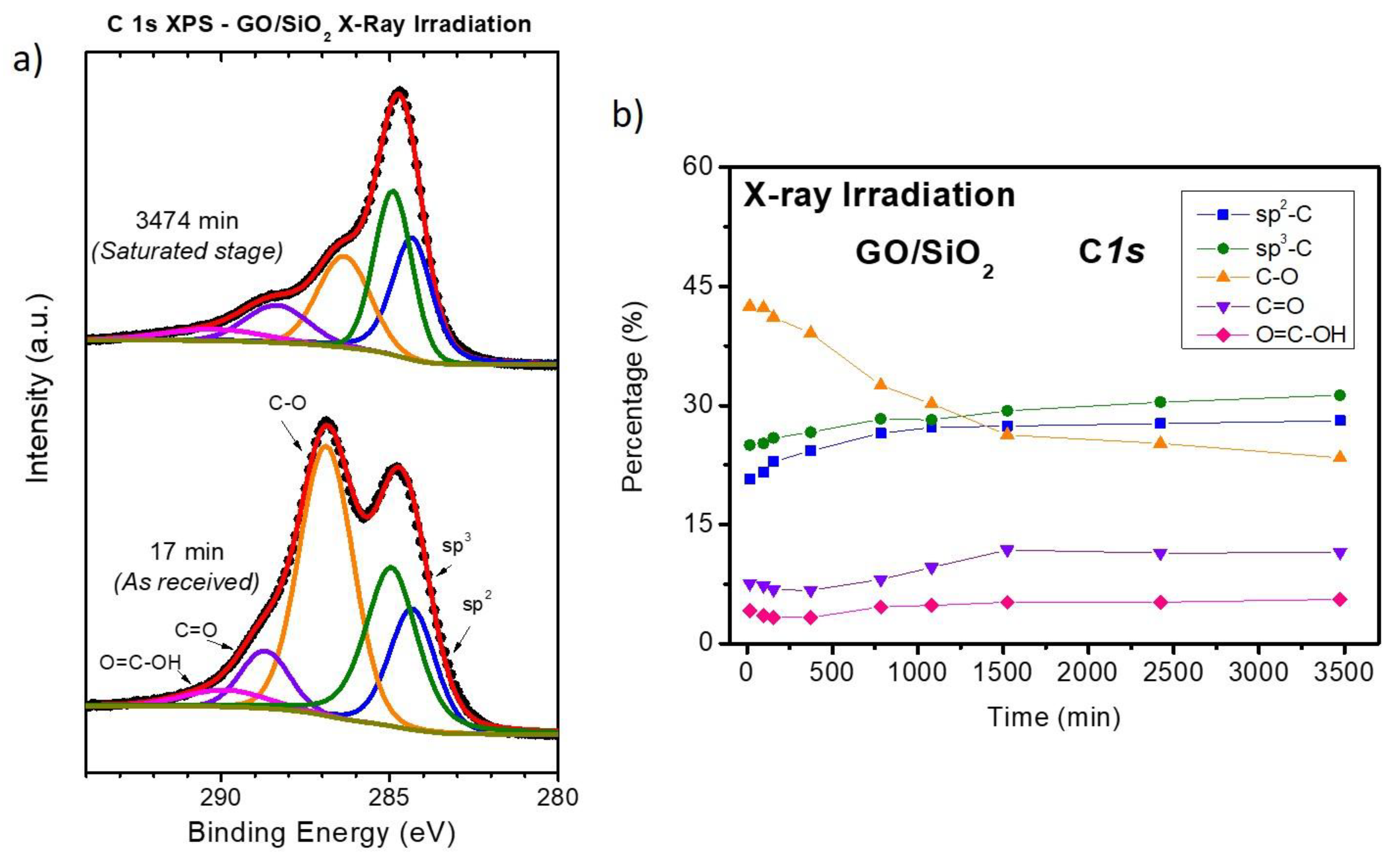
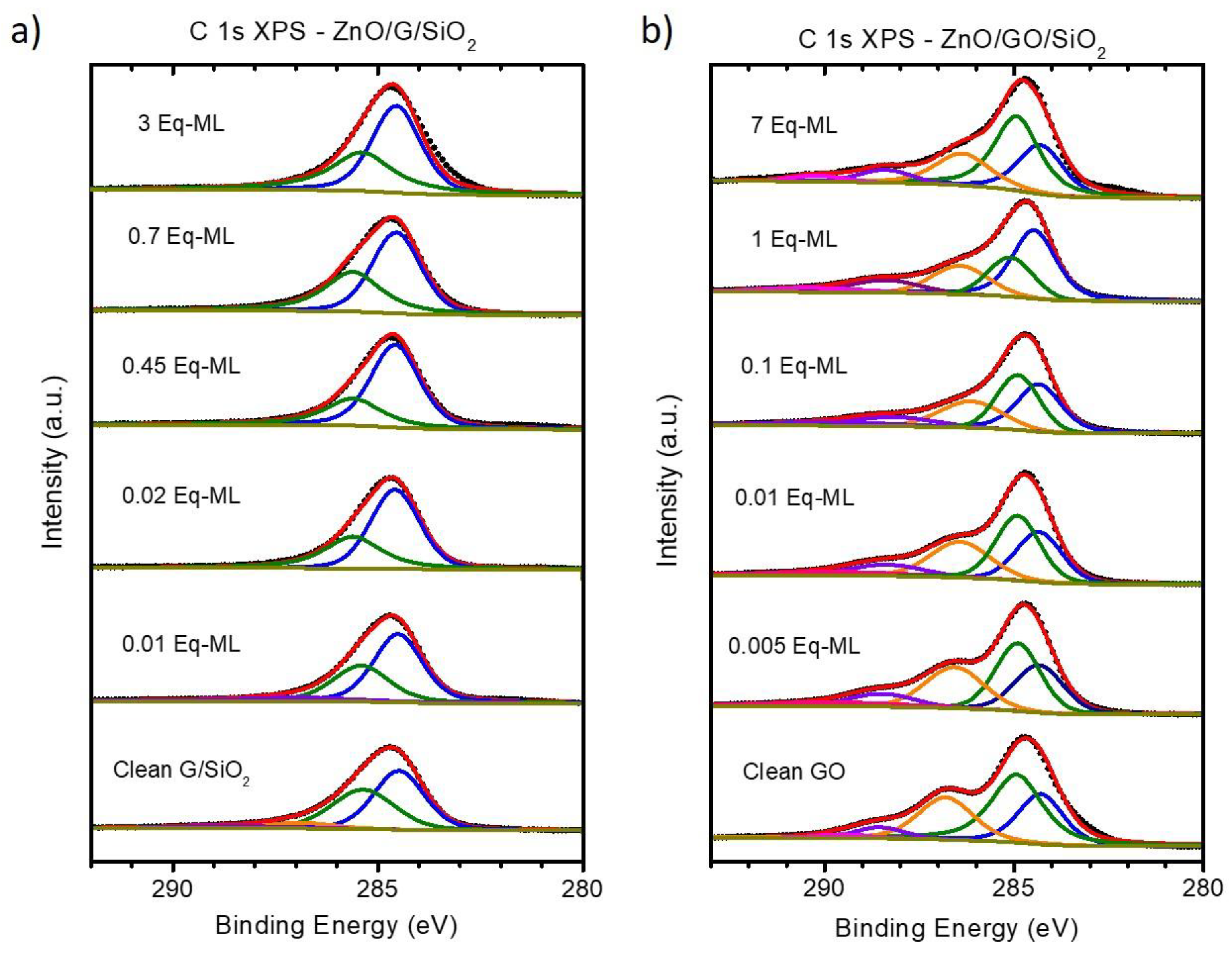
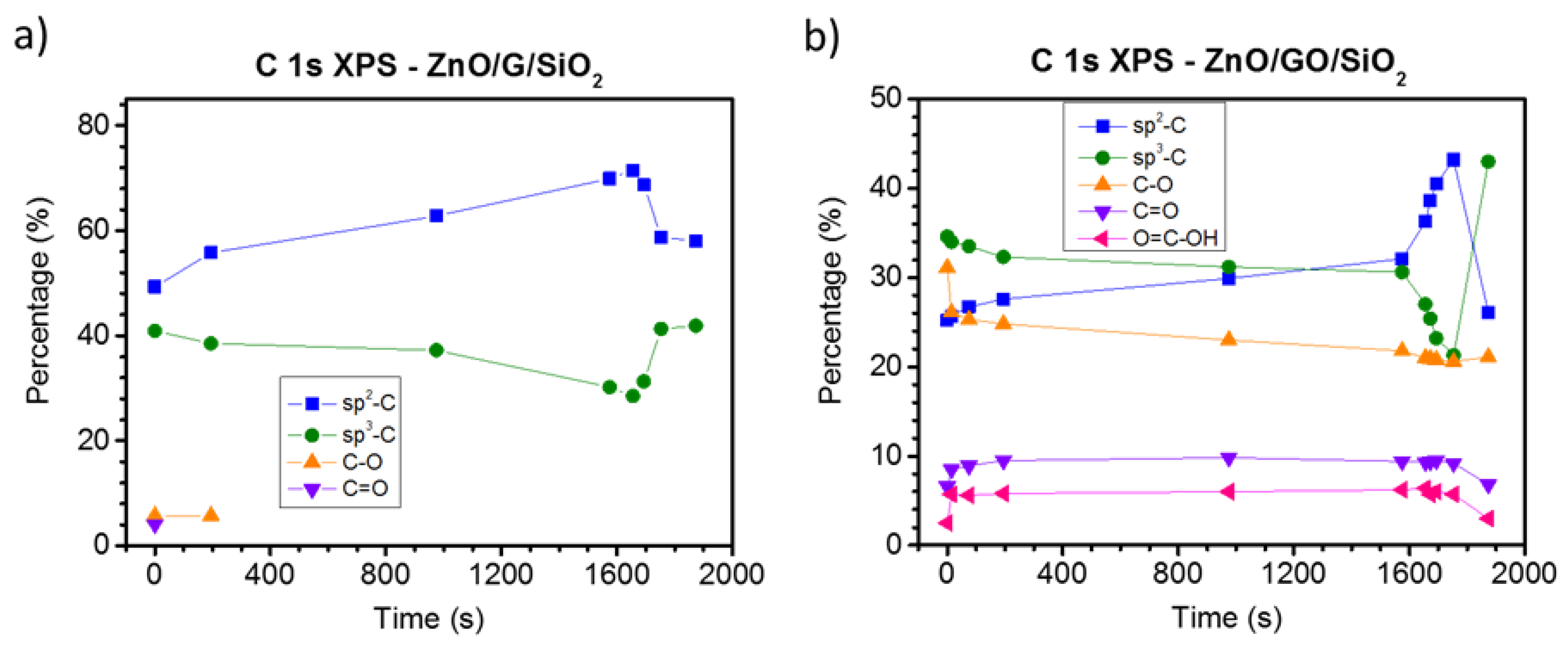
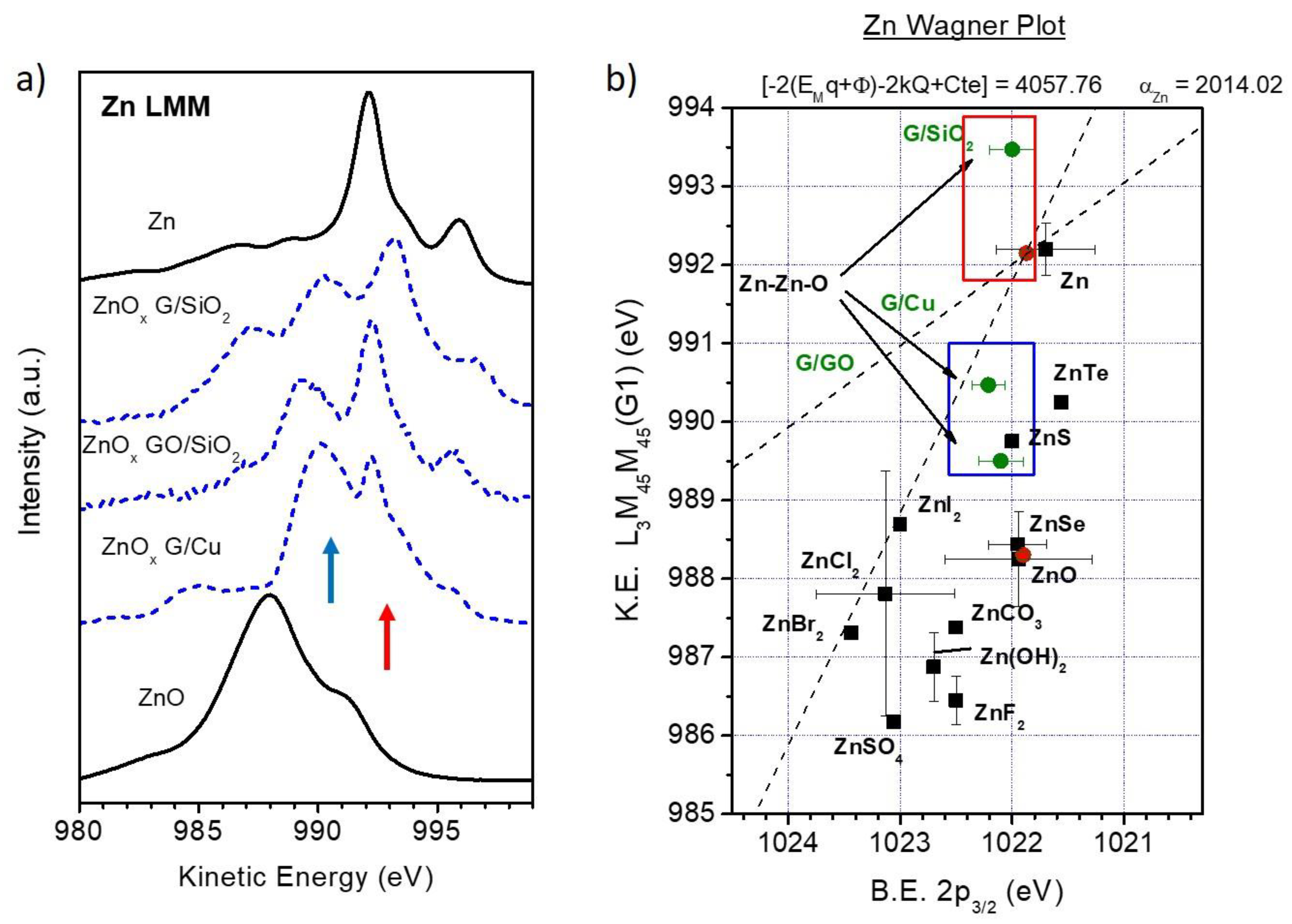
3.2. Chemical Analysis: The Interaction of ZnO with Graphene and GO
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nguyen-Phan, T.D.; Pham, V.H.; Shin, E.W.; Pham, H.D.; Kim, S.; Chung, J.S.; Seung, E.J.K.; Hur, H. The role of graphene oxide content on the adsorption-enhanced photocatalysis of titanium dioxide/graphene oxide composites. Chem. Eng. J. 2011, 170, 226–232. [Google Scholar] [CrossRef]
- Hu, M.; Yao, Z.; Wang, X. Graphene-Based Nanomaterials for Catalysis. Ind. Eng. Chem. Res. 2017, 56, 13, 3477–3502. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Wang, Y.; Hahn, Y.B. Graphene and its derivatives for solar cells application. Nano Energy 2018, 47, 51–65. [Google Scholar] [CrossRef]
- Milić, J.V.; Arora, N.; Dar, M.I.; Zakeeruddin, S.M.; Grätzel, M. Reduced Graphene Oxide as a Stabilizing Agent in Perovskite Solar Cells. Adv. Mater. Interfaces 2018, 5, 1800416. [Google Scholar] [CrossRef]
- Chidembo, A.; Aboutalebi, S.H.; Konstantinov, K.; Salari, M.; Winton, B.; Yamini, S.A.; Nevirkovets, I.P.; Liu, H.K. Globular reduced graphene oxide-metal oxide structures for energy storage applications. Energy Environ. Sci. 2012, 5, 5236–5240. [Google Scholar] [CrossRef] [Green Version]
- Wuab, Z.S.; Zhoua, G.; Yin, L.C.; Ren, W.; Li, F.; Cheng, H.M. Graphene/metal oxide composite electrode materials for energy storage. Nano Energy 2012, 1, 107–131. [Google Scholar] [CrossRef]
- Justino, C.I.L.; Gomes, A.R.; Freitas, A.C.; Duarte, A.C.; Rocha-Santos, T.A.P. Graphene based sensors and biosensors. Trac-Trend Anal. Chem. 2017, 91, 53–66. [Google Scholar] [CrossRef]
- Chiu, N.F.; Fan, S.Y.; Yang, C.D.; Huang, T.Y. Carboxyl-functionalized graphene oxide composites as SPR biosensors with enhanced sensitivity for immunoaffinity detection. Biosens. Bioelectron. 2017, 89, 370–376. [Google Scholar] [CrossRef]
