Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48


Research Institute of Energy Process, National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba West, 16-1 Onogawa, Tsukuba, Ibaraki 305-8569, Japan
Reactions 2020, 1(2), 195-209; https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020014
Submission received: 27 November 2020
/
Revised: 11 December 2020
/
Accepted: 17 December 2020
/
Published: 20 December 2020
(This article belongs to the Special Issue Catalytic Conversion of Carbonaceous Materials to Fuels and Chemicals)
Abstract
:A low-density polyethylene was hydrocracked to liquid hydrocarbons in autoclave reactors over catalysts containing Pt- and Al-modified MCM-48. Two kinds of Al-modified MCM-48 were synthesized for the reaction: Al-MCM-48 was synthesized using a sol–gel method by mixing Al(iso-OC3H7)3 with Si(OC2H5)4 and surfactant in a basic aqueous solution before hydrothermal synthesis, and Al/MCM-48 was synthesized using a post-modification method by grafting Al3+ ions on the surface of calcined Al/MCM-48. X-ray diffraction (XRD) patterns indicated that both Al-MCM-48 and Al/MCM-48 had a cubic mesoporous structure. The Brunauer–Emmett–Teller (BET) surface areas of Al-MCM-48 and Al/MCM-48 were larger than 1000 m2/g. 27Al Magic Angle Spinning-NMR (MAS NMR) indicated that Al3+ in Al-MCM-48 was located inside the framework of mesoporous silica, but Al3+ in Al/MCM-48 was located outside the framework of mesoporous silica. The results of ammonia temperature-programmed desorption (NH3-TPD) showed that the acidic strength of various samples was in the order of H-Y > Al/MCM-48 > Al-MCM-48 > MCM-48. After 4 MPa H2 was charged in the autoclave at room temperature, 1 wt % Pt/Al/MCM-48 catalyst showed a high yield of C9−C15 jet fuel range hydrocarbons of 85.9% in the hydrocracking of polyethylene at 573 K for 4 h. Compared with the reaction results of Pt/Al/MCM-48, the yield of light hydrocarbons (C1−C8) increased over Pt/H-Y, and the yield of heavy hydrocarbons (C16−C21) increased over Pt/Al-MCM-48 in the hydrocracking of polyethylene. The yield of C9−C15 jet fuel range hydrocarbons over the used catalyst did not decrease compared to the fresh catalyst in the hydrocracking of polyethylene to jet fuel range hydrocarbons over Pt/Al/MCM-48.
1. Introduction
The consumption of fossil fuels in transportation causes an increase of total CO2 emission in the world. The use of biomass-derived fuels (called biofuels) instead of fossil fuels is an effort to decrease CO2 emissions because the biomass absorbs CO2 during the growth process [1]. Bioethanol and biodiesel (fatty acid methyl esters) are the main transportation biofuels produced in the world at present [2,3]. However, these oxygen-containing biofuels are not suitable for current jet engines, which have been designed using hydrocarbons as fuel. Currently, jet fuels are almost entirely produced from crude oil taking into account flight safety. Biofuels with a chemical composition of hydrocarbons (called drop-in biofuels) have been researched because they are suitable for the current engines. Biomass-to-liquid fuel (BTL) and hydrotreatment processes are the main methods for producing drop-in biofuels at present. In the BTL process, the woody biomass is converted to syngas by gasification, and the formed syngas is then converted to mixed hydrocarbons by Fisher-Tropsch (F-T) reaction [4,5]. In the hydrotreatment process, vegetable or algae oils are converted to hydrocarbons by catalytic deoxygenation in hydrogen atmosphere [6,7,8,9]. Recently, a process of converting alcohol to jet fuel range hydrocarbons (called the ATJ process) has attracted considerable attention in the world [10]. Ethanol obtained from the fermentation process is the main feedstock in the ATJ process. Because bioethylene is easily obtained from dehydration of bioethanol, the oligomerization of ethylene is a method in the ATJ process [11,12]. However, the oligomerization of ethylene forms a large amount of light hydrocarbons (<C9), which decreases the yield of C9−C15 jet fuel range hydrocarbons from the ATJ process. Because polyethylene is already produced from ethylene polymerization on a large scale in industries, bioethylene obtained from dehydration of bioethanol can be used to produce biopolyethylene using the current industrial process. Moreover, the cracking of polyethylene to jet fuel range hydrocarbons has been researched in recent times [13,14,15,16]. Hence, the cracking of biopolyethylene (obtained from bioethylene) to jet fuel range hydrocarbons is an important reaction in the ATJ process [17,18]. As a result, the development of highly activity catalysts for the cracking of polyethylene to jet fuel range hydrocarbons is a key technology in the ATJ process.
