20 kDa PEGylated Adrenomedullin as a New Therapeutic Candidate for Inflammatory Bowel Disease

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results
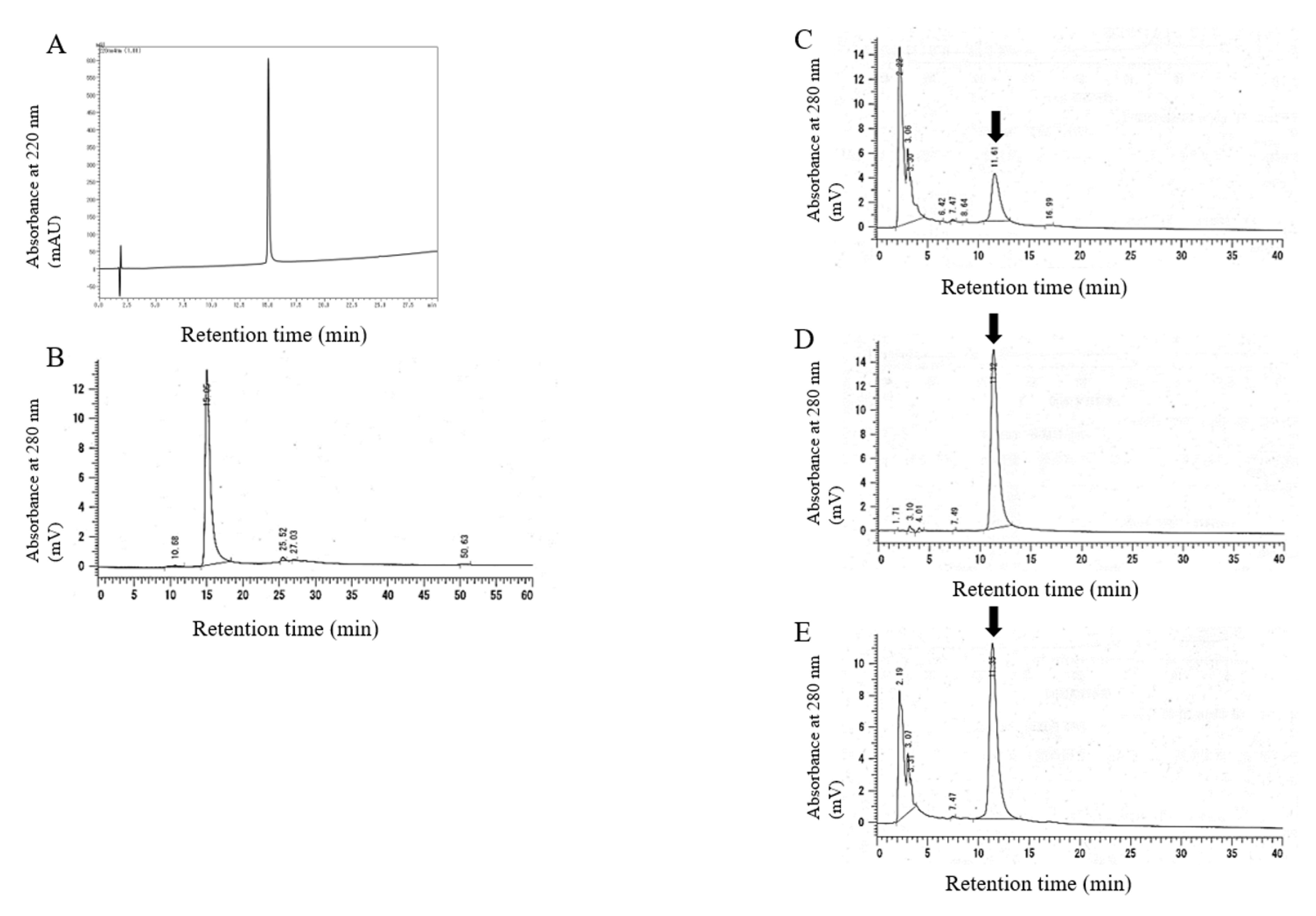
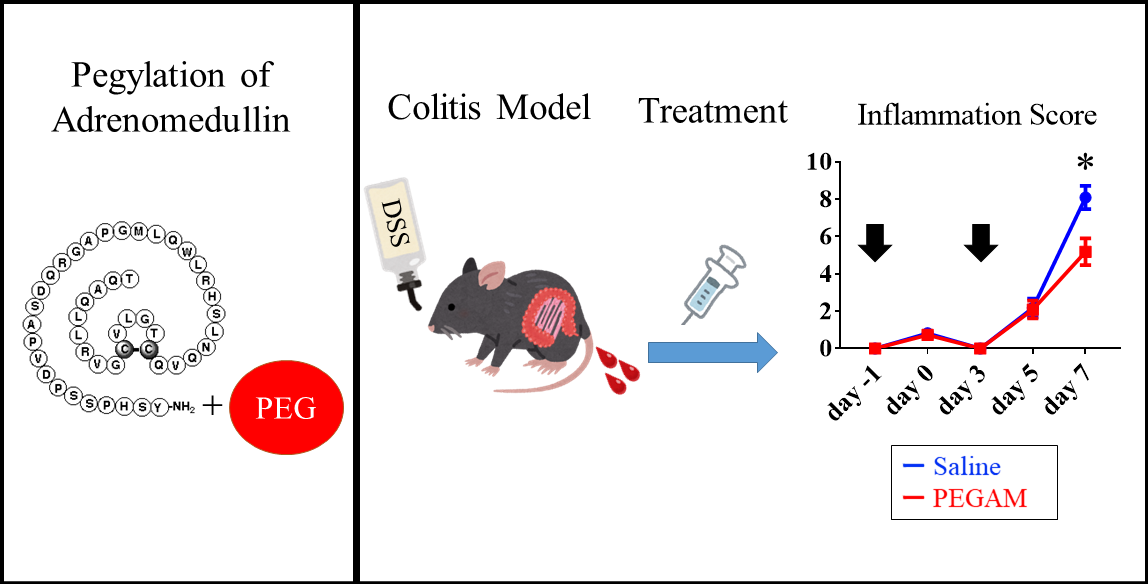
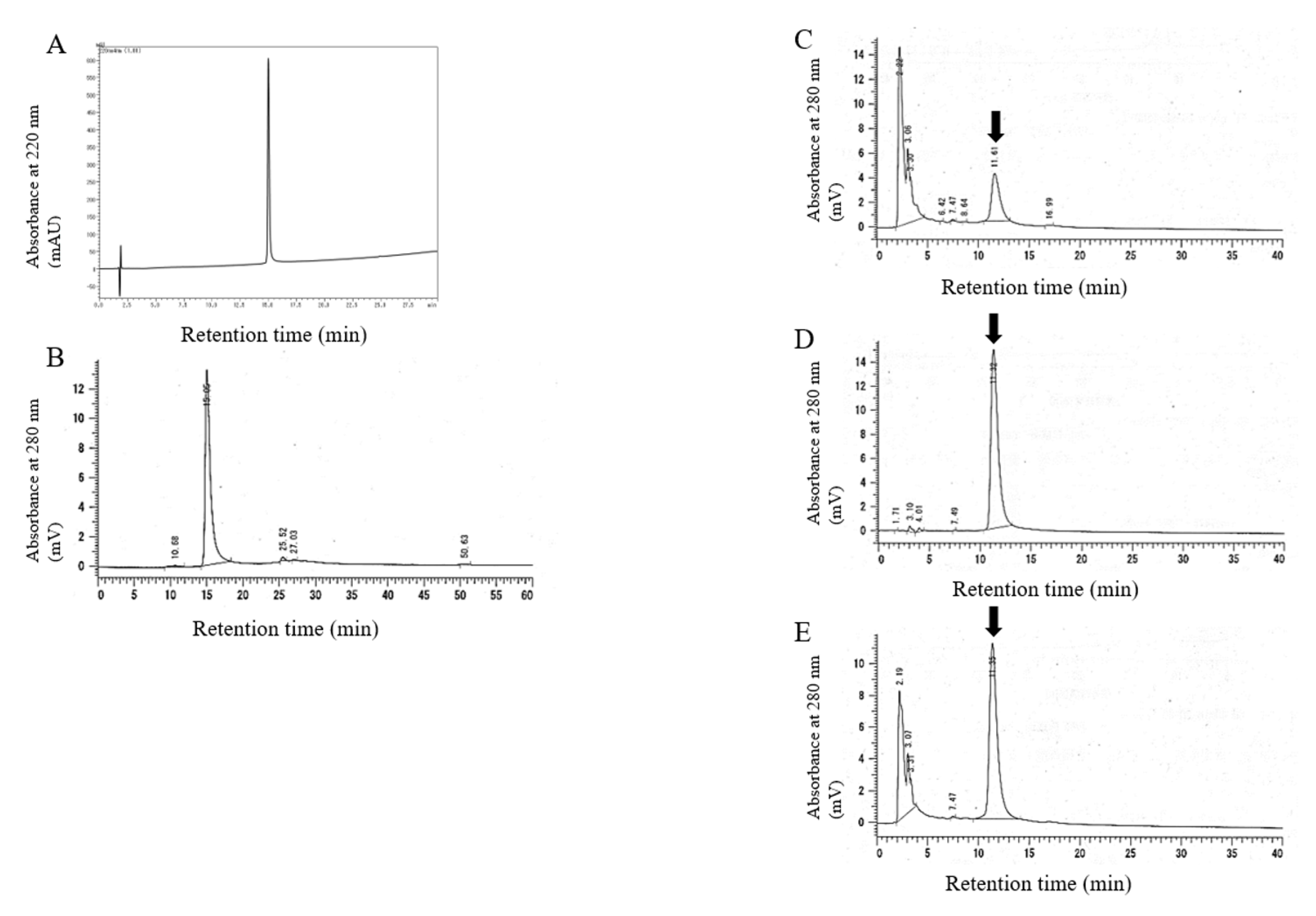
2.1. Synthesis and Characterization of 20kPEG-AM
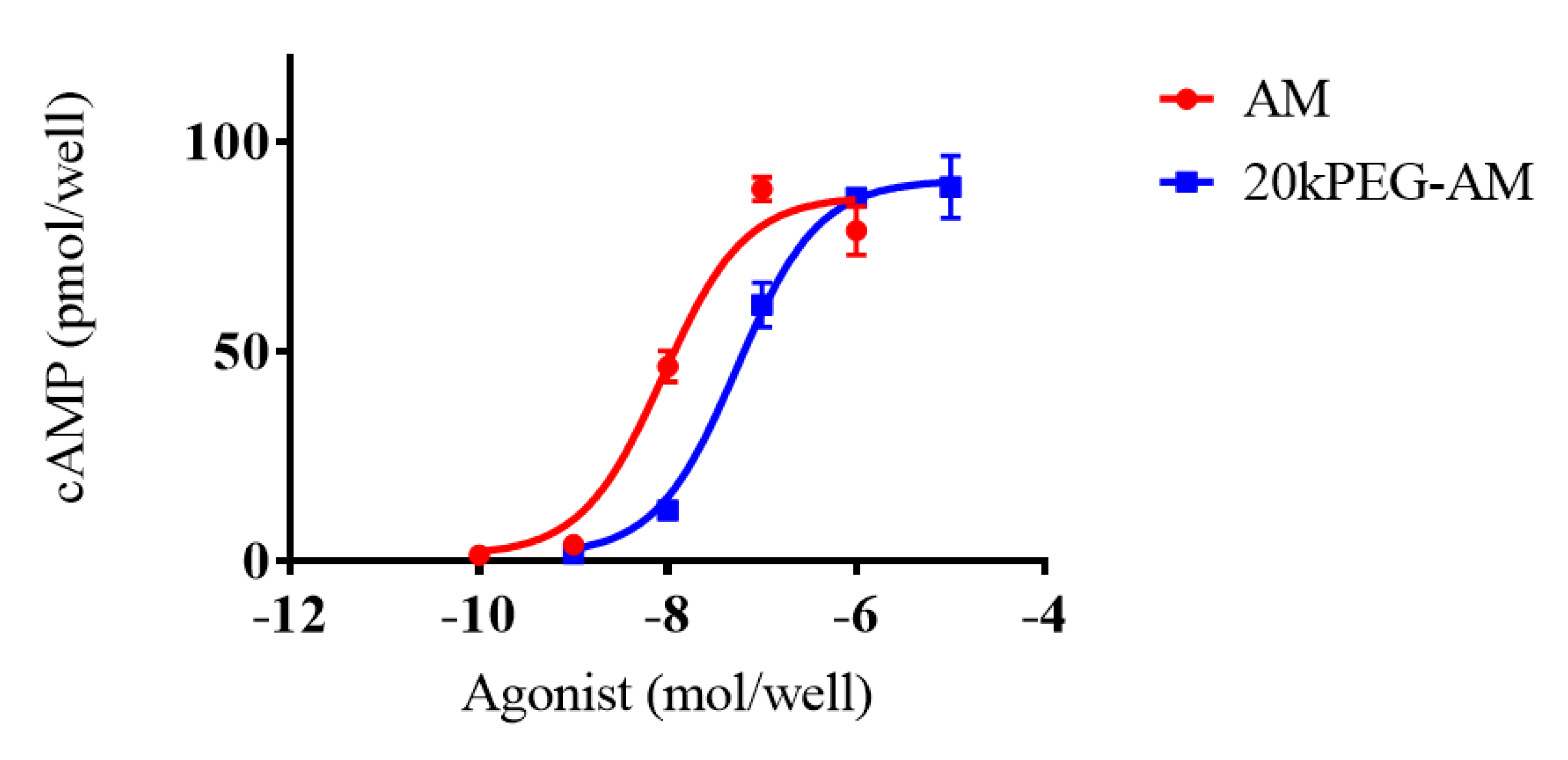
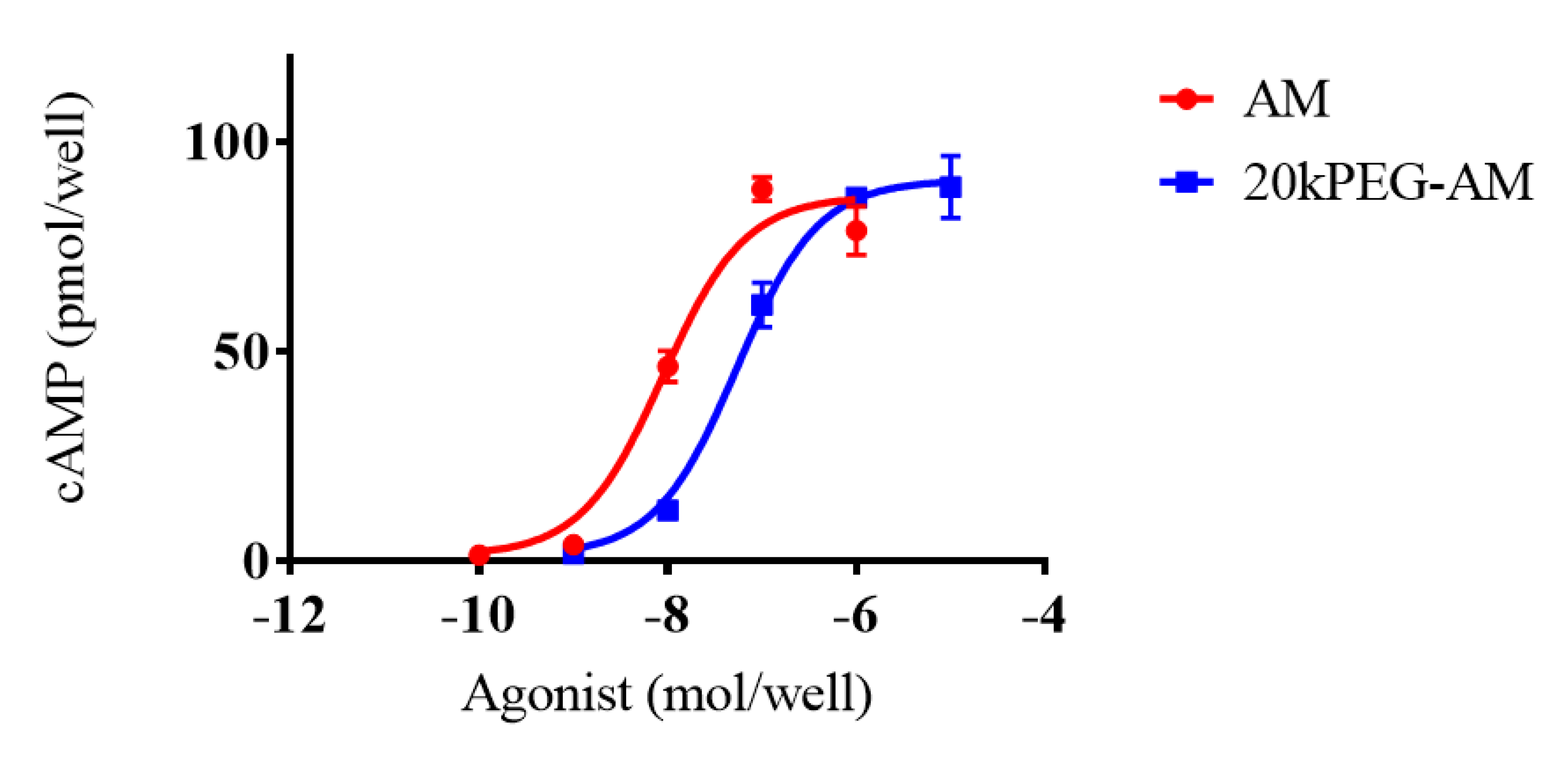
2.2. Cell Culture Experiments
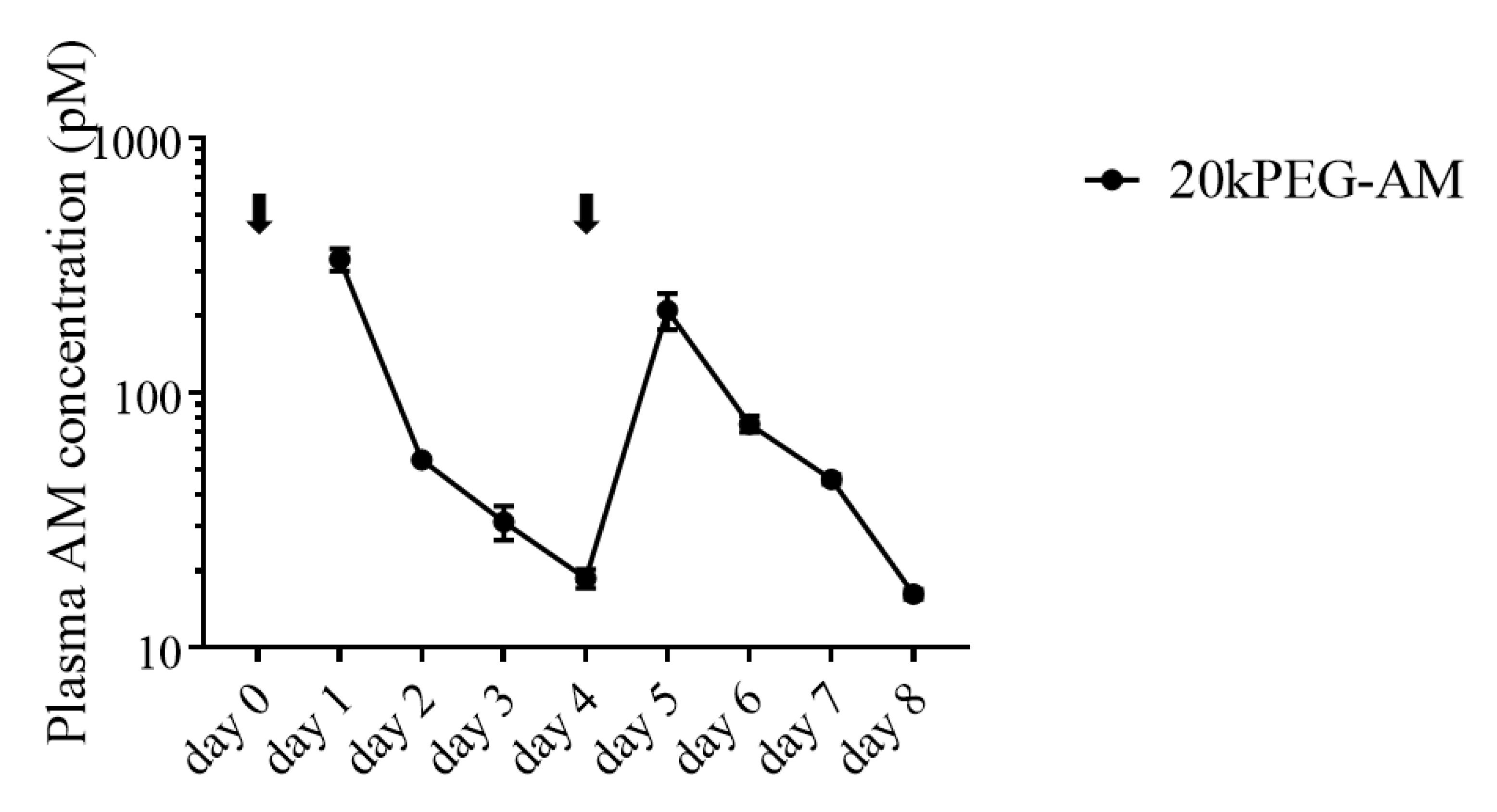
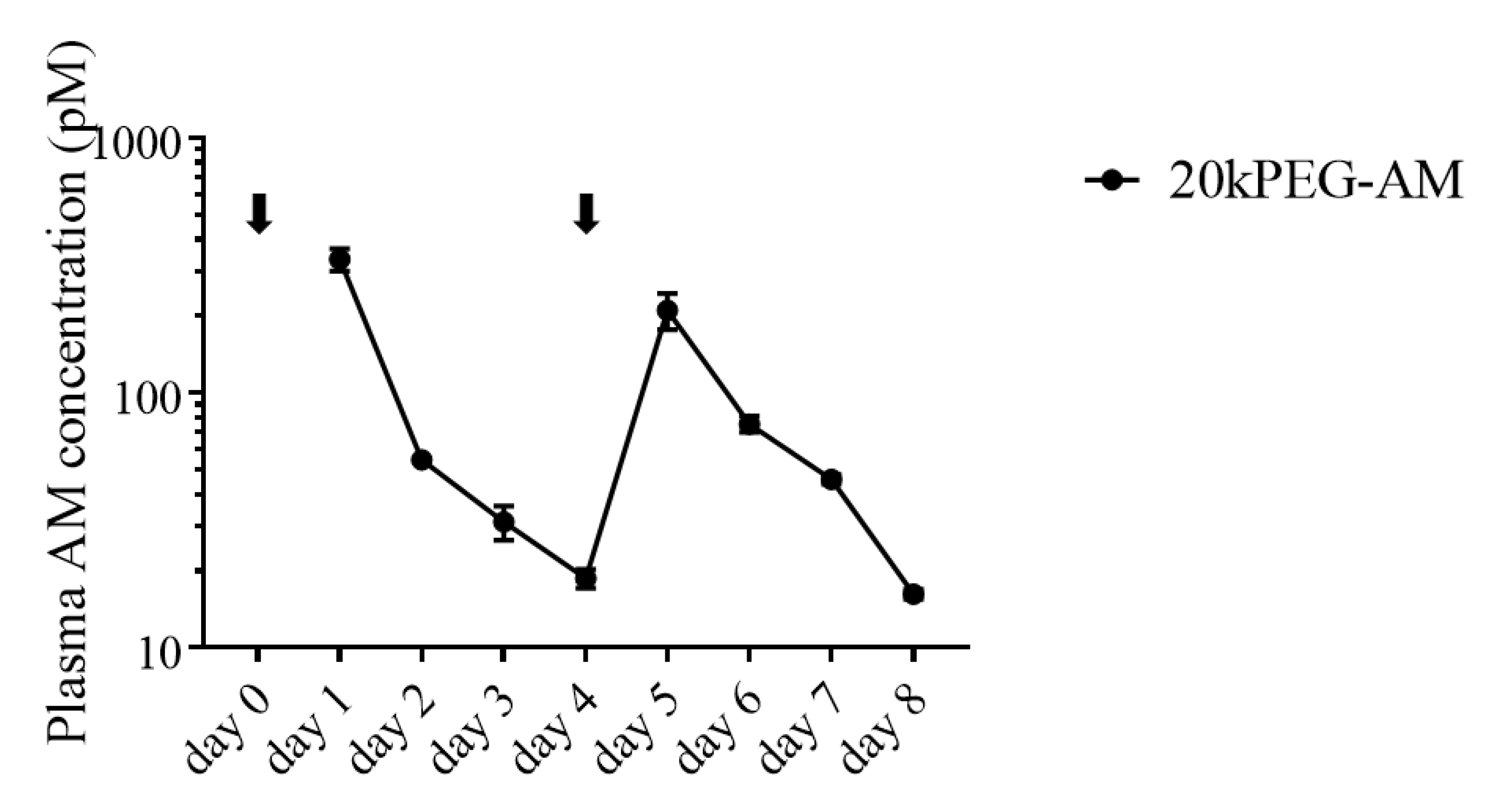
2.3. Plasma Levels of 20kPEG-AM Following Subcutaneous Administration in Mice
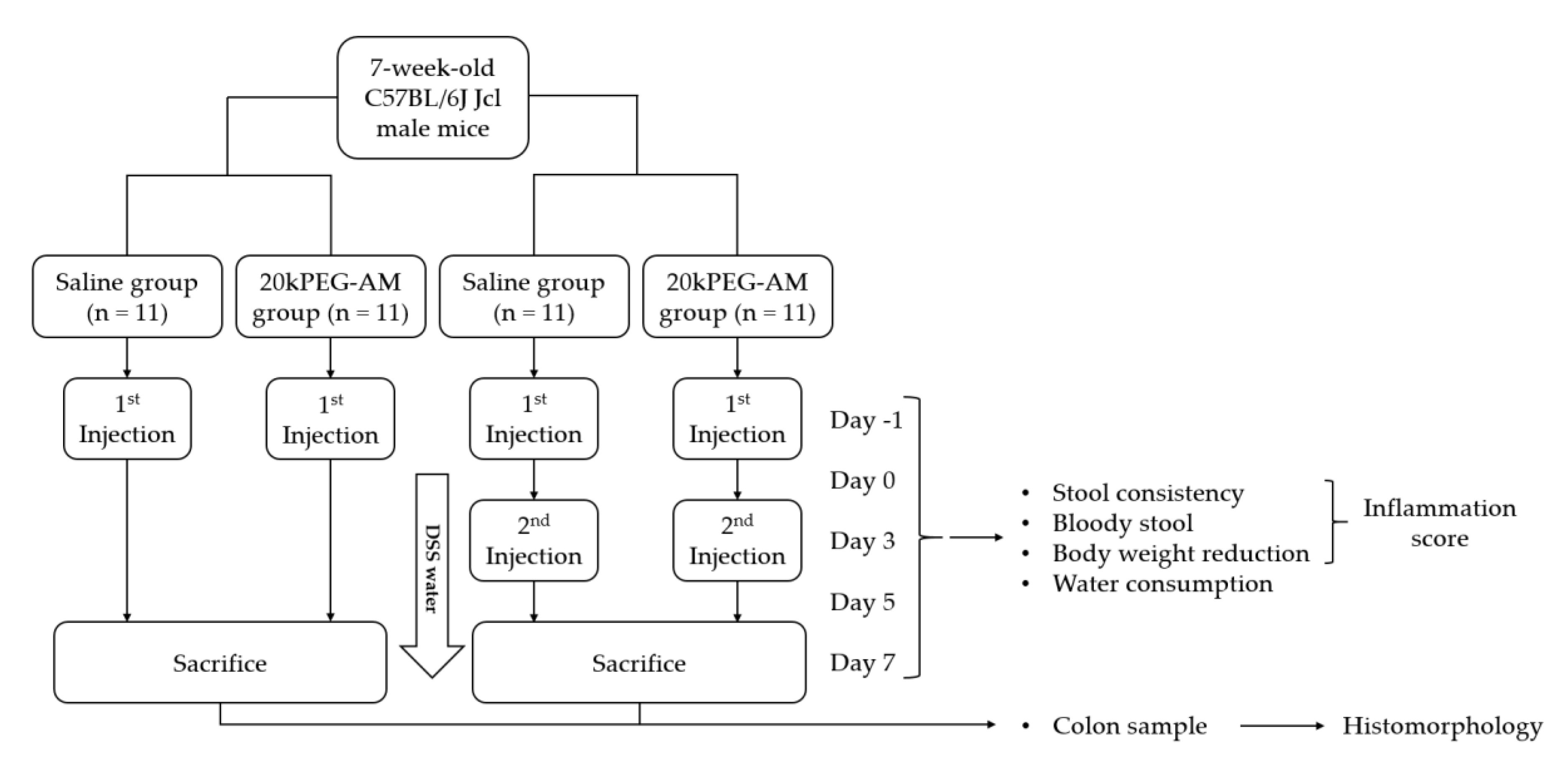
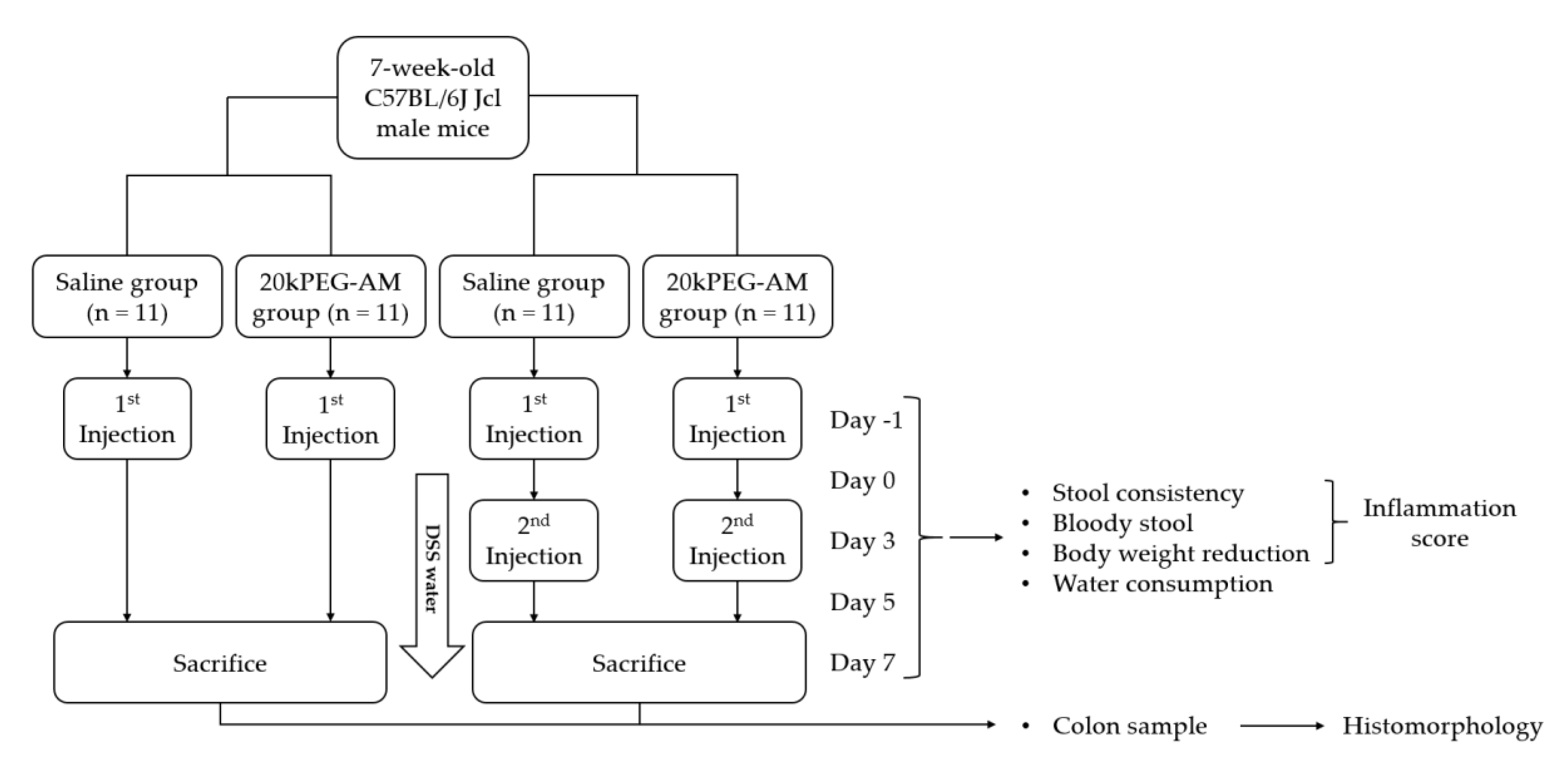
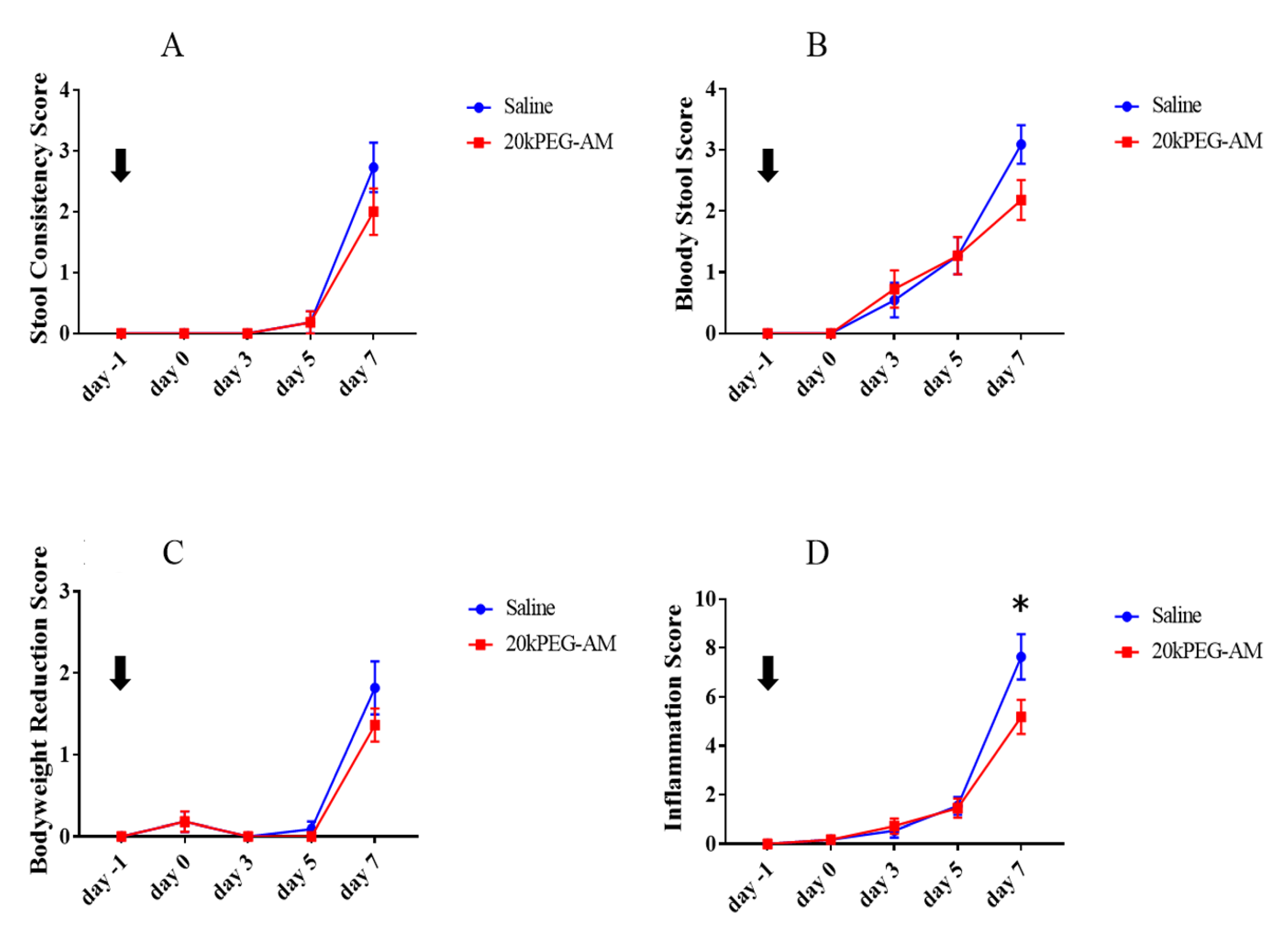
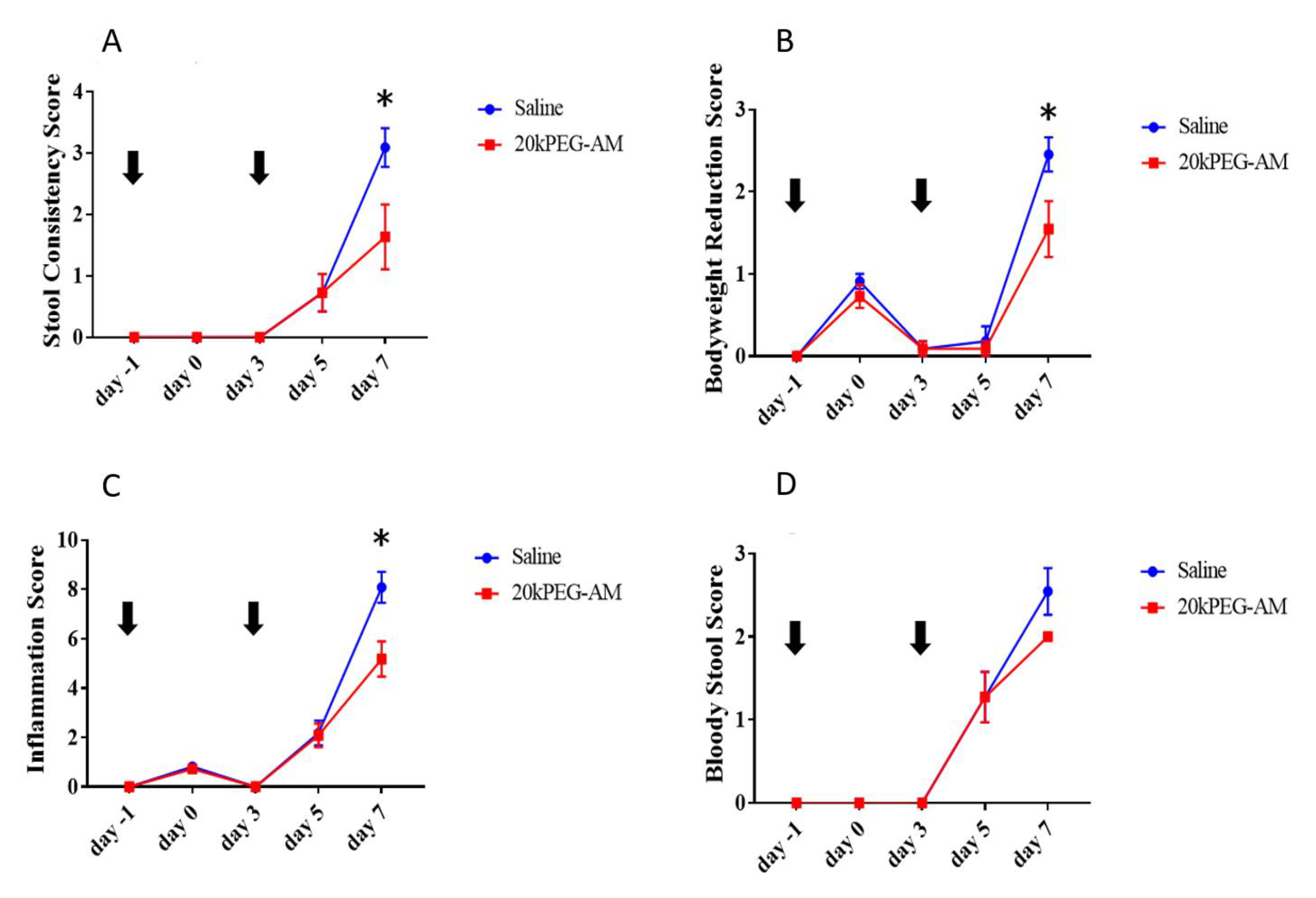
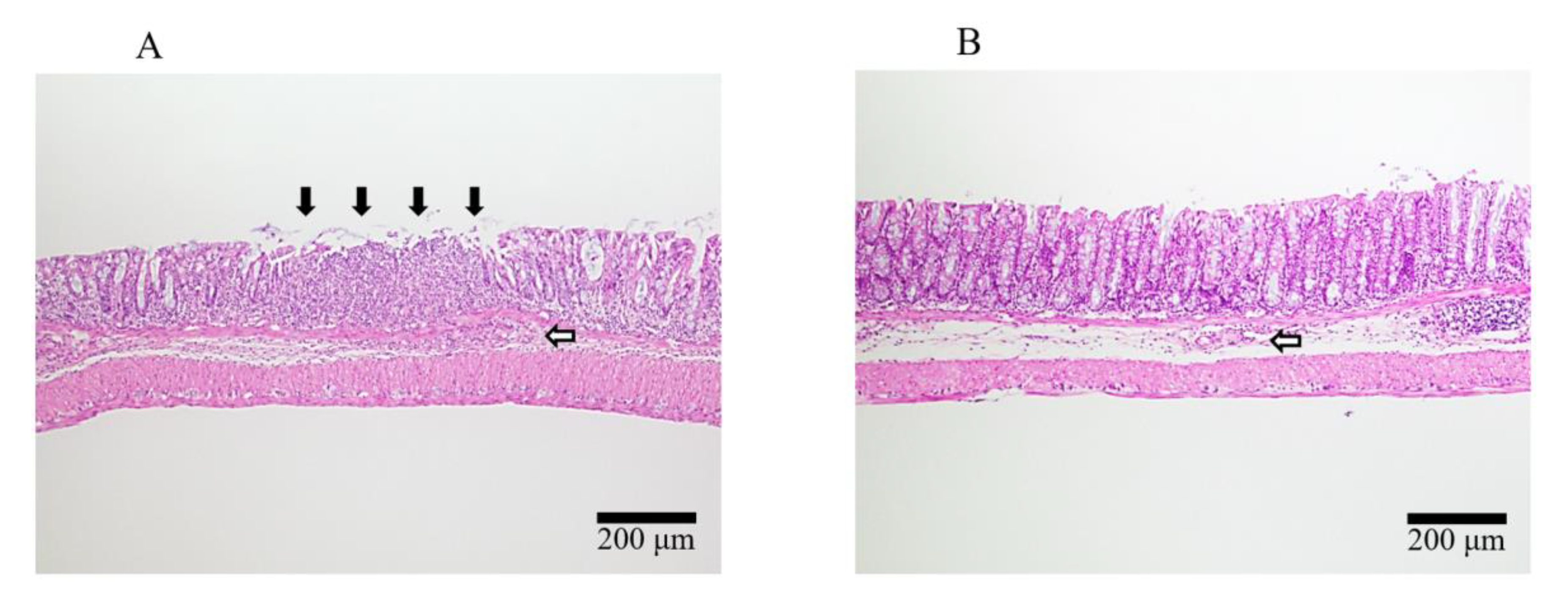
2.4. Induction of DSS Colitis and Treatment with 20kPEG-AM
3. Discussion
4. Materials and Methods
4.1. Analysis of 20kPEG-AM
4.2. Cell Culture Experiments
4.3. Animal Experiments
4.3.1. Plasma Concentration of 20kPEG-AM Following Subcutaneous Administration in Mice
4.3.2. Induction of DSS Colitis and Treatment with 20kPEG-AM
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kitamura, K.; Kangawa, K.; Kawamoto, M.; Ichiki, Y.; Nakamura, S.; Matsuo, H.; Eto, T. Adrenomedullin: A novel hypotensive peptide isolated from human pheochromocytoma. Biochem. Biophys. Res. Commun. 