
β-Lactoglobulin Adsorption Layers at the Water/Air Surface: 5. Adsorption Isotherm and Equation of State Revisited, Impact of pH

 ,
,  ,
,  ,
, 

Abstract
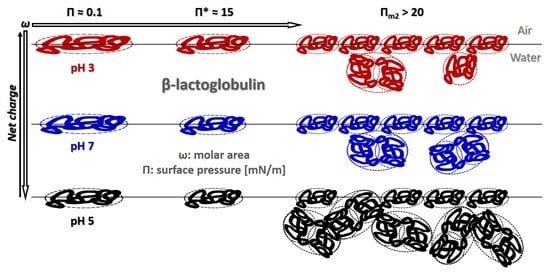
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Solutions
2.2. Experimental Methods: Tensiometry and Surface Dilational Rheometry
2.3. Theoretical Model
3. Results and Discussion
- the surface pressure isotherm, Π(c);
- the adsorption isotherm, Γ(c) [24]; and
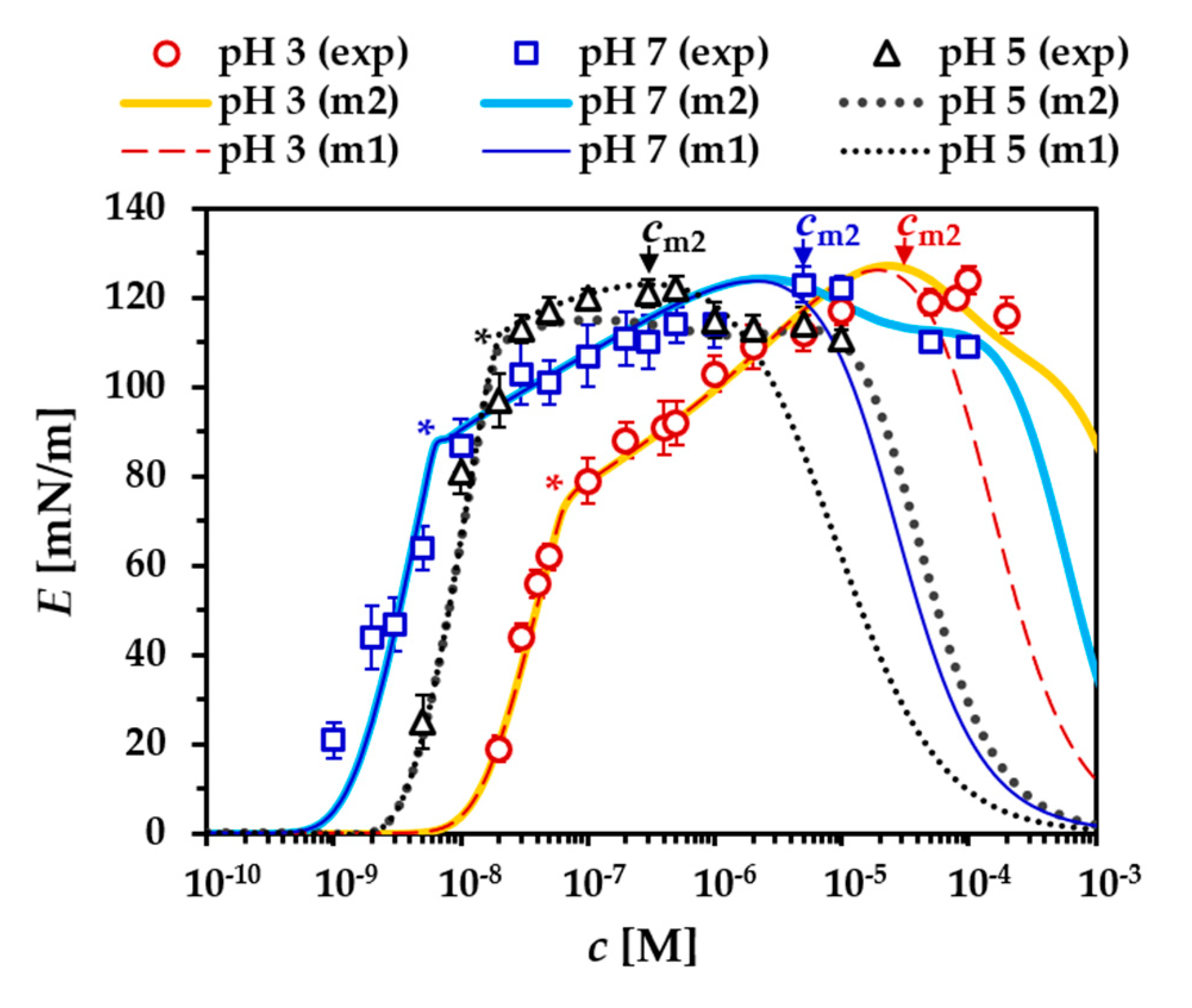
- the dependency of the dilational modulus on the protein concentration, E(c)f,g.
- 4.
- the equation of state, Π(Γ);
- 5.
- the distribution of the average molar area over the protein concentration, ω(c) [24], the surface pressure, ω(Π) and the surface excess, ω(Γ); and
- 6.
- the dependencies of the dilational modulus on the surface pressure, E(Π)f,g, and on the surface excess, E(Γ)f,g.
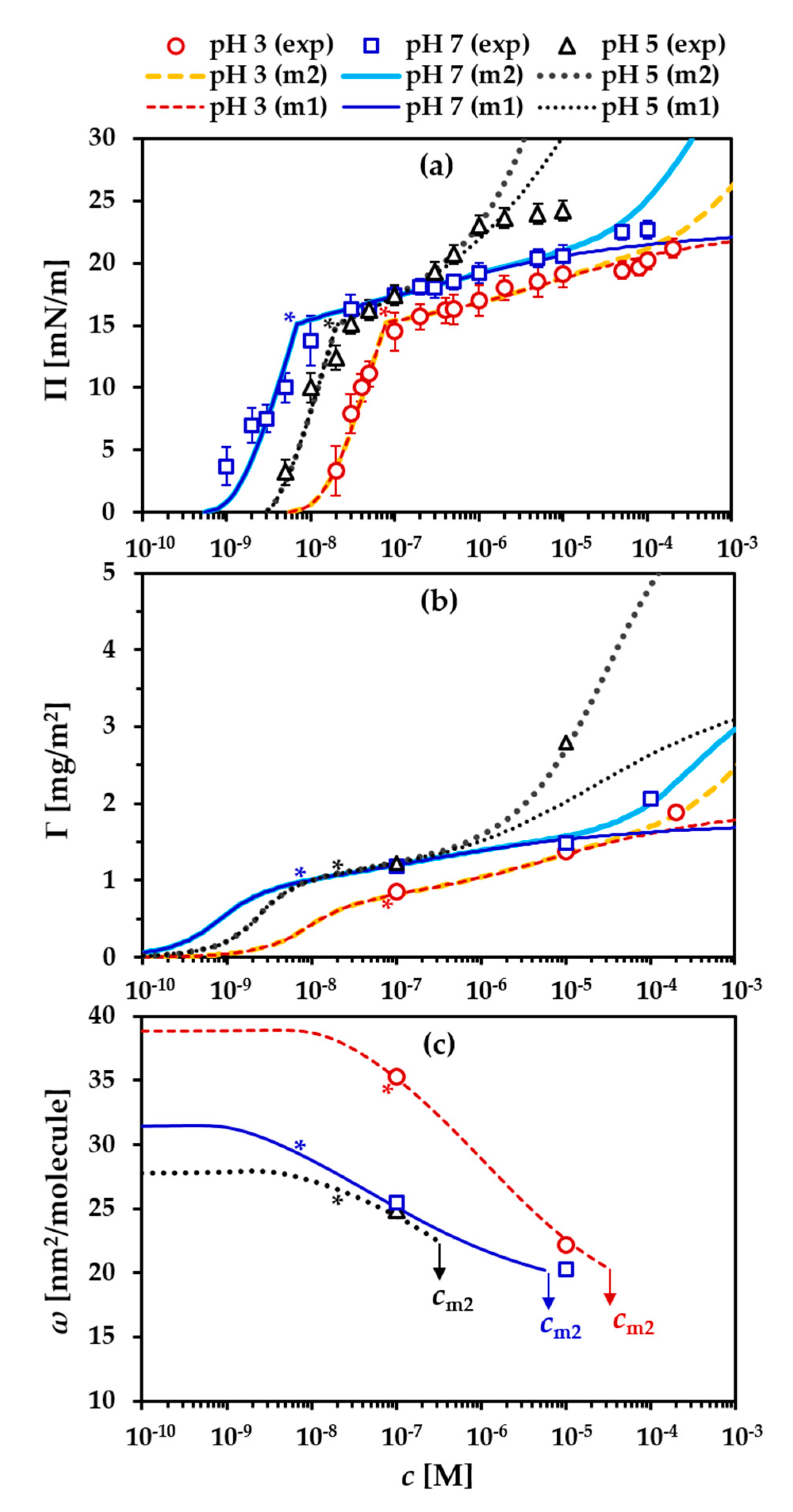
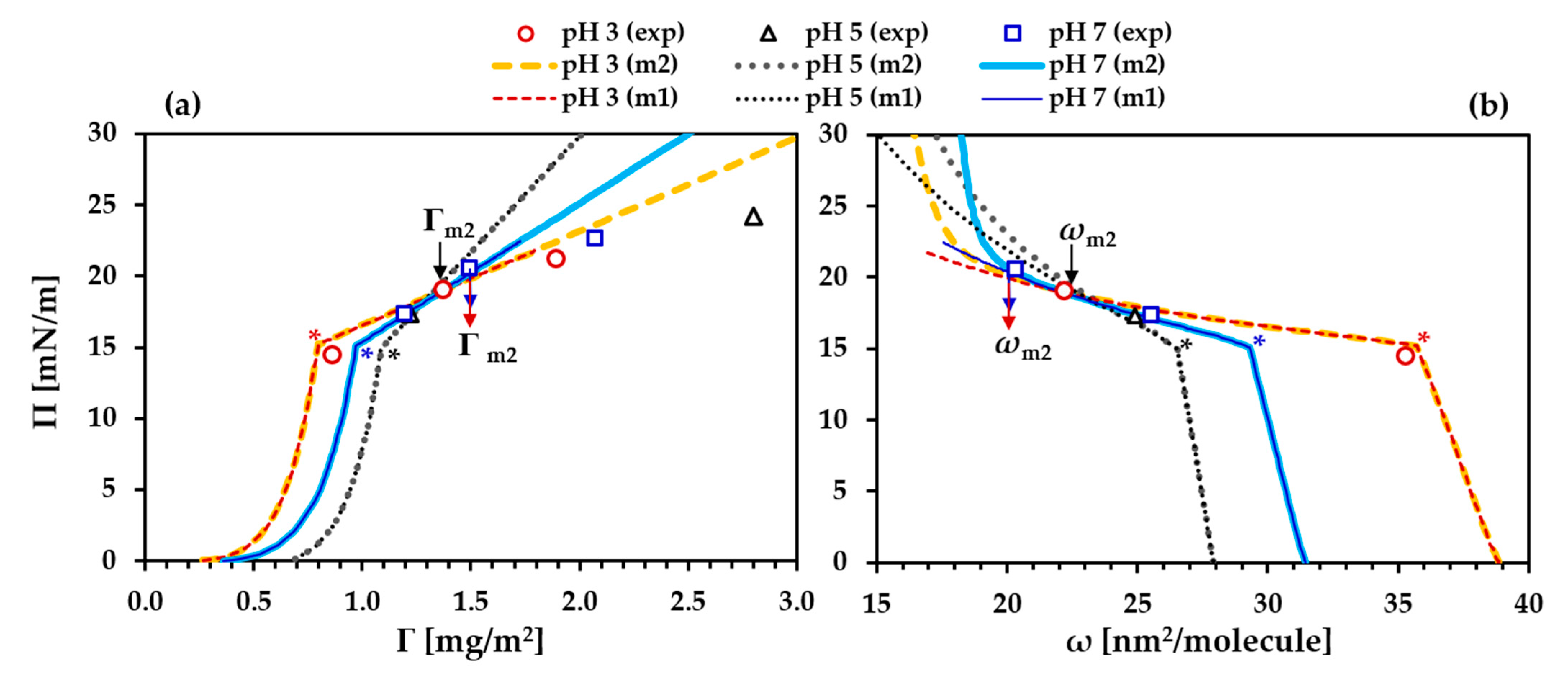
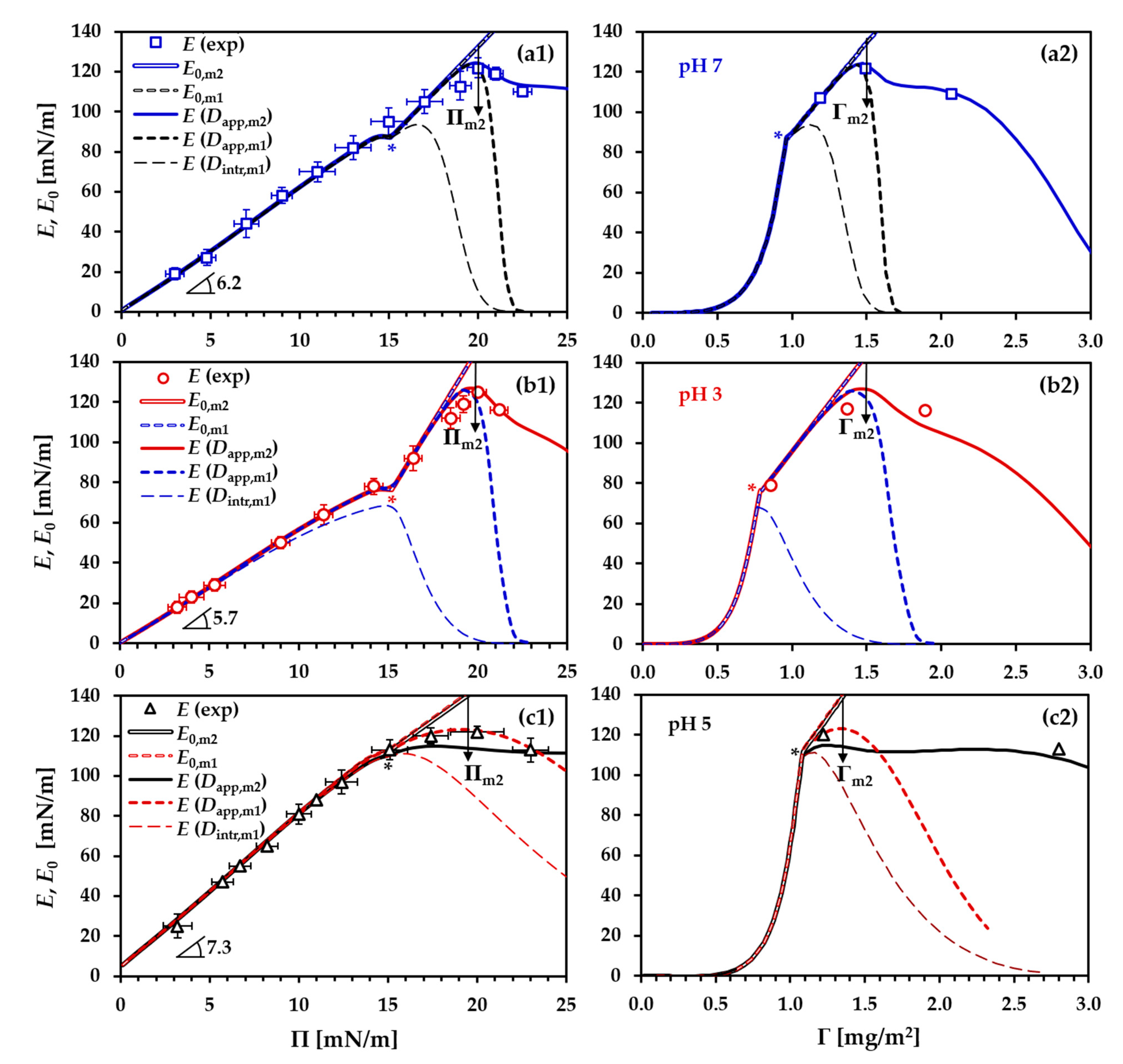
- At the very beginning, an approximate value for Π* should be set in order to divide the precritical and postcritical regions in the simulation curves. In the present case, the appropriate values for Π* are around 15 mN/m, where the three experimental Π(c) isotherms (for pH 3, pH 5 and pH 7) exhibit a kink;
- In accordance with Equations (1)–(7), the parameters ω1, ωn and bj are essential for optimizing the model simulations in a way to fit the experimental Γ(c) isotherm and the corresponding dependency ω(c) and, at the same time, the experimental Π(c) isotherm. In the initial iteration, this step aimed at locating the precritical region of the Π(c) isotherm along the c-axis while maintaining a good fit to the Γ(c) isotherm. At other fixed parameters, increase of the local adsorption constant, bj, shifts the precritical region of the Π(c) isotherm toward lower c; the individual effects of ω1 or ωn on the model simulations are illustrated in Figure S2b,c in the Supporting Information. In the final iterations, the values of these parameters were optimized to serve the best global fit to all processed dependencies. It must be stressed here that the input values of ω1 and ωn are boundary values. In the calculation procedure, the average molar area ω is allowed to vary between these minimal and maximal values, but the actual molar areas, computed as dependent on other parameters according to Equations (1)–(7), appear as output data (see Figure S3 in the Supporting Information). Those boundary values only guide the resulting best fit, and, therefore, a proper analysis should be based on the output data. Some characteristic values (ωΠ=0.1, ω* and ωm2 (for definitions, see the legend in Table 1)) outputted from the best fits will be discussed further below;
- The molar area increment ω0 was allowed to vary between 2.0 × 105 and 4.5 × 105 m2/mol [21,28,30,32,33], which corresponds to areas per increment between ≈0.33 nm2 and ≈0.75 nm2, respectively. We could mention here that Joos [15] used a limiting value of the solvent area per molecule of 0.10 nm2 (≈6 × 104 m2/mol); however, in our analysis we refrain from using such low values for ω0. Assuming a weak effect of pH on this parameter, we aimed at setting a constant value in the processing of the data packs at the three different pH. The effect of ω0 variation on the adsorption isotherm Γ(c) is less pronounced compared to that on the Π(c), Π(Γ) and E0(Π) dependencies (Figure S2d in the Supporting Information). For the latter one, optimizing ω0 was essential for description of the experimental E(Π) data. The best results were found using the lower values for ω0 so that this parameter was fixed at 2.0 × 105 m2/mol. It can be mentioned here that this value is very close to the molar area of a layer of adsorbed water molecules on mica surfaces (≈1.8 × 105 m2/mol or ≈0.30 nm2/molecule) [64];