Mesoporous silica materials are attractive materials in the field of heterogeneous catalysis because they possess a large Brunauer–Emmett–Teller (BET) surface area, uniform mesopores, and high thermal stability [19]. MCM-48 is a kind of mesoporous silica with cubic pore structures [20]. Compared to the hexagonal mesoporous silica MCM-41 (with one-dimensional pore channel), the cubic mesopore structure in MCM-48 has two independent, three-dimensional pore channels [21]. Hence, it is difficult to block the mesopores of MCM-48 during the reaction [21]. MCM-48-based materials have recently been used in the pyrolysis of biomass to liquid fuels [22,23,24].
In this study, the hydrocracking of polyethylene to jet fuel range hydrocarbons was achieved using bifunctional catalysts containing Pt- and Al-modified MCM-48. A 1 wt % Pt/Al/MCM-48 catalyst showed a high yield of C9−C15 jet fuel range hydrocarbons of 85.9% in the hydrocracking of polyethylene at 573 K for 4 h. The catalytic performance of the 1 wt % Pt/Al/MCM-48 catalysts was higher than those of the catalysts reported in the literature [13,14,15,16].
2. Materials and Methods
2.1. Catalyst Synthesis
MCM-48 was synthesized using a sol–gel method. Si(OC2H5)4 was used as a silicon source, cetyltrimethylammonium bromide (CTAB) was used as a surfactant, and NaOH was used as a OH− source [21]. First, a solution with a molar ratio of 1 Si/0.5 CTAB/0.5 OH−/170 H2O was stirred in a beaker to form a gel at room temperature. The formed gel was moved to a Teflon-coated autoclave, and the autoclave was then heated at 373 K for 7 days. After the hydrothermal synthesis process, the solid material was filtrated and then washed with distilled water. The solid material was dried in air at 383 K for 12 h to obtain as-synthesized MCM-48. Na-type MCM-48 was obtained by calcining the as-synthesized MCM-48 in air at 823 K for 4 h.
Al-MCM-48 was synthesized using Al(iso-OC3H7)3 as an aluminum source [25,26]. A gel with a molar ratio of 0.1 Al/1 Si/0.5 CTAB/0.5 OH−/170 H2O was synthesized. The gel was heated in a Teflon-coated autoclave at 373 K for 7 days. After the hydrothermal synthesis process, the solid material in the autoclave was filtrated and then washed with distilled water. As-synthesized Al-MCM-48 was obtained by drying the solid material at 383 K for 12 h. Na-type Al-MCM-48 was obtained by calcining the as-synthesized Al-MCM-48 at 823 K for 4 h.
Al/MCM-48 was synthesized using a post-modification method by grafting Al3+ ions on the calcined MCM-48 [27,28]. In a 200 mL beaker, a calculated amount of Al(iso-OC3H7)3 was dissolved in 100 mL of isopropyl alcohol to form a solution. A Na-type MCM-48 sample that had been calcined at 823 K for 4 h was added to the beaker (with an Al/Si molar ratio of 1/10) under stirring. Then, 25 mL H2O was added to the beaker at room temperature with stirring to precipitate aluminum oxide. After stirring at room temperature for 3 h, a solid material was obtained by filtration. The solid material was dried at 383 K for 12 h and calcined at 823 K for 4 h to form Na-type Al/MCM-48.