1993, 192, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Kangawa, K.; Eto, T. Adrenomedullin and PAMP: Discovery, structures, and cardiovascular functions. Microsc. Res. Tech. 2002, 57, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kitamura, K. Adrenomedullin peptides. In Handbook of Biologically Active Peptides; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1361–1368. [Google Scholar]
- Nagata, S.; Yamasaki, M.; Kitamura, K. Polyethylene glycol-conjugated human adrenomedullin as a possible treatment for vascular dementia. Peptides 2019, 121, 170133. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kitamura, K. Bench-to-bedside pharmacology of adrenomedullin. Eur. J. Pharm. 2015, 764, 140–148. [Google Scholar] [CrossRef]
- Nakamura, R.; Kato, J.; Kitamura, K.; Imamura, T.; Eto, T. Potential of adrenomedullin as a therapeutic tool for left ventricular remodeling after myocardial infarction. Nihon Rinsho 2004, 62 (Suppl. 9), 302–306. [Google Scholar]
- Kinoshita, Y.; Arita, S.; Murazoe, H.; Kitamura, K.; Ashizuka, S.; Inagaki-Ohara, K. Subcutaneously administered adrenomedullin exerts a potent therapeutic effect in a murine model of ulcerative colitis. Hum. Cell 2019, 32, 12–21. [Google Scholar] [CrossRef]