- Han, T.H.; Kim, H.; Kwon, S.J.; Lee, T.W. Graphene-based flexible electronic devices. Mater. Sci. Eng. R-Rep. 2017, 118, 1–43. [Google Scholar] [CrossRef]
- Liang, Y.; Zhao, F.; Cheng, Z.; Deng, Y.; Xiao, Y.; Cheng, H.; Zhang, P.; Huang, Y.; Shao, H.; Qu, L. Electric power generation via asymmetric moisturizing of graphene oxide for flexible, printable and portable electronics. Energy Environ. Sci. 2018, 11, 1730–1735. [Google Scholar] [CrossRef]
- Edalati, K.; Shakiba, A.; Vahdati-Khaki, J.; Zebarjad, S.M. Low-temperature hydrothermal synthesis of ZnO nanorods: Effects of zinc salt concentration, various solvents and alkaline mineralizers. Mater. Res. Bull. 2016, 74, 374–379. [Google Scholar] [CrossRef]
- Sphanel, L.; Anderson, M.A. Semiconductor clusters in the sol-gel process: Quantized aggregation, gelation, and crystal growth in concentrated zinc oxide colloids. J. Am. Chem. Soc. 1991, 113, 2826–2833. [Google Scholar] [CrossRef]
- Hasnidawani, J.N.; Azlina, H.N.; Norita, H.; Bonnia, N.N.; Ratim, S.; Ali, E.S. Synthesis of ZnO Nanostructures Using Sol-Gel Method. Procedia Chem. 2016, 19, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, D.Y.; Lee, S. Low-Power Graphene/ZnO Schottky UV Photodiodes with Enhanced Lateral Schottky Barrier Homogeneity. Nanomaterials 2019, 9, 799. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Li, Z. ZnO thin films produced by magnetron sputtering. Ceram. Int. 2004, 30, 1155–1159. [Google Scholar] [CrossRef]
- Al Asmar, R.; Zaouk, D.; Bahouth, P.; Podleki, J.; Foucaran, A. Characterization of electron beam evaporated ZnO thin films and stacking ZnO fabricated by e-beam evaporation and RF magnetron sputtering for the realization of resonators. Microelectron. Eng. 2006, 83, 393–398. [Google Scholar] [CrossRef]
- Özgür, Ü.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doğan, S.; Avrutin, V.; Cho, S.J.; Morkoç, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98, 041301. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Gilmore, C.M.; Horwitz, J.S.; Piqué, A.; Murata, H.; Kushto, G.P.; Schlaf, R.; Kafafi, Z.H.; Chrisey, D.B. Transparent conducting aluminum-doped zinc oxide thin films for organic light-emitting devices. Appl. Phys. Lett. 2000, 76, 259. [Google Scholar] [CrossRef]