Na-type MCM-48, Al-MCM-48, and Al/MCM-48 were converted to NH4-type samples by ion exchange. One gram of Na-type sample was stirred in 100 mL of NH4NO3 aqueous solution (1 M) at 333 K for 2 h to obtain a NH4-type sample. The NH4-type sample was dried at 383 K for 12 h and calcined at 823 K for 4 h to form a H-type sample.
Na-Y zeolite with a SiO2/Al2O3 ratio of 5.5 was bought from Tosho Chemical Industries, Ltd. NH4-Y was obtained by stirring Na-Y in an aqueous solution of NH4NO3 (1 M) in a beaker. H-Y was obtained by calcination of NH4-Y in air at 823 K for 4 h.
Pt-supported samples (Pt/Al-MCM-48, Pt/Al/MCM-48, and Pt/H-Y) were synthesized by an impregnation method. A H-type sample was stirred in a H2[PtCl4] aqueous solution, and the water was removed by evaporating in vacuum at 368 K. The obtained solid sample was dried at 383 K for 12 h and calcined at 823 K for 4 h. The Pt loadings were 1 wt % in the Pt-supported samples.
2.2. Catalyst Characterization
A MAC Science MXP-18 diffractometer equipped with a Cu Kα radiation (Xray Science Corp., Tokyo, Japan) was used to measure the powder X-ray diffraction pattern (XRD) for the solid samples. The measurement was carried out under the conditions of 40 kV and 50 mA.
27Al MAS NMR spectra were measured using a JEOL ECA-400 multinuclear solid-state magnetic resonance spectrometer (Japan Electron Optics Laboratory, Tokyo, Japan) at a magnetic field of 104 MHz. The chemical shifts were recorded with respect to [Al(H2O)6]3+.
Ammonia temperature-programmed desorption (NH3-TPD) was measured using a BELCAT-B instrument equipped with a thermal conductivity detector (TCD) and a mass spectrometer (Micro BEL Corp., Osaka, Japan). First, 0.1 g solid sample was pretreated at 673 K for 1 h in a 50 mL min−1 He flow. The temperature was decreased to 373 K, and NH3 molecules were then introduced to the solid sample. After eliminating the weakly adsorbed NH3 molecules by evacuation at 373 K for 1 h, NH3-TPD was measured from 373 to 873 K with a temperature increase rate of 8 K min−1.
N2 adsorption–desorption isotherms were measured at 77 K using a Belsorp 28SA automatic adsorption instrument (Micro BEL Corp., Osaka, Japan). The surface areas were calculated using a Brunauer–Emmett–Teller (BET) method, and the pore sizes were calculated using a Barrett–Joyner–Halenda (BJH) method.
The elemental analysis for the actual amount of Pt in the sample was carried out using a Thermo Jarrell Ash IRIS/AP instrument (Spectra Lab Scientific Inc., Markham, ON, Canada).
CO chemisorption was measured using a Shimadzu ASAP 2000 apparatus (Shimadzu Corp., Kyoto, Japan). The Pt particle size was calculated using the amount of irreversible adsorbed CO. The CO uptake was estimated by the extrapolation to zero pressure of the linear part of the isotherms. The difference between the total amount of adsorbed CO (COtot) and the reversible part of adsorbed CO (COrev) gave the irreversible part of adsorbed CO (COirr). The Pt particle size was calculated from the ratio of COirr to total Pt by the following equation:
where α is a geometrical parameter, M is the atomic weight of Pt, a is the effective area occupied by a Pt atom in the surface, ρ is the density of Pt, and N0 is Avogadro’s number. In this study, α was taken as 6 on the assumption of the spherical particle, and a was taken as 12.5 nm−2 according to the literature [29].
d = α(M/aρN0)·(COirr/Pt)−1
2.3. Catalytic Reaction
Low-density polyethylene (particle size: 0.5 mm; purity: >99%) was bought from Scientific Polymer Products, Inc., Tokyo, Japan. The gas cylinders of H2, N2, He, and Ar with purities larger than 99.995% were bought from Takachiho Chemical Industrial Co., Ltd., Tokyo, Japan.
