
Transcriptomic Analysis of Staphylococcus equorum KM1031, Isolated from the High-Salt Fermented Seafood Jeotgal, under Salt Stress


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Culture Conditions
2.2. Extraction, Purification, Sequencing, and Analysis of RNA from S. equorum
2.3. Genomic Analysis of S. equorum
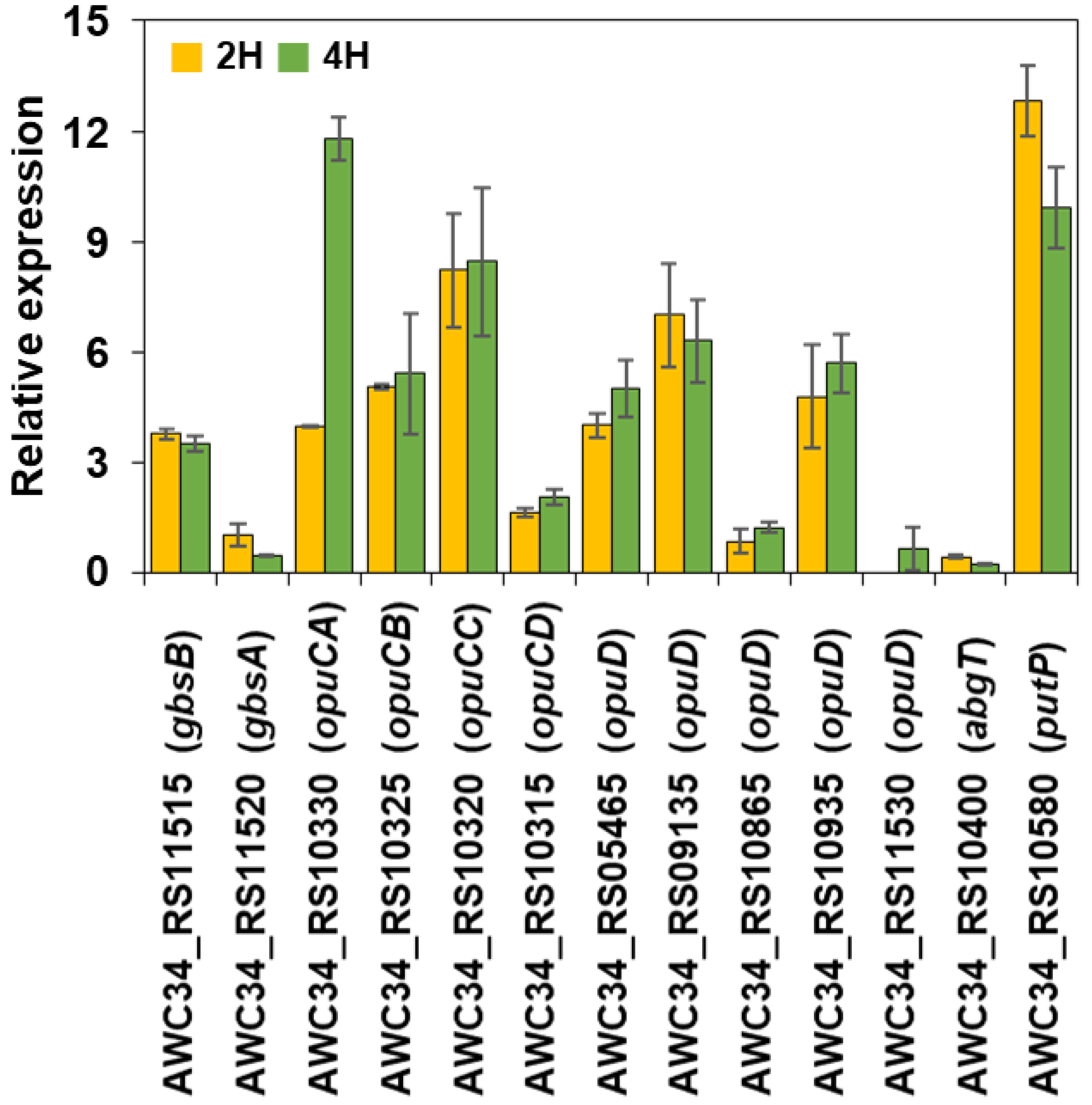
2.4. Quantitative Real-Time RT-PCR (qRT-PCR)
3. Results and Discussion
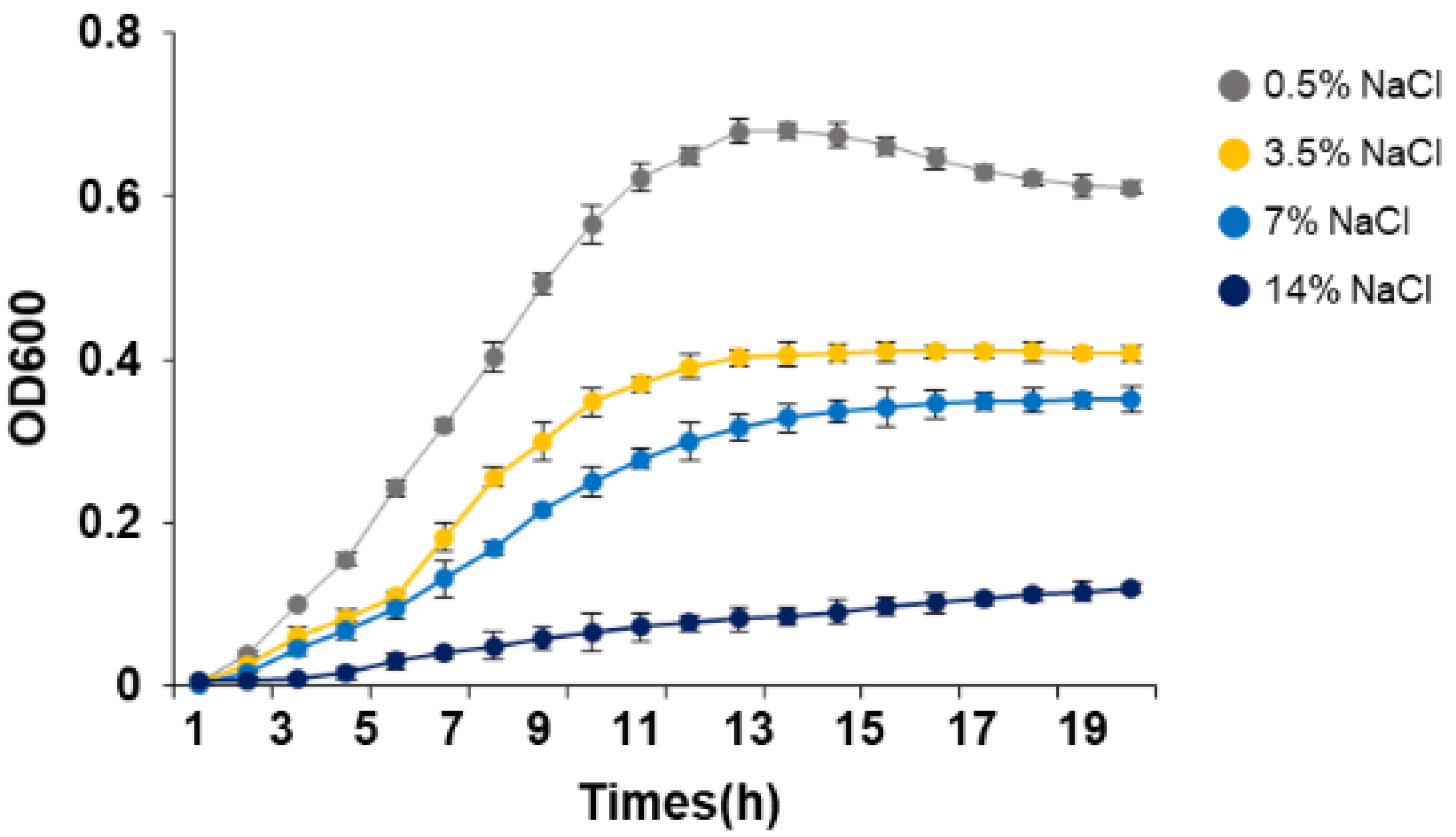
3.1. Growth of S. equorum under Salt Stress
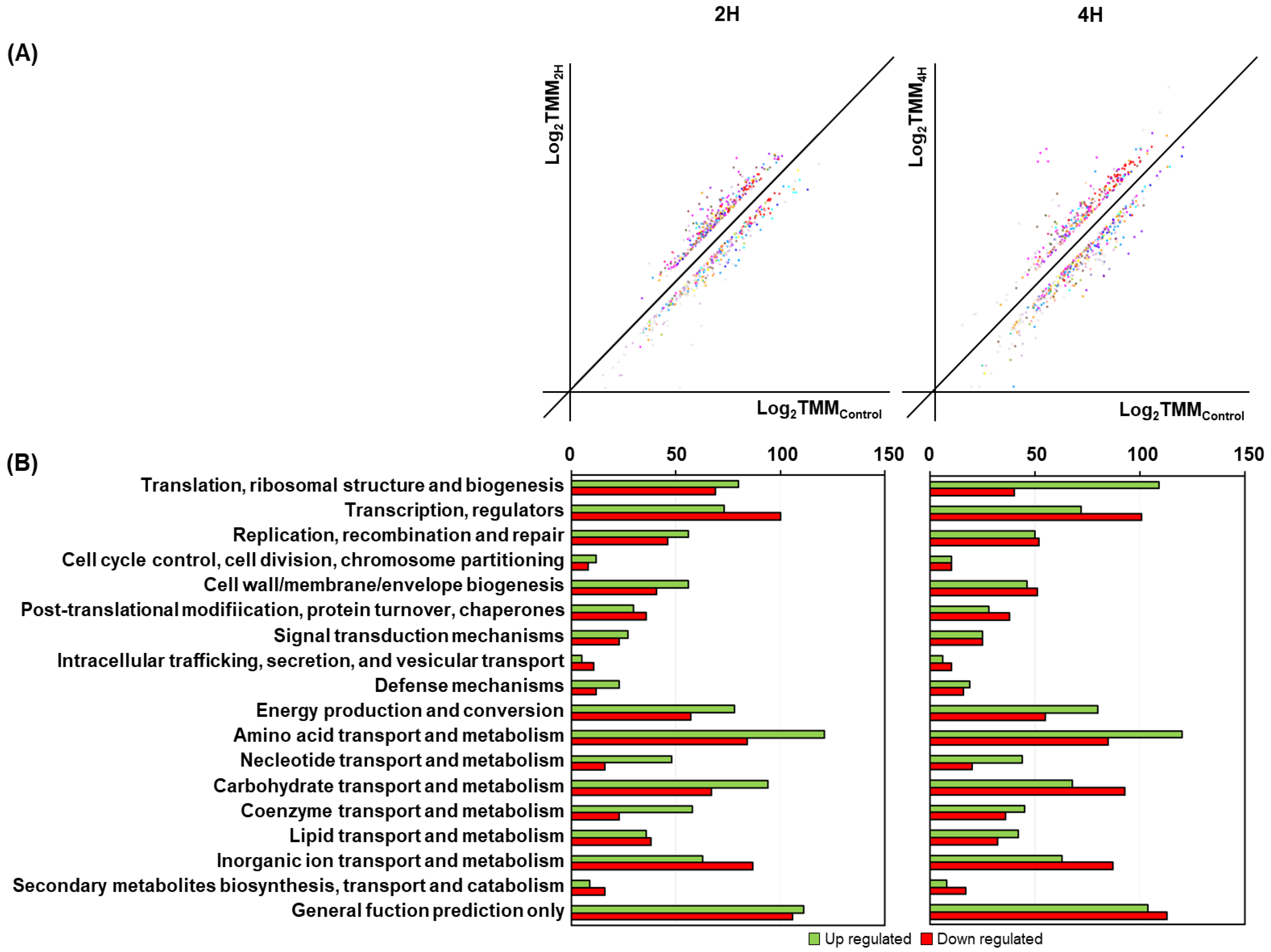
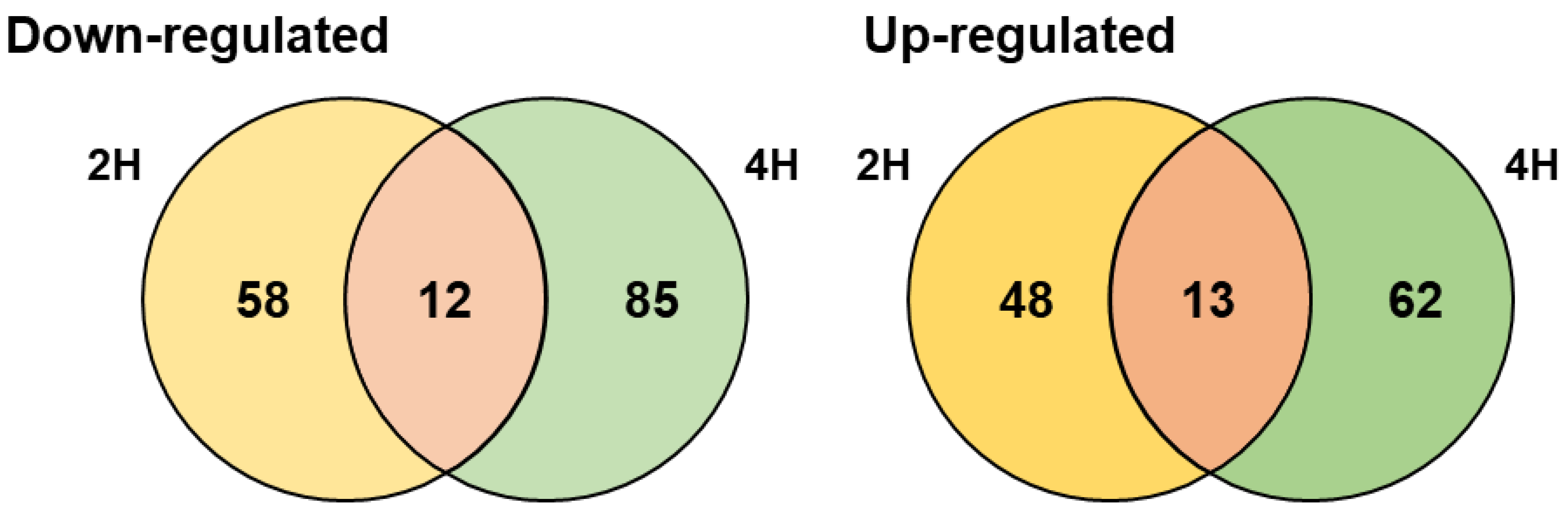
3.2. Comprehensive Transcriptomic Analysis of S. equorum under Salt Stress
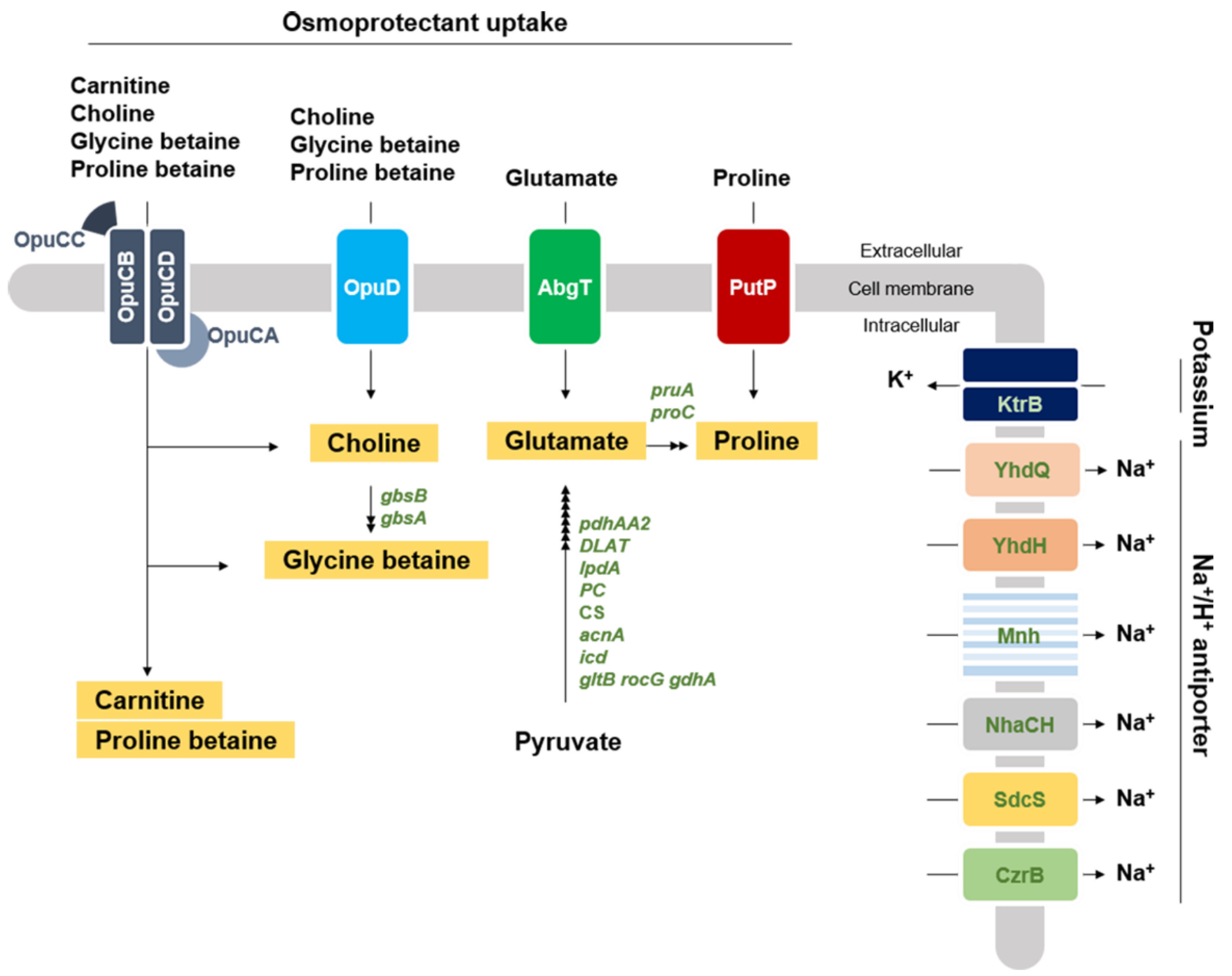
3.3. Genomic Analysis of Response of S. equorum to Salt
3.3.1. Osmoprotectant Uptake
3.3.2. Membrane Ion Transport
3.3.3. Osmoprotectant Synthesis
3.3.4. Regulation of the Opu System and Glycine Betaine Synthesis
3.4. Validation of RNA-Seq Data by qRT-PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marty, E.; Buchs, J.; Eugster-Meier, E.; Lacroix, C.; Meile, L. Identification of staphylococci and dominant lactic acid bacteria in spontaneously fermented Swiss meat products using PCR-RFLP. Food Microbiol. 2012, 29, 157–166. [Google Scholar] [CrossRef]
- Bockelmann, W.; Willems, K.P.; Neve, H.; Heller, K.H. Cultures for the ripening of smear cheeses. Int. Dairy J. 2005, 15, 719–732. [Google Scholar] [CrossRef]
- Corbiere Morot-Bizot, S.; Leroy, S.; Talon, R. Staphylococcal community of a small unit manufacturing traditional dry fermented sausages. Int. J. Food Microbiol. 2006, 108, 210–217. [Google Scholar] [CrossRef]