- Rousset, J.; Saucedo, E.; Lincot, D. Extrinsic Doping of Electrodeposited Zinc Oxide Films by Chlorine for Transparent Conductive Oxide Applications. Chem. Mater. 2009, 21, 3, 534–540. [Google Scholar] [CrossRef]
- Hoffman, R.L. ZnO-channel thin-film transistors: Channel mobility. J. Appl. Phys. 2004, 95, 5813. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Xu, R.; Tang, K.; Shi, W.; Xia, Y. Growth of p-type ZnO films and fabrication of ZnO photodiode-based UV detectors. Semicond. Sci. Technol. 2009, 24, 075025. [Google Scholar] [CrossRef]
- Sekine, N.; Chou, C.H.; Kwan, W.L.; Yang, Y. ZnO nano-ridge structure and its application in inverted polymer solar cell. Org. Electron. 2009, 10, 1473–1477. [Google Scholar] [CrossRef]
- Zhu, L.; Zeng, W. Room-temperature gas sensing of ZnO-based gas sensor: A review. Sens. Actuator A 2017, 267, 242–261. [Google Scholar] [CrossRef]
- Mattevi, C.; Kima, H.; Chhowalla, M. A review of chemical vapour deposition of graphene on copper. J. Mater. Chem. 2011, 21, 3324. [Google Scholar] [CrossRef]
- Chen, J.H.; Jang, C.; Xiao, S.; Ishigami, M.; Fuhrer, M.S. Intrinsic and extrinsic performance limits of graphene devices on SiO2. Nat. Nanotechnol. 2008, 3, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, D.G.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- Tavakoli, M.M.; Aashuri, H.; Simchi, A.; Fan, Z. Hybrid zinc oxide/graphene electrodes for depleted heterojunction colloidal quantum-dot solar cells. Phys. Chem. Chem. Phys. 2015, 17, 24412–24419. [Google Scholar] [CrossRef]