The reaction was carried out in a batch-type reaction system using a 100 mL stainless-steel autoclave equipped with a stirrer. The catalyst was pretreated in 50 mL min−1 H2 flow at 673 K (increasing from room temperature to 673 K at a rate of 10 K min−1) for 1 h. Then, 1 g of the reduced catalyst and 10 g of polyethylene were put in the autoclave reactor. After 4.0 MPa H2 was introduced in the autoclave at room temperature, the autoclave reactor was heated from room temperature to reaction temperature with a temperature increase rate of 10 K min−1 under stirring (400 rpm). The pressure in the autoclave reactor increased with increasing temperature and finally reached about 8.0 MPa at 573 K. During the reaction at 573 K, the pressure in the autoclave slightly changed because of the consumption of H2 and the formation of light hydrocarbons.
The autoclave cooled down to 343 K as soon as the reaction finished. Then, the gas in the autoclave was moved to a plastic gas bag at 343 K. The volume of total gas in the gas bag was measured using a WS-1 integration flow meter (Shinagawa Corp., Tokyo, Japan) at 343 K. The concentrations of various components in the gas bag at 343 K were analyzed using a Shimadzu GC-2014 gas chromatography (GC) equipped with a flame ionization detector (FID) (Shimadzu Corp., Kyoto, Japan). A PoraBOND Q capillary column was set in the GC-FID to separate various components in the gas products. The yield of each hydrocarbon was calculated using its concentration in the total gas (obtained from GC-FID) and the volume of total gas (at 343 K).
After taking out the gas products from the autoclave reactor at 343 K, the autoclave was cooled down to room temperature. Then, the liquid products and the solid materials were taken out from the autoclave reactor.
The liquid products were obtained by filtrating out the solid materials. After the weight of liquid products was measured, a sample containing 10 wt % liquid products in CH2Cl2 solution was prepared for analysis. A Shimadzu GC-2014 type GC-FID and a Shimadzu GCMS-QP2010 Ultra type GC-MS were used for analyzing the liquid sample. The same type of UA-DX30 capillary columns were equipped on GC-FID and GC-MS, and the same temperature programs were used for GC-FID and GC-MS analyses. The temperature program contained three steps: holding at 313 K for 3 min, then increasing from 313 to 613 K with a temperature increase rate of 20 K min−1, and then holding at 613 K for 12 min.
The components in the liquid product were identified by GC-MS with reference to the NIST-11 database, and the concentrations of various components were determined by GC-FID. The position of each normal alkane in the chromatograms of GC-MS and GC-FID were obtained using a pure reagent in CH2Cl2 solution. In GC-FID analysis, the value of the peak area corresponding to 1 wt % concentration of each n-alkane in the standard sample was used as a factor to calculate the amount of each n-alkane in the liquid products. The iso-alkanes and alkenes in the liquid products were identified by GC-MS, and their concentrations were determined by GC-FID analysis (using the factor of n-alkane with the same carbon number).
The solid materials after reaction contained unreacted polyethylene and solid products. The solid products were not analyzed in this study. The yield of each hydrocarbon with a carbon chain from C1 to C21 was calculated using the results of the gas analysis and the liquid analysis. Because the target of this study was jet fuel range hydrocarbons, the products were classified as three groups: C1–C8 (fuel gas and gasoline), C9–C15 (jet fuel), and C16–C21 (diesel). The yield of each group was obtained from the sum of the corresponding hydrocarbons.