- Blaiotta, G.; Pennacchia, C.; Villani, F.; Ricciardi, A.; Tofalo, R.; Parente, E. Diversity and dynamics of communities of coagulase-negative staphylococci in traditional fermented sausages. J. Appl. Microbiol. 2004, 97, 271–284. [Google Scholar] [CrossRef]
- Mauriello, G.; Casaburi, A.; Blaiotta, G.; Villani, F. Isolation and technological properties of coagulase negative staphylococci from fermented sausages of Southern Italy. Meat Sci. 2004, 67, 149–158. [Google Scholar] [CrossRef]
- Guan, L.; Cho, K.-H.; Lee, J.-H. Analysis of the cultivable bacterial community in jeotgal, a Korean salted and fermented seafood, and identification of its dominant bacteria. Food Microbiol. 2011, 28, 101–113. [Google Scholar] [CrossRef]
- An, D.; Kim, H.-R.; Jeong, D.-W.; Caldwell, J.M.; Lee, J.-H. Bacterial community monitoring of commercial kimchi produced in Korea and China with evidence of Bacilli spore formation during fermentation. Kor. J. Microbiol. Biotechnol. 2014, 42, 121–130. [Google Scholar] [CrossRef]
- Heo, S.; Lee, J.-H.; Jeong, D.-W. Food-derived coagulase-negative Staphylococcus as starter cultures for fermented foods. Food Sci. Biotechnol. 2020, 29, 1023–1035. [Google Scholar] [CrossRef]
- Sondergaard, A.K.; Stahnke, L.H. Growth and aroma production by Staphylococcus xylosus, S. carnosus and S. equorum—A comparative study in model systems. Int. J. Food Microbiol. 2002, 75, 99–109. [Google Scholar] [CrossRef]
- Stahnke, L.H. Aroma components from dried sausages fermented with Staphylococcus xylosus. Meat Sci. 1994, 38, 39–53. [Google Scholar] [CrossRef]