- Iesato, Y.; Yuda, K.; Chong, K.T.; Tan, X.; Murata, T.; Shindo, T.; Yanagi, Y. Adrenomedullin: A potential therapeutic target for retinochoroidal disease. Prog. Retin Eye Res. 2016, 52, 112–129. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, B.-H.; Yu, Y.-R.; Tang, C.-S.; Qi, Y.-F. Adrenomedullin protects against fructose-induced insulin resistance and myocardial hypertrophy in rats. Peptides 2011, 32, 1415–1421. [Google Scholar] [CrossRef]
- Ashizuka, S.; Inatsu, H.; Kita, T.; Kitamura, K. Adrenomedullin Therapy in Patients with Refractory Ulcerative Colitis: A Case Series. Dig. Dis. Sci. 2016, 61, 872–880. [Google Scholar] [CrossRef] [Green Version]
- Ashizuka, S.; Kuroishi, N.; Nakashima, K.; Inatsu, H.; Kita, T.; Kitamura, K. Adrenomedullin: A Novel Therapy for Intractable Crohn’s Disease with a Loss of Response to Infliximab. Intern. Med. 2019, 58, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Kubo, K.; Tokashiki, M.; Kuwasako, K.; Tamura, M.; Tsuda, S.; Kubo, S.; Yoshizawa-Kumagaye, K.; Kato, J.; Kitamura, K. Biological properties of adrenomedullin conjugated with polyethylene glycol. Peptides 2014, 57, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Yamasaki, M.; Kitamura, K. Anti-Inflammatory Effects of PEGylated Human Adrenomedullin in a Mouse DSS-Induced Colitis Model. Drug Dev. Res. 2017, 78, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-Life Extension of Biopharmaceuticals using Chemical Methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef]