3. Results and Discussion
3.1. Characterization of Catalysts
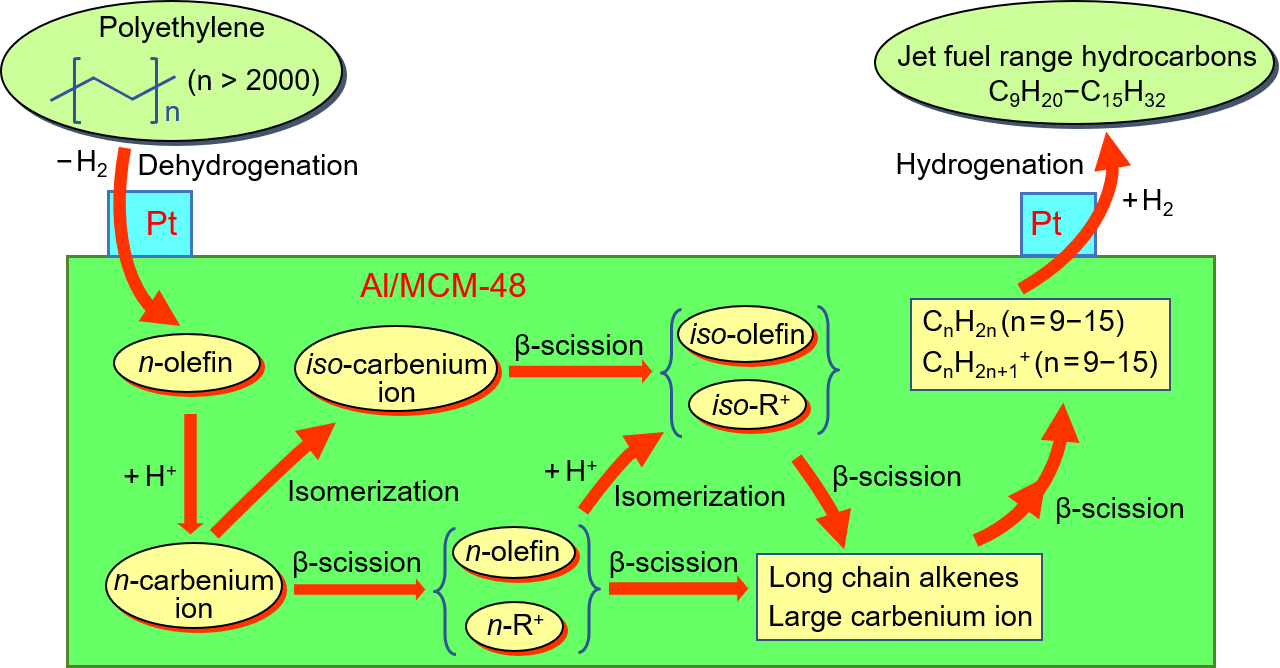
Figure 1 shows the XRD patterns of various samples after calcination at 823 K for 4 h. All samples exhibited only the phase of mesoporous SiO2, and the peak of the Al2O3 phase could not be observed in the XRD patterns. MCM-48 showed an extremely strong peak at 2.2 degrees, a medium-strong peak at 2.5 degrees, and five weak peaks at the range of 4–6 degrees. The peak at 2.2 degrees corresponded to the (2 1 1) plane and the peak at 2.5 degrees corresponded to the (2 2 0) plane. Five weak peaks at the range of 4–6 degrees corresponded to the (3 2 1), (4 0 0), (4 2 0), (3 3 2), and (4 2 2) planes [21]. This XRD pattern indicated that the regular cubic mesoporous structure existed in the MCM-48 [20]. The XRD patterns of Al-MCM-48 and Al/MCM-48 were similar to that of MCM-48. The d211 spacing values calculated by the degree of the (2 1 1) plane were 43.5, 43.4, and 40.4 Å for MCM-48, Al/MCM-48, and Al-MCM-48, respectively. The Al-MCM-48 sample synthesized by mixing Al3+ ions in the gel before hydrothermal synthesis showed a lower value of d211 spacing compared to that of MCM-48. This implies that the small Al3+ ions entered the framework of mesoporous silica in Al-MCM-48. On the other hand, the Al/MCM-48 sample synthesized by grafting Al3+ ions on calcined MCM-48 showed a d211 spacing value similar to that of MCM-48, implying that the small Al3+ ions could not enter the mesoporous framework by a post-synthesis method in Al/MCM-48.