- Zhang, T.F.; Wu, G.A.; Wang, J.Z.; Yu, Y.Q.; Zhang, D.Y.; Wang, D.D.; Jiang, J.B.; Wang, J.M.; Luo, L.B. A sensitive ultraviolet light photodiode based on graphene-on-zinc oxide Schottky junction. Nanophotonics 2017, 6, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Zhao, C.; Xing, L.; Xiang, J.; Cui, L.; Jiao, J.; Sai, H.; Li, Z.; Li, F. Formation of uniform reduced graphene oxide films on modified PET substrates using drop-casting method. Particuology 2014, 17, 66–73. [Google Scholar] [CrossRef]
- Puah, P.Y.; Moh, P.Y.; Lee, P.C.; How, S.E. Spin-coated graphene oxide as a biomaterial for Wharton’s Jelly derived mesenchymal stem cell growth: A preliminary study. Adv. Perform. Mater. 2018, 33, 835–843. [Google Scholar] [CrossRef]
- Jadhav, S.T.; Rajoba, S.J.; Patil, S.A.; Han, S.H.; Jadhav, L.D. Temperature-Dependent Photoluminescence of Graphene Oxide. J. Electron. Mater. 2016, 45, 379–385. [Google Scholar] [CrossRef]
- Holm, A.; Wrasman, C.J.; Kao, K.C.; Riscoe, A.R.; Cargnello, M.; Frank, C.W. Langmuir–Blodgett Deposition of Graphene Oxide—Identifying Marangoni Flow as a Process that Fundamentally Limits Deposition Control. Langmuir 2018, 34, 33, 9683–9691. [Google Scholar] [CrossRef]
- Jilani, S.M.; Banerji, P. Graphene Oxide−Zinc Oxide Nanocomposite as Channel Layer for Field Effect Transistors: Effect of ZnO Loading on Field Effect Transport. Appl. Mater. Interfaces 2014, 6, 16941–16948. [Google Scholar] [CrossRef]
- Kumar, B.; Lee, K.Y.; Park, H.K.; Chae, S.J.; Lee, Y.H.; Kim, S.W. Controlled Growth of Semiconducting Nanowire, Nanowall, and Hybrid Nanostructures on Graphene for Piezoelectric Nanogenerators. ACS Nano 2011, 5, 4197–4204. [Google Scholar] [CrossRef]
- Zheng, L.; Cheng, X.; Cao, D.; Wang, G.; Wang, Z.; Xu, D.; Xia, C.; Shen, L.; Yu, Y.; Shen, D. Improvement of Al2O3 Films on Graphene Grown by Atomic Layer Deposition with Pre-H2O Treatment. ACS Appl. Mater. Interfaces 2014, 6, 7014–7019. [Google Scholar] [CrossRef] [PubMed]
- Schilirò, E.; Nigro, R.L.; Roccaforte, F.; Deretzis, I.; La Magna, A.; Armano, A.; Agnello, S.; Pecz, B.; Ivanov, I.G.; Yakimova, R.; et al. Seed-Layer-Free Atomic Layer Deposition of Highly Uniform Al2O3 Thin Films onto Monolayer Epitaxial Graphene on Silicon Carbide. Adv. Mater. Interfaces 2019, 6, 1900097. [Google Scholar] [CrossRef]