- Inui, T.; Bodi, J.; Kubo, S.; Nishio, H.; Kimura, T.; Kojima, S.; Maruta, H.; Muramatsu, T.; Sakakibara, S. Solution synthesis of human midkine, a novel heparin-binding neurotrophic factor consisting of 121 amino acid residues with five disulphide bonds. J. Pept. Sci. 1996, 2, 28–39. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Bailon, P.; Berthold, W. Polyethylene glycol-conjugated pharmaceutical proteins. Pharm. Sci. Technol. Today 1998, 1, 352–356. [Google Scholar] [CrossRef]
- Kozlowski, A.; Harris, J.M. Improvements in protein PEGylation: Pegylated interferons for treatment of hepatitis C. J. Control Release 2001, 72, 217–224. [Google Scholar] [CrossRef]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [Green Version]
- Contrera, J.F.; Matthews, E.J.; Kruhlak, N.L.; Benz, R.D. Estimating the safe starting dose in phase I clinical trials and no observed effect level based on QSAR modeling of the human maximum recommended daily dose. Regul Toxicol. Pharm. 2004, 40, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Milton Harris, J.; Martin, N.E.; Modi, M. Pegylation: A novel process for modifying pharmacokinetics. Clin. Pharm. 2001, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Karasawa, T.; Inaba, C.; Ishimura, K.; Tadokoro, K.; Koshikawa, S.; Yoshihara, F.; Nagaya, N.; Sakio, H.; Kangawa, K.; et al. Effects of long-term intravenous administration of adrenomedullin (AM) plus hANP therapy in acute decompensated heart failure: A pilot study. Circ. J. 2009, 73, 892–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, Y.; Miyazaki, S.; Yasuda, S.; Nagaya, N.; Noguchi, T.; Yamada, N.; Morii, I.; Kawamura, A.; Doi, K.; Miyatake, K.; et al. The first clinical pilot study of intravenous adrenomedullin administration in patients with acute myocardial infarction. J. Cardiovasc. Pharm. 2010, 56, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Duffey, M.E.; Hainau, B.; Ho, S.; Bentzel, C.J. Regulation of epithelial tight junction permeability by cyclic AMP. Nature 1981, 294, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.B.; Taylor, C.T.; Parkos, C.A.; Colgan, S.P. Epithelial permeability induced by neutrophil transmigration is potentiated by hypoxia: Role of intracellular cAMP. J. Cell Physiol. 1998, 176, 76–84. [Google Scholar] [CrossRef]
- Lawrence, D.W.; Comerford, K.M.; Colgan, S.P. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am. J. Physiol. Cell Physiol. 2002, 282, C1235–C1245. [Google Scholar] [CrossRef] [Green Version]