- For the exponential coefficient α, it was possible to use relatively close values in the cases of pH 3 (α = 2.7) and pH 7 (α = 2.2), but for pH 5, a substantially higher value (α = 3.8) was required. The effects of α on the model simulations are illustrated in Figure S2e in the Supporting Information;
- The effects of variation of the interaction parameter a on the model simulations are illustrated in Figure S2f in the Supporting Information. It should be noted that the simulations of the modulus E and those of the precritical region of the Π(Γ) equation of state are very sensitive to this parameter;
- The aggregation number na was optimized in respect to the postcritical region of the surface pressure isotherm Π(c) and to pin the local maximum of the E(Π) dependency observed at Π = 19–20 mN/m;
- Finally, the adsorption constant for the secondary layer b2 was set to follow the experimental adsorption isotherm Γ(c), and then Π* and na were tuned in order to obtain best fits with the surface pressure isotherm Π(c) and the rheology data.
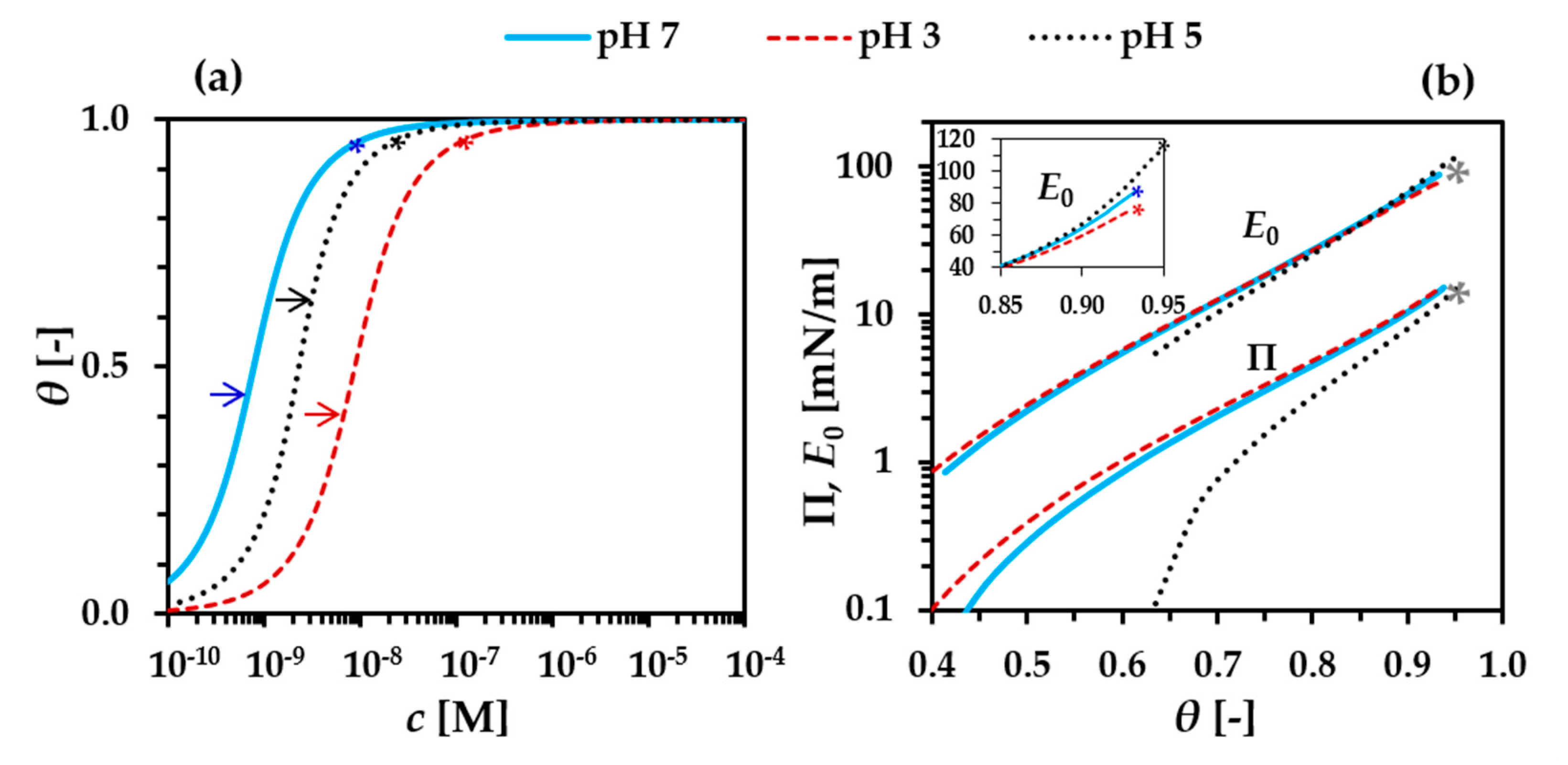
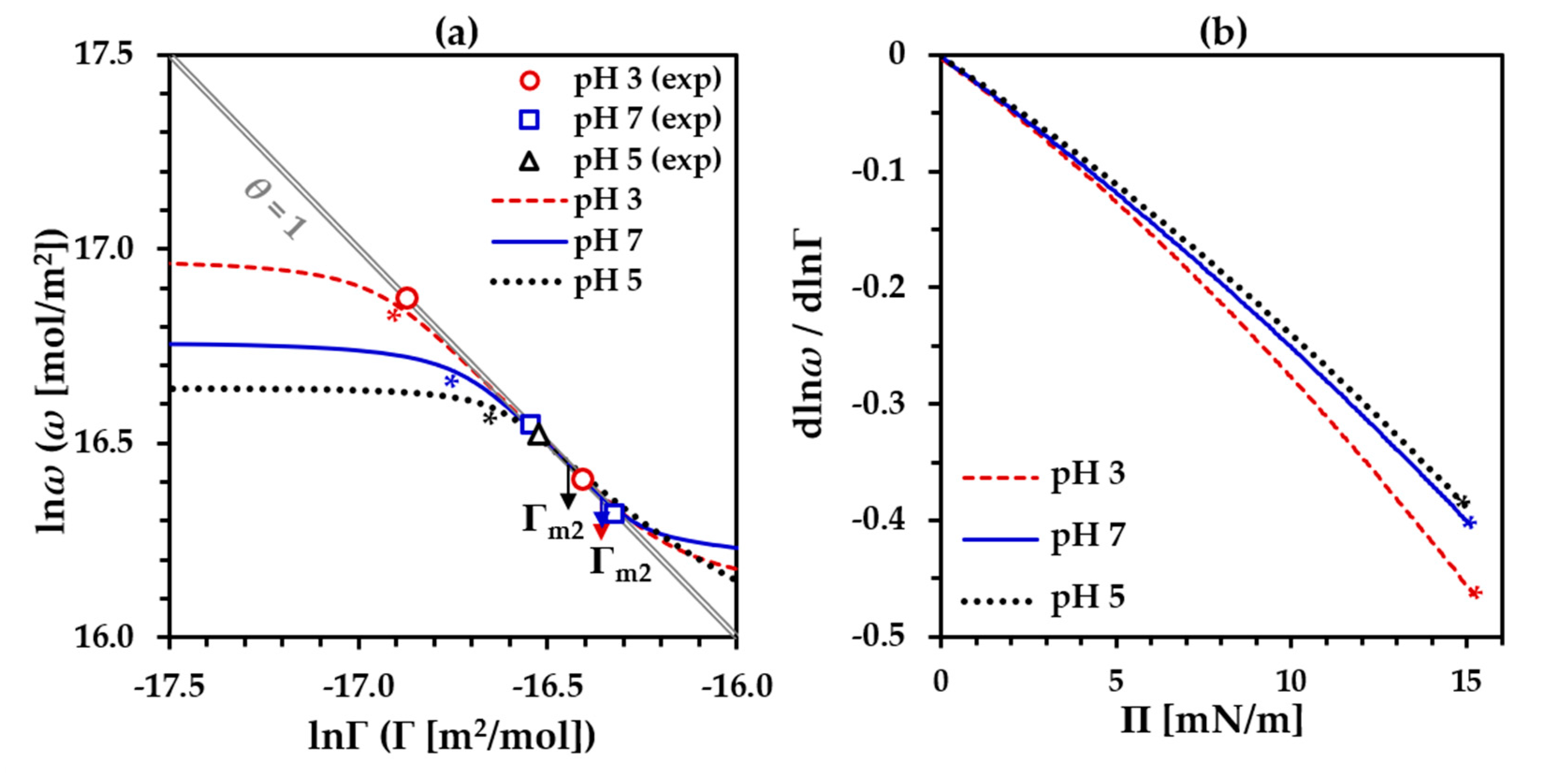
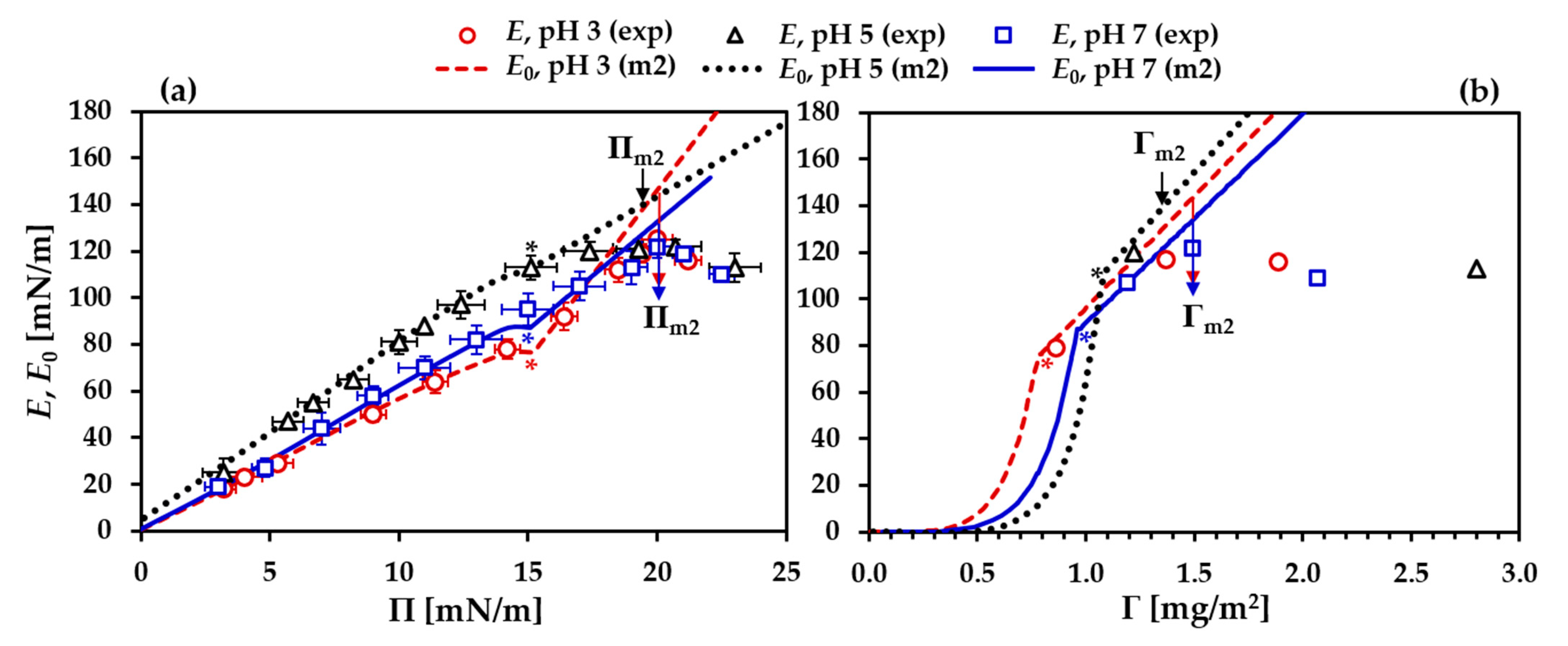
3.1. Precritical Region of the BLG Adsorption Layer
3.2. Postcritical Region of Monolayer
3.3. Formation of a Secondary Layer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vincent, B. Early (pre-DLVO) studies of particle aggregation. Adv. Colloid Interface Sci. 2012, 170, 56–67. [Google Scholar] [CrossRef]
- Gochev, G.; Platikanov, D.; Miller, R. Chronicles of foam films. Adv. Colloid Interface Sci. 2016, 233, 115–125. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Lucassen-Reynders, E.H. Adsorption of single and mixed ionic surfactants at fluid interfaces. Adv. Colloid Interface Sci. 2002, 96, 295–323. [Google Scholar] [CrossRef]
- Ivanov, I.B.; Ananthapadmanabhan, K.P.; Lips, A. Adsorption and structure of the adsorbed layer of ionic surfactants. Adv. Colloid Interface Sci. 2006, 123–126, 189–212. [Google Scholar] [CrossRef]
- Liggieri, L.; Mileva, E.; Miller, R. The surface layer as the basis for foam formation and stability. In Foam Films and Foams: Fundamentals and Applications; Exerowa, D., Gochev, G., Platikanov, D., Liggieri, L., Miller, R., Eds.; CRC Press: Boca Raton, FL, USA, 2018; Chapter 1. [Google Scholar]
- de Gennes, P.G. Polymers at an interface; a simplified view. Adv. Colloid Interface Sci. 1987, 27, 189–209. [Google Scholar] [CrossRef]
- Fleer, G.J.; Cohen Stuart, M.A.; Scheutjens, J.M.H.M.; Cosgrove, T.; Vincent, B. Polymers Interfaces; Chapman and Hall: London, UK, 1993. [Google Scholar]
- Lyklema, J. Adsorption of polymers and polyelectrolytes. In Fundamentals of Colloid and Interface Science; Lyklema, J., Ed.; Academic Press: London, UK, 1995; Volume 2, Chapter 5. [Google Scholar]
- Benjamins, J.; de Feijter, J.A.; Evans, M.T.A.; Graham, D.E.; Phillips, M.C. Dynamic and static properties of proteins adsorbed at the air/water interface. Faraday Discuss. Chem. Soc. 1975, 59, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.E.; Phillips, M.C. Proteins at Liquid Interfaces II. Adsorption Isotherms. J. Colloid Interface Sci. 1979, 70, 415–426. [Google Scholar] [CrossRef]
- Douillard, R.; Lefebvre, J.; Tran, V. Applicability of Gibbs’ law to protein adsorption isotherms. Colloids Surfaces A 1993, 78, 109–113. [Google Scholar] [CrossRef]