- Deetae, P.; Bonnarme, P.; Spinnler, H.E.; Helinck, S. Production of volatile aroma compounds by bacterial strains isolated from different surface-ripened French cheeses. Appl. Microbiol. Biotechnol. 2007, 76, 1161–1171. [Google Scholar] [CrossRef]
- Fulladosa, E.; Garriga, M.; Martin, B.; Guardia, M.D.; Garcia-Regueiro, J.A.; Arnau, J. Volatile profile and microbiological characterization of hollow defect in dry-cured ham. Meat Sci. 2010, 86, 801–807. [Google Scholar] [CrossRef]
- Place, R.B.; Hiestand, D.; Gallmann, H.R.; Teuber, M. Staphylococcus equorum subsp. linens, subsp. nov., a starter culture component for surface ripened semi-hard cheeses. Syst. Appl. Microbiol. 2003, 26, 30–37. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Jung, G.; Lee, J.-H. Cultivable bacterial community analysis of saeu-jeotgal, a Korean high-salt-fermented seafood, during ripening. Microbiol. Biotechnol. Lett. 2016, 44, 293–302. [Google Scholar] [CrossRef]
- Jung, J.; Choi, S.; Jeon, C.-O.; Park, W. Pyrosequencing-based analysis of the bacterial community in Korean traditional seafood, ojingeo jeotgal. J. Microbiol. Biotechnol. 2013, 23, 1428–1433. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Heo, S.; Ryu, S.; Blom, J.; Lee, J.-H. Genomic insights into the virulence and salt tolerance of Staphylococcus equorum. Sci. Rep. 2017, 7, 5383. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Han, S.; Lee, J.-H. Safety and technological characterization of Staphylococcus equorum isolates from jeotgal, a Korean high-salt-fermented seafood, for starter development. Int. J. Food Microbiol. 2014, 188, 108–115. [Google Scholar] [CrossRef]