- MacManus, C.F.; Campbell, E.L.; Keely, S.; Burgess, A.; Kominsky, D.J.; Colgan, S.P. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J. 2011, 25, 1856–1864. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Narumi, K.; Tsuji, S.; Tsubokawa, T.; Nakaya, M.A.; Wakayama, T.; Zuka, M.; Ohshima, T.; Yamagishi, M.; Okada, T. Impact of adrenomedullin on dextran sulfate sodium-induced inflammatory colitis in mice: Insights from in vitro and in vivo experimental studies. Int. J. Colorectal Dis. 2011, 26, 1453–1462. [Google Scholar] [CrossRef]
- Ashizuka, S.; Inagaki-Ohara, K.; Kuwasako, K.; Kato, J.; Inatsu, H.; Kitamura, K. Adrenomedullin treatment reduces intestinal inflammation and maintains epithelial barrier function in mice administered dextran sulphate sodium. Microbiol. Immunol. 2009, 53, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Herrero, S.; Larrayoz, I.M.; Narro-Íñiguez, J.; Rubio-Mediavilla, S.; Martínez, A. Lack of Adrenomedullin Aggravates Acute TNBS-Induced Colitis Symptoms in Mice, Especially in Females. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Herrero, S.; Larrayoz, I.M.; Narro-Íñiguez, J.; Villanueva-Millán, M.J.; Recio-Fernández, E.; Pérez-Matute, P.; Oteo, J.A.; Martínez, A. Lack of Adrenomedullin Results in Microbiota Changes and Aggravates Azoxymethane and Dextran Sulfate Sodium-Induced Colitis in Mice. Front. Physiol. 