- Hong, H.K.; Jo, J.; Hwang, D.; Lee, J.; Kim, N.Y.; Son, S.; Kim, J.H.; Jin, M.J.; Jun, Y.C.; Erni, R.; et al. Atomic Scale Study on Growth and Heteroepitaxy of ZnO Monolayer on Graphene. Nano Lett. 2017, 17, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Morales, C.; Black, A.; Urbanos, F.J.; Granados, D.; Méndez, J.; del Campo, A.; Yubero, F.; Soriano, L. Study of the Interface of the Early Stages of Growth under Quasi-Equilibrium Conditions of ZnO on Graphene/Cu and Graphite. Adv. Mater. Interfaces 2019, 6, 1801689. [Google Scholar] [CrossRef]
- Morales, C.; Urbanos, F.J.; del Campo, A.; Leinen, D.; Granados, D.; Prieto, P.; Aballe, L.; Foerster, M.; Soriano, L. Chemical and electronic inhomogeneities of Graphene on polycrystalline Cu and its influence on the early stages of growth of ZnO. Carbon 2020. Under review. [Google Scholar]
- Morales, C.; Urbanos, F.J.; del Campo, A.; Leinen, D.; Granados, D.; Rodríguez, M.A.; Soriano, L. Electronic Decoupling of Graphene from Copper Induced by Deposition of ZnO: A Complex Substrate/Graphene/Deposit/Environment Interaction. Adv. Mater. Interfaces 2020, 1902062. [Google Scholar] [CrossRef]
- Black, A.; Jiménez, F.; Benardo-Gavito, R.; Casado, S.; Granados, D.; Vázquez de Parga, A.L. Growth and characterization of 7,7,8,8-tetracyano-quinodimethane crystals on chemical vapor deposition graphene. J. Cryst. Growth 2016, 453, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Fernández, D.; Méndez, J.; Yubero, F.; Domínguez-Cañizares, G.; Gutiérrez, A.; Soriano, L. Study of the early stages of growth of Co oxides on oxide substrates. Surf. Interface Anal. 2014, 46, 975–979. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D.; Chastain, J. (Eds.) Handbook of X-ray Photoelectron Spectroscopy, Perkin–Elmer Corporation Physical Electronics Division; Perkin-Elmer Corporation, Physical Electronics Division: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Tanuma, S.; Powell, C.J.; Penn, D.R. Proposed formula for electron inelastic mean free paths based on calculations for 31 materials. Surf. Sci. Lett. 1987, 192, L849–L857. [Google Scholar] [CrossRef]