- Lee, J.-H.; Heo, S.; Jeong, D.-W. Genomic insights into Staphylococcus equorum KS1039 as a potential starter culture for the fermentation of high-salt foods. BMC Genom. 2018, 19, 136. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Na, H.; Ryu, S.; Lee, J.-H. Complete genome sequence of Staphylococcus equorum KS1039 isolated from saeu-jeotgal, Korean high-salt-fermented seafood. J. Biotechnol. 2016, 219, 88–89. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Kevin, F.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Wood, J.M.; Bremer, E.; Csonka, L.N.; Kraemer, R.; Poolman, B.; van der Heide, T.; Smith, L.T. Osmosensing and osmoregulatory compatible solute accumulation by bacteria. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 130, 437–460. [Google Scholar] [CrossRef]
- Hoffmann, T.; Bremer, E. Guardians in a stressful world: The Opu family of compatible solute transporters from Bacillus subtilis. Biol. Chem. 2017, 398, 193–214. [Google Scholar] [CrossRef]
- Boch, J.; Kempf, B.; Schmid, R.; Bremer, E. Synthesis of the osmoprotectant glycine betaine in Bacillus subtilis: Characterization of the gbsAB genes. J. Bacteriol. 1996, 178, 5121–5129. [Google Scholar] [CrossRef]
- Nau-Wagner, G.; Opper, D.; Rolbetzki, A.; Boch, J.; Kempf, B.; Hoffmann, T.; Bremer, E. Genetic control of osmoadaptive glycine betaine synthesis in Bacillus subtilis through the choline-sensing and glycine betaine-responsive GbsR repressor. J. Bacteriol. 2012, 194, 2703–2714. [Google Scholar] [CrossRef]