2016, 7, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, N.; Maemura, K.; Hirata, I.; Murano, M.; Sasaki, S.; Katsu, K. Inhibition of dextran sulphate sodium (DSS)-induced colitis in mice by intracolonically administered antibodies against adhesion molecules (endothelial leucocyte adhesion molecule-1 (ELAM-1) or intercellular adhesion molecule-1 (ICAM-1)). Clin. Exp. Immunol. 1999, 117, 462. [Google Scholar] [CrossRef]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kühl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid (Expected Residues/Molecule) Residues/Molecule | |||
|---|---|---|---|
| Asp (6) 6.00 | Thr (3) 2.92 | Ser (4) 3.63 | Glu (6) 5.99 |
| Gly (4) 3.96 | Ala (2) 1.99 | Cys (2) 1.85 | Val (2) 2.01 |
| Met (1) 0.93 | Ile (2) 1.96 | Leu (2) 2.00 | Tyr (3) 2.84 |
| Phe (4) 3.97 | Lys (4) 3.98 | NH3 (10) 10.72 | |
| His (1) 1.03 | Arg (4) 4.01 | Pro (2) 2.03 | |
| Score | Stool Consistency | Bloody Stool | Bodyweight Reduction |
|---|---|---|---|
| 0 | Normal | No blood | No reduction |
| 1 | 1–5% reduction | ||
| 2 | Soft | Fecal occult blood | 5–10% reduction |
| 3 | 10–20% reduction | ||
| 4 | Diarrhea | Mucus and bloody stool |
| Inflammatory Cell Infiltrate | Intestinal Architecture | ||||
|---|---|---|---|---|---|
| Severity | Extent | Score 1 | Epithelial Changes | Mucosal Architecture | Score 2 |
| Mild | Mucosa | 1 | Focal erosions | 1 | |