- Touggard, S. Quases Software. Available online: www.QUASES.com (accessed on 31 July 2017).
- Horcas, I.; Fernández, R.; Gómez Rodríguez, J.M.; Colchero, J.; Gómez Herrero, J.; Baró, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. [Google Scholar] [CrossRef]
- Mendialdu, J.; Casanov, R.; Barbaux, Y. XPS studies of V2O5, V6O13, VO2 and V2O3. J. Electron Spectrosc. Relat. Phenom. 1995, 71, 249–261. [Google Scholar] [CrossRef]
- Baer, D.R.; Engelhard, M.H.; Lea, A.S. Introduction to Surface Science Spectra data on electron and x-ray damage: Sample degradation during XPS and AES measurements. Surf. Sci. Spectra 2003, 10, 47. [Google Scholar] [CrossRef]
- Buchwalter, L.P.; Czornyj, G. Poly (methyl methacrylate) degradation during x-ray photoelectron spectroscopy analysis. J. Vac. Sci. Technol. A 1990, 8, 781. [Google Scholar] [CrossRef]
- Sato, M.; Furusawa, T.; Hotta, T.; Watanabe, H.; Suzuki, N. Effect of oxide thickness on the degradation of organic silane monolayers on silicon wafer surface during XPS measurement. Surf. Interface Anal. 2006, 38, 838–841. [Google Scholar] [CrossRef]
- Recek, N.; Primc, G.; Vesel, A.; Mozetic, M.; Avila, J.; Razado-Colambo, I.; Asensio, M. Degradation of Albumin on Plasma-Treated Polystyrene by Soft X-ray Exposure. Polymers 2016, 8, 244. [Google Scholar] [CrossRef] [Green Version]
- Blanton, T.N.; Majumdar, D. Characterization of X-ray irradiated graphene oxide coatings using X-ray diffraction, X-ray photoelectron spectroscopy, and atomic force microscopy. Powder Diffr. 2013, 28, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Silipigni, L.; Cutroneo, M.; Salvato, G.; Torrisi, L. In-situ soft X-ray effects on graphene oxide films. Radiat. Eff. Defects Solids 2018, 173, 740–750. [Google Scholar] [CrossRef]