- Young, J.W.; Locke, J.C.; Elowitz, M.B. Rate of environmental change determines stress response specificity. Proc. Natl. Acad. Sci. USA 2013, 110, 4140–4145. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Gene Locus | Gene | E.C. No. | Log2 (Fold-Change) * | COG | Product | |
|---|---|---|---|---|---|---|---|
| 2 h/con | 4 h/con | ||||||
| Transporter | |||||||
| Osmoprotectant uptake | |||||||
| AWC34_RS10320 | opuCC | 0.04 | 1.08 | E | Osmoprotectant ABC transporter substrate-binding protein | ||
| AWC34_RS10325 | opuCB | −0.31 | 1.32 | ABC transporter permease | |||
| AWC34_RS10315 | opuCD | −0.47 | 1.03 | E | ABC transporter permease | ||
| AWC34_RS10330 | opuCA | 7.6.2.9 | 0.19 | 1.15 | EP | Betaine/proline/choline family ABC transporter ATP-binding protein | |
| AWC34_RS05465 | opuD | 1.88 | 2.31 | P | BCCT family transporter | ||
| AWC34_RS09135 | opuD | 2.21 | 2.02 | P | BCCT family transporter | ||
| AWC34_RS10865 | opuD | 0.85 | 3.72 | P | BCCT family transporter | ||
| AWC34_RS10935 | opuD | 0.95 | 0.95 | P | BCCT family transporter | ||
| AWC34_RS11530 | opuD | 3.22 | 2.6 | P | BCCT family transporter | ||
| AWC34_RS10400 | abgT | 1.09 | 0.55 | H | p-aminobenzoyl-glutamate transporter family transporter | ||
| AWC34_RS10580 | putP | 1.48 | 4.61 | E | Sodium/proline symporter PutP | ||
| AWC34_RS04230 | ktrA | −0.36 | −1.68 | P | TrkA family potassium uptake protein | ||
| AWC34_RS03930 | ktrB | −1.46 | −0.58 | P | Ktr system potassium uptake protein D | ||
| Na+/H+ antiporter | |||||||
| AWC34_RS03865 | yibQ | −0.13 | 0.15 | Putative Na+/H+ antiporter YjbQ | |||