- Lucassen-Reynders, E.H. Competitive adsorption of emulsifiers 1. Theory for adsorption of small and large molecules. Colloids Surf. A 1994, 91, 79–88. [Google Scholar] [CrossRef]
- Damodaran, D.; Razumovsky, L. Role of surface area-to-volume ratio in protein adsorption at the air–water interface. Surf. Sci. 2008, 602, 307–315. [Google Scholar] [CrossRef]
- Ter-Minassian-Saraga, L. Protein denaturation on adsorption and water activity at interfaces: An analysis and suggestion. J. Colloid Interface Sci. 1981, 80, 393–401. [Google Scholar] [CrossRef]
- Joos, P. Approach for an equation of state for adsorbed protein surfaces. Biochim. Biophys. Acta Biomembr. 1975, 375, 1–9. [Google Scholar] [CrossRef]
- Uraizee, F.; Narsimhan, N. A Surface Equation of State for Globular Proteins at the Air-Water Interface. J. Colloid Interface Sci. 1991, 146, 169–178. [Google Scholar] [CrossRef]
- Douillard, R.; Daoud, M.; Aguié-Béghin, V. Polymer thermodynamics of adsorbed protein layers. Curr. Opin. Colloid Interface Sci. 2003, 8, 380–386. [Google Scholar] [CrossRef]
- Frumkin, A. Die Kapillarkurve der höheren Fettsäuren und die Zustandsgleichung der Oberflächenschicht (translated by authors: The capillary curve of long-chain fatty acids and the equation of state of the surface layer). Z. Physik. Chem. (Leipzig) 1925, 116, 466–484. [Google Scholar]
- Joos, P.; Serrien, G. The principle of Braun-Le Châtelier at surfaces. J. Colloid Interface Sci. 1991, 145, 291–294. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Lucassen-Reynders, E.H.; Miller, R. Description of the adsorption behaviour of proteins at water/fluid interfaces in the framework of a two-dimensional solution model. Adv. Colloid Interface Sci. 2003, 106, 237–259. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Miller, R. Equilibrium and Dynamic Characteristics of Protein Adsorption Layers at Gas–Liquid Interfaces: Theoretical and Experimental Data. Colloid J. 2005, 67, 393–404. [Google Scholar] [CrossRef]
- Grigoriev, D.O.; Fainerman, V.B.; Makievski, A.V.; Krägel, J.; Wüstneck, R.; Miller, R. β-Casein bilayer adsorption at the solution/air interface: Experimental evidences and theoretical description. J. Colloid Interface Sci. 2002, 253, 257–264. [Google Scholar] [CrossRef]
- Holt, S.A.; McGillivray, D.J.; Poon, S.; White, J.W. Protein deformation and surfactancy at an interface. J. Phys. Chem. B 2000, 104, 7431–7438. [Google Scholar] [CrossRef]
- Gochev, G.; Scoppola, E.; Campbell, R.; Noskov, B.A.; Miller, R.; Schneck, E. β-Lactoglobulin Adsorption Layers at the Water/Air Surface: 3. Neutron Reflectivity Study on the Effect of pH. J. Phys. Chem. B 2019, 123, 10877–10889. [Google Scholar] [CrossRef]
- Miller, R.; Fainerman, V.B.; Makievski, A.V.; Krägel, J.; Grigoriev, D.O.; Kazakov, V.N.; Sinyachenko, O.V. Dynamics of protein and mixed protein/surfactant adsorption layers at the water/fluid interface. Adv. Colloid Interface Sci. 2000, 86, 39–82. [Google Scholar] [CrossRef]
- Miller, R.; Fainerman, V.B.; Makievski, A.V.; Leser, M.; Michel, M.; Aksenenko, E.V. Determination of protein adsorption by comparative drop and bubble profile analysis tensiometry. Colloids Surf. B 2004, 36, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Wüstneck, R.; Fainerman, V.B.; Aksenenko, E.; Kotsmar, C.; Pradines, V.; Krägel, J.; Miller, R. Surface dilatational behavior of β-casein at the solution/air interface at different pH values. Colloids Surf. A 2012, 404, 17–24. [Google Scholar] [CrossRef]
- Gochev, G.; Retzlaff, I.; Aksenenko, E.V.; Fainerman, V.B.; Miller, R. Adsorption isotherm and equation of state for β-Lactoglobulin layers at the air/water surface. Colloids Surf. A 2013, 422, 33–38. [Google Scholar] [CrossRef]