- Yang, D.; Velamakanni, A.; Bozoklu, G.; Park, S.; Stoller, M.; Piner, R.D.; Stankovich, S.; Jung, I.; Field, D.A.; Ventrice, C.A., Jr.; et al. Chemical analysis of graphene oxide films after heat and chemical treatments by X-ray photoelectron and Micro-Raman spectroscopy. Carbon 2009, 47, 145–152. [Google Scholar] [CrossRef]
- Haubner, K.; Murawski, J.; Olk, P.; Eng, L.M.; Ziegler, C.; Adolphi, B.; Jaehne, E. The route to functional graphene oxide. Chem. Phys. Chem. 2010, 11, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Hontoria-Lucas, C.; López-Peinado, A.J.; López-González, J.D.; Rojas-Cervantes, M.L.; Martín-Aranda, R.M. Study of oxygen-containing groups in a series of graphite oxides: Physical and chemical characterization. Carbon 1995, 33, 1585–1592. [Google Scholar] [CrossRef]
- Webb, M.J.; Palmgren, P.; Pal, P.; Karis, O.; Grennberg, H. A simple method to produce almost perfect graphene on highly oriented pyrolytic graphite. Carbon 2011, 49, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Cunge, G.; Ferrah, D.; Petit-Etienne, C.; Davydova, A.; Okuno, H.; Kalita, D.; Bouchiat, V.; Renault, O. Dry efficient cleaning of poly-methylmethacrylate residues from graphene with high-density H2 and H2-N2 plasmas. J. Appl. Phys. 2015, 118, 123302. [Google Scholar] [CrossRef]
- Morales, C.; Díaz-Fernández, D.; Mossanek, R.J.O.; Abbate, M.; Méndez, J.; Pérez-Dieste, V.; Escudero, C.; Rubio-Zuazo, J.; Prieto, P.; Soriano, L. Controlled ultra-thin oxidation of graphite promoted by cobalt oxides: Influence of the initial 2D CoO wetting layer. Appl. Surf. Sci. 2020, 509, 145118. [Google Scholar] [CrossRef]
- Rössler, N.; Kotsis, K.; Staemmler, V. Ab initio calculations for the Zn 2s and 2p core level binding energies in Zn oxo compounds and ZnO. Phys. Chem. Chem. Phys. 2006, 8, 697. [Google Scholar] [CrossRef] [PubMed]
- Leontiev, S.A.; Koshcheev, S.V.; Devyatov, V.G.; Cherkashin, A.E.; Mikheeva, P. Detailed XPS and UPS studies of the band structure of zinc oxide. J. Struct. Chem. 1997, 38, 725. [Google Scholar] [CrossRef]