| AWC34_RS00935 | yhdH | −1.73 | −1.66 | P | Sodium-dependent transporter | ||
| AWC34_RS03605 | mnhA1 | 0.57 | 0.69 | Na+/H+ antiporter subunit A | |||
| AWC34_RS03600 | mnhB1 | 0.35 | 0.99 | P | Na+/H+ antiporter subunit B | ||
| AWC34_RS03595 | mnhC1 | 0.01 | 0.93 | P | Na+/H+ antiporter subunit C | ||
| AWC34_RS03590 | mnhD1 | 1.6.5.3 | 0.46 | 0.69 | CP | Na+/H+ antiporter subunit D | |
| AWC34_RS03585 | mnhE1 | 0.24 | 0.63 | P | Na+/H+ antiporter subunit E | ||
| AWC34_RS03580 | mnhF1 | −0.35 | 0.66 | P | Na+/H+ antiporter subunit F1 | ||
| AWC34_RS03575 | mnhG1 | −0.18 | 0.47 | P | Na+/H+ antiporter subunit G1 | ||
| AWC34_RS02340 | mnhA2 | −0.15 | 0.38 | Putative antiporter subunit mnhA2 | |||
| AWC34_RS02345 | mnhB2 | −0.15 | 0.78 | Na+/H+ antiporter subunit B | |||
| AWC34_RS02350 | mnhC2 | 0.02 | 0.46 | P | Na+/H+ antiporter Mnh2 subunit C | ||
| AWC34_RS02355 | mnhD2 | −0.3 | 0.55 | CP | Na+/H+ antiporter subunit D | ||
| AWC34_RS02360 | mnhE2 | −0.7 | 0.45 | P | Na+/H+ antiporter subunit E | ||
| AWC34_RS02365 | mnhF2 | −0.71 | 0.53 | P | Na+/H+ antiporter Mnh2 subunit F | ||
| AWC34_RS02370 | mnhG2 | −0.58 | 0.17 | P | Na+/H+ antiporter subunit G | ||
| AWC34_RS09570 | nhaC | 0.57 | −0.7 | C | Na+/H+ antiporter NhaC | ||
| AWC34_RS09730 | nhaC | −1.13 | 0.74 | C | Na+/H+ antiporter NhaC | ||
| AWC34_RS10840 | nhaC | −0.12 | 1.01 | C | Na+/H+ antiporter NhaC | ||
| AWC34_RS02375 | nhaH | 0.17 | −0.25 | P | Na+/H+ antiporter NhaH | ||
| AWC34_RS08235 | sdcS | 0.29 | −0.39 | P | Sodium-dependent dicarboxylate transporter | ||