- Dan, A.; Wüstneck, R.; Krägel, J.; Aksenenko, E.V.; Fainerman, V.B.; Miller, R. Interfacial adsorption and rheological behavior of β-casein at the water/hexane interface at different pH. Food Hydrocoll. 2014, 34, 193–201. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Aksenenko, E.V.; Krägel, J.; Miller, R. Thermodynamics, interfacial pressure isotherms and dilational rheology of mixed protein-surfactant adsorption layers. Adv. Colloid Interface Sci. 2016, 233, 200–222. [Google Scholar] [CrossRef]
- Won, J.Y.; Gochev, G.G.; Ulaganathan, V.; Krägel, J.; Aksenenko, E.V.; Fainerman, V.B.; Miller, R. Dilational visco-elasticity of BLG adsorption layers at the solution/tetradecane interface–Effect of pH and ionic strength. Colloids Surf. A 2017, 521, 204–210. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Kovalchuk, V.I.; Aksenenko, E.V.; Zinkovych, I.I.; Makievski, A.V.; Nikolenko, M.V.; Miller, R. Dilational Viscoelasticity of Proteins Solutions in Dynamic Conditions. Langmuir 2018, 34, 6678–6686. [Google Scholar] [CrossRef]
- Kovalchuk, V.I.; Aksenenko, E.V.; Trukhin, D.V.; Makievski, A.V.; Fainerman, V.B.; Miller, R. Effect of amplitude on the surface dilational visco-elasticity of protein solutions. Colloids Interfaces 2018, 2, 57. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.; Aksenenko, E.V.; Zinkovych, I.I.; Fainerman, V.B. Adsorption of proteins at the aqueous solution/alkane interface: Co-adsorption of protein and alkane. Adv. Colloid Interface Sci. 2015, 222, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Dan, A.; Gochev, G.; Krägel, J.; Aksenenko, E.V.; Fainerman, V.B.; Miller, R. Interfacial rheology of mixed layers of food proteins and surfactants. Curr. Opin. Colloid Interface Sci. 2013, 18, 302–310. [Google Scholar] [CrossRef]
- Lucassen-Reynders, E.H.; Fainerman, V.B.; Miller, R. Surface Dilational Modulus or Gibbs’ Elasticity of Protein Adsorption Layers. J. Phys. Chem. B 2004, 108, 9173–9176. [Google Scholar] [CrossRef]
- Lucassen-Reynders, E.H.; Benjamins, J.; Fainerman, V.B. Dilational rheology of protein films adsorbed at fluid interfaces. Curr. Opin. Colloid Interface Sci. 2010, 15, 264–270. [Google Scholar] [CrossRef]
- Maldonado-Valderrama, J.; Miller, R.; Fainerman, V.B.; Wilde, P.J.; Morris, V.J. Effect of gastric conditions on β-lactoglobulin interfacial networks: Influence of the oil phase on protein structure. Langmuir 2010, 26, 15901–15908. [Google Scholar] [CrossRef] [PubMed]
- Alahverdjieva, V.S.; Grigoriev, D.O.; Ferri, J.K.; Fainerman, V.B.; Aksenenko, E.V.; Leser, M.; Michel, M.; Miller, R. Adsorption behaviour of hen egg-white lysozyme at the air/water interface. Colloids Surfaces A 2008, 323, 167–174. [Google Scholar] [CrossRef]
- Engelhardt, K.; Lexis, M.; Gochev, G.; Konnerth, C.; Miller, R.; Willenbacher, N.; Peukert, W.; Braunschweig, B. pH Effects on the Molecular Structure of β-Lactoglobulin Modified Air–Water Interfaces and Its Impact on Foam Rheology. Langmuir 2013, 29, 11646–11655. [Google Scholar] [CrossRef]
- Gochev, G.; Retzlaff, I.; Exerowa, D.; Miller, R. Electrostatic stabilization of foam films from β-lactoglobulin solutions. Colloids Surfaces A 2014, 460, 272–279. [Google Scholar] [CrossRef]
- Lech, F.J.; Delahaije, R.J.B.M.; Meinders, M.B.J.; Gruppen, H.; Wierenga, P.A. Identification of critical concentrations determining foam ability and stability of β-lactoglobulin. Food Hydrocoll. 2016, 57, 46–54. [Google Scholar] [CrossRef]
- Ulaganathan, V.; Retzlaff, I.; Won, J.; Gochev, G.; Gehin-Delval, C.; Leser, M.; Noskov, B.A.; Miller, R. β-Lactoglobulin adsorption layers at the water/air surface: 1. Adsorption kinetics and surface pressure isotherm: Effect of pH and ionic strength. Colloids Surf. A 2017, 519, 153–160. [Google Scholar] [CrossRef]