- Mikheeva, P.; Zhidomirov, G.M.; Ruzankin, S.F.; Leontiev, S.A.; Devyatov, V.G.; Koshcheev, S.V.; Cherkashin, A.E. Modeling the photoelectron spectra of the valence O2p-band of zinc oxide by the Xα-scattered wave method. J. Struct. Chem. 1997, 38, 732. [Google Scholar] [CrossRef]
- Deroubaix, G.; Marcus, P. X-ray photoelectron spectroscopy analysis of copper and zinc oxides and sulphides. Surf. Interface Anal. 1992, 18, 39–46. [Google Scholar] [CrossRef]
- Aronniemi, M.; Sainio, J.; Lahtinen, J. Aspects of using the factor analysis for XPS data interpretation. Surf. Sci. 2007, 601, 479–489. [Google Scholar] [CrossRef]
- Measurement Services Division of the National Institute of Standards and Technology (NIST), Wagner Plot. Available online: https://srdata.nist.gov/xps/main_search_menu.aspx (accessed on 5 June 2018).
- Wagner, C.D.; Joshi, A. The auger parameter, its utility and advantages: A review. J. Electron Spectrosc. Relat. Phenom. 1988, 47, 283. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Alvarez, M.A.; Morales, C.; Méndez, J.; del Campo, A.; Urbanos, F.J.; Díaz, A.; Reséndiz, L.; Flege, J.I.; Granados, D.; Soriano, L. A Comparative Study of the ZnO Growth on Graphene and Graphene Oxide: The Role of the Initial Oxidation State of Carbon. C 2020, 6, 41. https://0-doi-org.brum.beds.ac.uk/10.3390/c6020041
Gomez-Alvarez MA, Morales C, Méndez J, del Campo A, Urbanos FJ, Díaz A, Reséndiz L, Flege JI, Granados D, Soriano L. A Comparative Study of the ZnO Growth on Graphene and Graphene Oxide: The Role of the Initial Oxidation State of Carbon. C. 2020; 6(2):41. https://0-doi-org.brum.beds.ac.uk/10.3390/c6020041
Chicago/Turabian StyleGomez-Alvarez, Miguel Angel, Carlos Morales, Javier Méndez, Adolfo del Campo, Fernando J. Urbanos, Aarón Díaz, Luis Reséndiz, Jan Ingo Flege, Daniel Granados, and Leonardo Soriano. 2020. "A Comparative Study of the ZnO Growth on Graphene and Graphene Oxide: The Role of the Initial Oxidation State of Carbon" C 6, no. 2: 41. https://0-doi-org.brum.beds.ac.uk/10.3390/c6020041