| AWC34_RS08920 | czrB | −0.26 | −0.68 | P | CDF family zinc efflux transporter CzrB | ||
| AWC34_RS03560 | −2.47 | −0.53 | R | Na+/H+ antiporter family protein | |||
| AWC34_RS09820 | −1.32 | 1 | C | Sodium:proton antiporter | |||
| AWC34_RS10590 | 0.38 | 0.32 | P | Sodium:proton antiporter | |||
| Synthesis | |||||||
| Glutamate | |||||||
| AWC34_RS04260 | pdhA | 1.2.4.1 | 1.21 | −0.14 | C | Pyruvate dehydrogenase (acetyl-transferring) E1 component subunit alpha | |
| AWC34_RS04265 | pdhA2 | 1.2.4.1 | 0.89 | −0.07 | C | Alpha-ketoacid dehydrogenase subunit beta | |
| AWC34_RS04270 | DLAT | 2.3.1.12 | 0.61 | 0.37 | C | 2-oxo acid dehydrogenase subunit E2 | |
| AWC34_RS04275 | lpdA | 1.8.1.4 | −0.49 | 0.23 | C | Dihydrolipoyl dehydrogenase | |
| AWC34_RS06290 | lpdA | 1.8.1.4 | 0.49 | −0.22 | C | Dihydrolipoyl dehydrogenase | |
| AWC34_RS04350 | PC | 6.4.1.1 | −0.57 | 0.34 | C | Pyruvate carboxylase | |
| AWC34_RS07130 | CS | 2.3.3.1 | 0.33 | 2.18 | C | Citrate synthase | |
| AWC34_RS05470 | acnA | 4.2.1.3 | 0.59 | 2.26 | C | Aconitate hydratase AcnA | |
| AWC34_RS07125 | icd | 1.1.1.42 | −0.32 | 1.92 | E | NADP-dependent isocitrate dehydrogenase | |
| AWC34_RS00985 | gltB | 1.4.1.14 | 0.92 | 1.45 | Glutamate synthase large subunit | ||
| AWC34_RS00990 | 1.4.1.14 | −0.11 | 1.83 | E | Glutamate synthase subunit beta | ||
| AWC34_RS01735 | rocG | 1.4.1.2 | 0.96 | 1.1 | E | Glu/Leu/Phe/Val dehydrogenase | |
| AWC34_RS03635 | gdhA | 1.4.1.3 | 1.44 | 1.36 | E | Glu/Leu/Phe/Val dehydrogenase | |
| Proline | |||||||
| AWC34_RS10920 | pruA | 1.2.1.88 | −0.14 | 0.27 | C | L-glutamate gamma-semialdehyde dehydrogenase | |