- Ulaganathan, V.; Retzlaff, I.; Won, J.; Gochev, G.; Gunes, D.; Gehin-Delval, C.; Leser, M.; Noskov, B.A.; Miller, R. β-Lactoglobulin adsorption layers at the water/air surface: 2. Dilational rheology: Effect of pH and ionic strength. Colloids Surf. A 2017, 521, 167–176. [Google Scholar] [CrossRef]
- Gochev, G.; Ulaganathan, V.; Retzlaff, I.; Gehin-Delval, C.; Gunez, D.Z.; Leser, M.E.; Kulozik, U.; Miller, R.; Braunschweig, B. β-Lactoglobulin adsorption layers at the water/air surface: 4. Impact on Stability of Foam Films and Foams. Minerals 2020, 10, 636. [Google Scholar] [CrossRef]
- Toro-Sierra, J.; Tolkach, A.; Kulozik, U. Fractionation of α-lactalbumin and β-lactoglobulin from whey protein isolate using selective thermal aggregation, an optimized membrane separation procedure and resolubilization techniques at pilot plant scale. Food Bioprocess Technol. 2013, 6, 1032–1043. [Google Scholar] [CrossRef]
- Clark, D.C.; Husband, F.; Wilde, P.; Cornec, M.; Miller, R.; Krägel, J.; Wüstneck, R.J. Evidence of extraneous surfactant adsorption altering adsorbed layer properties of β-lactoglobulin. Chem. Soc. Faraday Trans. 1995, 91, 1991–1996. [Google Scholar] [CrossRef]
- Javadi, A.; Mucic, N.; Karbaschi, M.; Won, J.Y.; Lotfi, M.; Dan, A.; Ulaganathan, V.; Gochev, G.; Makievski, A.V.; Kovalchuk, V.I.; et al. Characterization methods for liquid interfacial layers. Eur. Phys. J. Spec. Top. 2013, 222, 7–29. [Google Scholar] [CrossRef]
- Mackie, A.R.; Husband, F.A.; Holt, C.; Wilde, P.J. Adsorption of β-Lactoglobulin variants A and B to the air-water interface. Int. J. Food Sci. Technol. 1999, 34, 509–516. [Google Scholar] [CrossRef]
- Ridout, M.J.; Mackie, A.R.; Wilde, P.J. Rheology of Mixed β-Casein/β-Lactoglobulin Films at the Air-Water Interface. J. Agric. Food Chem. 2004, 52, 3930–3937. [Google Scholar] [CrossRef] [PubMed]
- Noskov, B.A. Protein conformational transitions at the liquid-Gas interface as studied by dilational surface rheology. Adv. Colloids Interface Sci. 2014, 206, 222–241. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Miller, R. Equation of State for Concentrated Protein Surface Layers at the Water/Air Interface. Langmuir 1999, 15, 1812–1816. [Google Scholar] [CrossRef]
- Douillard, R.; Lefebvre, J. Adsorption of proteins at the gas-liquid interface: Models for concentration and pressure isotherms. J. Colloid Interface Sci. 1990, 139, 488–499. [Google Scholar] [CrossRef]
- Douillard, R.; Daoud, M.; Lefebvre, J.; Minier, C.; Lecannu, G.; Coutret, J. State equation of β-casein at the air/water interface. J. Colloid Interface Sci. 1994, 163, 277–288. [Google Scholar] [CrossRef]
- Miller, R.; Aksenenko, E.V.; Fainerman, V.B.; Pison, U. Kinetics of adsorption of globular proteins at liquid/fluid interfaces. Colloids Surf. A 2001, 183–185, 381–390. [Google Scholar] [CrossRef]
- Lucassen, J.; Hansen, R.S. Damping of Waves on Monolayer-Covered Surfaces. II. Influence of Bulk-to-Surface Diffusional Interchange on Ripple Characteristics. J. Colloid Interface Sci. 1967, 23, 319–328. [Google Scholar] [CrossRef]
- Lucassen, J.; van den Tempel, M. Dynamic measurements of dilational properties of a liquid interface. Chem. Eng. Sci. 1972, 27, 1283–1291. [Google Scholar] [CrossRef]
- Lucassen, J.; van den Tempel, M. Longitudinal waves on visco-elastic surfaces. J. Colloid Interface Sci. 1972, 41, 491–500. [Google Scholar] [CrossRef]
- Joos, P. Dynamic Surface Phenomena; VSP: Utrecht, The Netherlands, 1999. [Google Scholar]