| Moderate | Mucosa and submucosa | 2 | Erosions | ± Focal ulcerations | 2 |
| Marked | Transmural | 3 | Extended ulcerations ± granulation tissue ± pseudopolyps | 3 | |
| Sum of scores 1 and 2: | 0–6 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miki, G.; Kuroishi, N.; Tokashiki, M.; Nagata, S.; Tamura, M.; Yoshiya, T.; Yoshizawa-Kumagaye, K.; Ashizuka, S.; Kato, J.; Yamasaki, M.; et al. 20 kDa PEGylated Adrenomedullin as a New Therapeutic Candidate for Inflammatory Bowel Disease. Gastrointest. Disord. 2020, 2, 366-377. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2040033
Miki G, Kuroishi N, Tokashiki M, Nagata S, Tamura M, Yoshiya T, Yoshizawa-Kumagaye K, Ashizuka S, Kato J, Yamasaki M, et al. 20 kDa PEGylated Adrenomedullin as a New Therapeutic Candidate for Inflammatory Bowel Disease. Gastrointestinal Disorders. 2020; 2(4):366-377. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2040033
Chicago/Turabian StyleMiki, Goro, Nobuko Kuroishi, Mariko Tokashiki, Sayaka Nagata, Masaji Tamura, Taku Yoshiya, Kumiko Yoshizawa-Kumagaye, Shinya Ashizuka, Joji Kato, Motoo Yamasaki, and et al. 2020. "20 kDa PEGylated Adrenomedullin as a New Therapeutic Candidate for Inflammatory Bowel Disease" Gastrointestinal Disorders 2, no. 4: 366-377. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2040033