| AWC34_RS13165 | pruA | 1.2.1.88 | −2.4 | −2.92 | Betaine-aldehyde dehydrogenase | ||
| AWC34_RS06225 | proC | 1.5.1.2 | 0.43 | 2.24 | E | Pyrroline-5-carboxylate reductase | |
| AWC34_RS11515 | gbsB | 1.1.99.1 | 1.93 | 0.93 | P | Soluble type III alcohol dehydrogenase | |
| AWC34_RS11520 | gbsA | 1.2.1.3 | 4.21 | 2.34 | C | Glycine betaine aldehyde dehydrogenase | |
| Regulator | |||||||
| AWC34_RS06515 | rpoD | - | 0.73 | 0.77 | K | RNA polymerase sigma factor RpoD | |
| AWC34_RS08530 | sigB | - | 0.71 | 0.73 | K | RNA polymerase sigma factor SigB | |
| AWC34_RS11525 | gbsR | - | 1.46 | 1.31 | K | GbsR/MarR family transcriptional regulator | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, S.; Park, J.; Lee, E.; Lee, J.-H.; Jeong, D.-W. Transcriptomic Analysis of Staphylococcus equorum KM1031, Isolated from the High-Salt Fermented Seafood Jeotgal, under Salt Stress. Fermentation 2022, 8, 403. https://0-doi-org.brum.beds.ac.uk/10.3390/fermentation8080403
Heo S, Park J, Lee E, Lee J-H, Jeong D-W. Transcriptomic Analysis of Staphylococcus equorum KM1031, Isolated from the High-Salt Fermented Seafood Jeotgal, under Salt Stress. Fermentation. 2022; 8(8):403. https://0-doi-org.brum.beds.ac.uk/10.3390/fermentation8080403
Chicago/Turabian StyleHeo, Sojeong, Junghyun Park, Eungyo Lee, Jong-Hoon Lee, and Do-Won Jeong. 2022. "Transcriptomic Analysis of Staphylococcus equorum KM1031, Isolated from the High-Salt Fermented Seafood Jeotgal, under Salt Stress" Fermentation 8, no. 8: 403. https://0-doi-org.brum.beds.ac.uk/10.3390/fermentation8080403