- Fainerman, V.B.; Lylyk, S.V.; Aksenenko, E.V.; Makievski, A.V.; Ravera, F.; Petkov, J.T.; Yorke, J.; Miller, R. Adsorption layer characteristics of Tritons surfactants. 3. Dilational visco-elasticity. Colloids Surf. A 2009, 334, 16–21. [Google Scholar] [CrossRef]
- Lucassen, J. Dynamic dilational properties of composite surfaces. Colloids Surf. 1992, 65, 139–149. [Google Scholar] [CrossRef]
- Benjamins, J. Static and Dynamic Properties of Proteins Adsorbed at Liquid Interfaces. Ph.D. Thesis, Wageningen University, Wageningen, The Netherlands, 2000. [Google Scholar]
- MacRitchie, F.; Alexander, A.E. Kinetics of adsorption of proteins at interfaces, Part I, the role of bulk diffusion in adsorption. J. Colloid Interface Sci. 1963, 18, 453–457. [Google Scholar] [CrossRef]
- Narayanaswamy, N.; Ward, C.A. Area Occupied by a Water Molecule Adsorbed on Silica at 298 K: Zeta Adsorption Isotherm Approach. J. Phys. Chem. C 2020, 124, 9269–9280. [Google Scholar] [CrossRef]
- Song, K.B.; Damodaran, S. Influence of electrostatic forces on the adsorption of succinylated β-lactoglobulin at the air-water interface. Langmuir 1991, 72, 2737–2742. [Google Scholar] [CrossRef]
- Wierenga, P.A.; Meinders, M.B.J.; Egmond, M.R.; Voragen, A.G.J.; de Jongh, H.H.J. Quantitative description of the relation between protein net charge and protein adsorption to air-water interfaces. J. Phys. Chem. B 2005, 109, 16946–16952. [Google Scholar] [CrossRef]
- MacRitchie, F.; Alexander, A.E. Kinetics of adsorption of proteins at interfaces, Part III, the role of electrical barriers in adsorption. J. Colloid Interface Sci. 1963, 18, 464–469. [Google Scholar] [CrossRef]
- Shimuzu, M.; Saito, M.; Yamauchi, K. Emulsifying and structural properties of β-lactoglobulin at different pHs. Agric. Biol. Chem. 1985, 49, 189–194. [Google Scholar]
- Wierenga, P.A.; Meinders, M.B.J.; Egmond, M.R.; Voragen, F.A.G.J.; De Jongh, H.H.J. Protein exposed hydrophobicity reduces the kinetic barrier for adsorption of ovalbumin to the air-water interface. Langmuir 2003, 19, 8964–8970. [Google Scholar] [CrossRef]
- Jara, F.L.; Sánchez, C.C.; Patino, J.M.R.; Pilosof, A.M.R. Competitive adsorption behavior of α-lactoglobulin β -lactalbumin, bovin serum albumin in presence of hydroxypropylmethylcellulose. Influence of pH. Food Hydrocoll. 2014, 35, 189–197. [Google Scholar] [CrossRef]
- Sengupta, T.; Damodaran, S. Role of dispersion interactions in the adsorption of proteins at oil-water and air-water interfaces. Langmuir 1998, 14, 6457–6469. [Google Scholar] [CrossRef]
- Sengupta, T.; Razumovsky, L.; Damodaran, S. Energetics of protein-interface interactions and its effect on protein adsorption. Langmuir 1999, 15, 6991–7001. [Google Scholar] [CrossRef]
- Sundaram, S.; Ferry, J.K.; Vollhardt, D.; Stebe, K.J. Surface phase behavior and surface tension evolution for lysozyme adsorption onto clean interfaces and into DPPC monolayers: Theory and Experiment. Langmuir 1998, 14, 1208. [Google Scholar] [CrossRef]
- Erickson, J.; Sundaram, S.; Stebe, K.J. Surface pressure evolution of lysozyme solutions. Langmuir 2000, 16, 5072–5078. [Google Scholar] [CrossRef]
- Wierenga, P.A.; Gruppen, H. New views on foams from protein solutions. Curr. Opin. Colloid Interface Sci. 2010, 15, 365–373. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Chaudhuri, S.; Roy, J.; Sinha, N.K.; Sen, A. Physicochemical investigations on buffalo β-lactoglobulin. Studies on sedimentation, diffusion, and hydrogen ion titration. Arch. Biochem. Biophys. 1971, 144, 6–15. [Google Scholar] [CrossRef]
- de Feijter, J.A.; Benjamins, J. Soft-particle model of compact macromolecules at interfaces. J. Colloid Interface Sci. 1982, 90, 289–293. [Google Scholar] [CrossRef]
- Timasheff, S.N.; Mescanti, L.; Basch, J.J.; Townend, R. Conformational transitions of bovine b-lactoglobulins A, B, and C. J. Biol. Chem. 1966, 241, 2496–2501. [Google Scholar] [CrossRef]
- Taulier, N.; Chalikian, T.V. Characterization of pH-induced transitions of β-lactoglobulin: Ultrasonic, densimetric, and spectroscopic studies. J. Mol. Biol. 2001, 314, 873–889. [Google Scholar] [CrossRef] [Green Version]
- Timasheff, S.N.; Townend, R. Structure of the β-Lactoglobulin tetramer. Nature 1964, 203, 517–519. [Google Scholar] [CrossRef]
- Vogtt, K.; Javid, N.; Alvarez, E.; Sefcik, J.; Bellissent-Funel, M.-C. Tracing nucleation pathways in protein aggregation by using small angle scattering methods. Soft Matter 2011, 7, 3906–3914. [Google Scholar] [CrossRef]
- Scheuble, N.; Lussi, M.; Geue, T.; Carrière, F.; Fischer, P. Blocking gastric lipase adsorption and displacement processes with viscoelastic biopolymer adsorption layers. Biomacromolecules 2016, 17, 3328–3337. [Google Scholar] [CrossRef]
- Bertsch, P.; Thoma, A.; Bergfreund, J.; Geue, T.; Fischer, P. Transient measurement and structure analysis of protein–polysaccharide multilayers at fluid interfaces. Soft Matter 2019, 15, 6362–6368. [Google Scholar] [CrossRef] [Green Version]
- Bergfreund, J.; Diener, M.; Geue, T.; Nussbaum, N.; Kummer, N.; Bertsch, P.; Nyström, G.; Fischer, P. Globular protein assembly and network formation at fluid interfaces: Effect of oil. Soft Matter 2021, 17, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Schestkowa, H.; Wollborn, T.; Westphal, A.; Wagemans, A.M.; Fritsching, U.; Drusch, S. Conformational state and charge determine the interfacial stabilization process of beta-lactoglobulin at preoccupied interfaces. J. Colloid Interface Sci. 2019, 536, 300–309. [Google Scholar] [CrossRef]
- Kieserling, H.; Pankow, A.; Keppler, J.K.; Wagemans, A.M.; Drusch, S. Conformational state and charge determine the interfacial film formation and film stability of β-lactoglobulin. Food Hydrocoll. 2021, 114, 106561. [Google Scholar] [CrossRef]
- de la Torre, J.G.; Huertas, M.L.; Carrasco, B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000, 78, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Beretta, S.; Chirico, G.; Baldini, G. Short-range interactions of globular proteins at high ionic strengths. Macromolecules 2000, 33, 8663–8670. [Google Scholar] [CrossRef]
- Jung, D.M.; Ebeler, S.E. Investigation of Binding Behavior of α- and β-lonones to β-Lactoglobulin at different pH values using a diffusion-based NOE pumping technique. J. Agric. Food Chem. 2003, 51, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Verheul, M.; Pedersen, J.S.; Roefs, S.P.F.M.; de Kruif, K.G. Association behavior of native β-lactoglobulin. Biopolymers 1999, 49, 11–20. [Google Scholar] [CrossRef]
- Gottschalk, M.; Nilsson, H.; Roos, H.; Halle, B. Protein self-association in solution: The bovine β-lactoglobulin dimer and octamer. Protein Sci. 2003, 12, 2404–2411. [Google Scholar] [CrossRef]
- Aymard, P.; Durand, D.; Nicolai, T. The effect of temperature and ionic strength on the dimerisation of β-lactoglobulin. Int. J. Biol. Macromol. 1996, 19, 213–221. [Google Scholar] [CrossRef]
- Sakai, K.; Sakurai, K.; Sakai, M.; Hoshino, M.; Goto, Y. Conformation and stability of thiol-modified bovine β-lactoglobulin. Prot. Sci. 2000, 9, 1719–1729. [Google Scholar]
- MacRitchie, F. Reversibility of protein adsorption. In Proteins at Liquid Interfaces, Volume 7 in: Studies in Interface Science; Möbius, D., Miller, R., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Chapter 4. [Google Scholar]
- Fainerman, V.B.; Miller, R.; Ferri, J.K.; Watzke, H.; Leser, M.E.; Michel, M. Reversibility and irreversibility of adsorption of surfactants and proteins at liquid interfaces. Adv. Colloid Interface Sci. 2006, 123–126, 163–171. [Google Scholar] [CrossRef]
- Perriman, A.W.; Henderson, M.J.; Holt, S.A.; White, J.W. Effect of the air-water interface on the stability of β-lactoglobulin. J. Phys. Chem. B 2007, 111, 13527–13537. [Google Scholar] [CrossRef]
- Mercadante, D.; Melton, L.D.; Norris, G.E.; Loo, T.S.; Williams, M.A.K.; Dobson, R.C.J.; Jameson, G.B. Bovine β-lactoglobulin is dimeric under imitative physiological conditions: Dissociation equilibrium and rate constants over the pH range of 2.5–7.5. Biophys. J. 2012, 103, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Parameters | [unit] | pH 7 | pH 3 | pH 5 | |||
|---|---|---|---|---|---|---|---|
| Input parameters | |||||||
| Π* (Critical surface pressure) | [mN/m] | 15.1 | 15.2 | 15.0 | |||
| ω1 (Minimum molar area) or (Area per molecule) | [m2/mol] [nm2/molecule] | 9.7 × 106 16.1 | 8.4 × 106 14.0 | 4.5 × 106 7.5 | |||
| ωn (Maximum molar area) or (Area per molecule) | [m2/mol] [nm2/molecule] | 2.40 × 107 39.8 | 2.95 × 107 49.0 | 2.02 × 107 33.5 | |||
| ω0 (Area increment) or (Area per increment) | [m2/mol] [nm2/increment] | 2 × 105 0.33 | 2 × 105 0.33 | 2 × 105 0.33 | |||
| bj (Local adsorption constant, monolayer) | [m3/mol] | 2400 | 50 | 27 | |||
| α (Exponential coefficient) | [-] [-] [-] | 2.2 | 2.7 | 3.8 | |||
| a (Frumkin interaction parameter) | 0.70 | 0.66 | 0.92 | ||||
| na (Aggregation number) | 11 | 16 | 7 | ||||
| m (Number of layers) | [-] | 1 | 2 | 1 | 2 | 1 | 2 |
| b2 (Adsorption constant, 2nd layer) | [m3/mol] | 0 | 3 | 0 | 0.6 | 0 | 50 |
| Output parameters a | |||||||
| n (Number of adsorbed states) | [-] | 72 | 106 | 79 | |||
| cΠ=0.1 c * cm2 | [M] [M] [M] | 7 × 10−10 7 × 10−9 5 × 10−6 | 7 × 10−9 8 × 10−8 3 × 10−5 | 3 × 10−9 2 × 10−8 3 × 10−7 | |||
| ΓΠ=0.1 Γ* Γm2 | [mg/m2] [mg/m2] [mg/m2] | 0.42 0.97 | 0.31 0.80 | 0.69 1.09 | |||
| 1.50 b | 1.50 b | 1.35 b | |||||
| ωΠ=0.1 ω* ωm2 | [nm2/molecule] [nm2/molecule] [nm2/molecule] | 31.4 29.3 | 38.9 35.7 | 27.9 26.5 | |||
| 20.4 ± 0.2 b | 20.4 ± 0.2 b | 22.4 ± 0.2 b | |||||
| θΠ=0.1 θ* | [-] [-] | 0.44 | 0.40 | 0.63 | |||
| 0.94 | 0.94 | 0.95 | |||||
| E0* | [mN/m] [mN/m] [mN/m] | 87 | 76 | 113 | |||
| Πm2 E0,m2 | 20 | 20 | 19.5 | ||||
| 135 | 144 | 140 | |||||
| K* (Relative compression at ω*) c Km2 (Relative compression at ωm2) c | [%] [%] | 7 35 d | 8 48 d | 5 20 d | |||
| pH 7 | pH 3 | pH 5 | ||||
|---|---|---|---|---|---|---|
| m1 | m2 | m1 | m2 | m1 | m2 | |
| Dapp [m2/s] | 8.0 × 10−15 | 1.2 × 10−14 | 1.3 × 10−15 | 1.8 × 10−15 | 1.0 × 10−11 | 3.8 × 10−11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gochev, G.G.; Kovalchuk, V.I.; Aksenenko, E.V.; Fainerman, V.B.; Miller, R. β-Lactoglobulin Adsorption Layers at the Water/Air Surface: 5. Adsorption Isotherm and Equation of State Revisited, Impact of pH. Colloids Interfaces 2021, 5, 14. https://0-doi-org.brum.beds.ac.uk/10.3390/colloids5010014
Gochev GG, Kovalchuk VI, Aksenenko EV, Fainerman VB, Miller R. β-Lactoglobulin Adsorption Layers at the Water/Air Surface: 5. Adsorption Isotherm and Equation of State Revisited, Impact of pH. Colloids and Interfaces. 2021; 5(1):14. https://0-doi-org.brum.beds.ac.uk/10.3390/colloids5010014
Chicago/Turabian StyleGochev, Georgi G., Volodymyr I. Kovalchuk, Eugene V. Aksenenko, Valentin B. Fainerman, and Reinhard Miller. 2021. "β-Lactoglobulin Adsorption Layers at the Water/Air Surface: 5. Adsorption Isotherm and Equation of State Revisited, Impact of pH" Colloids and Interfaces 5, no. 1: 14. https://0-doi-org.brum.beds.ac.uk/10.3390/colloids5010014