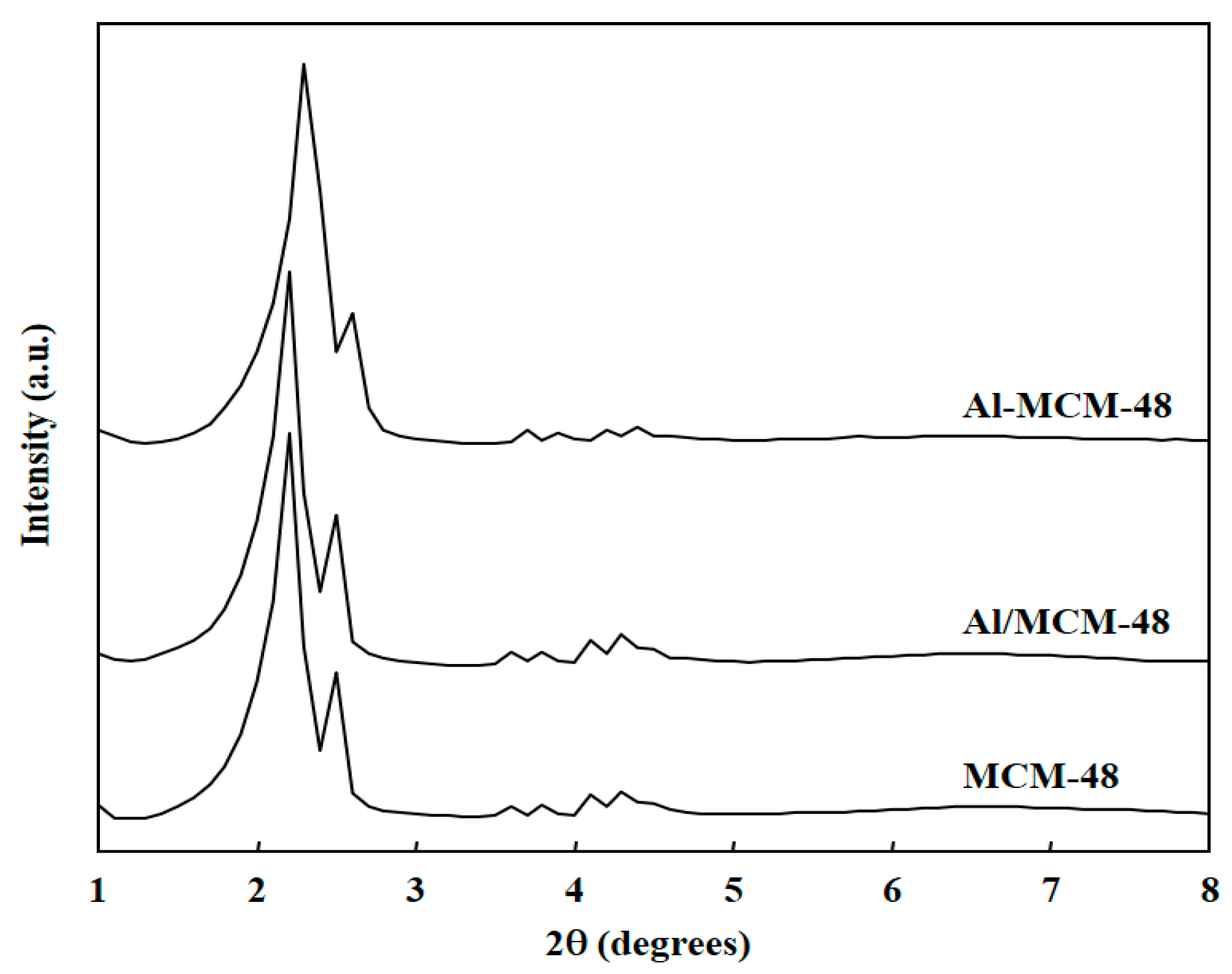
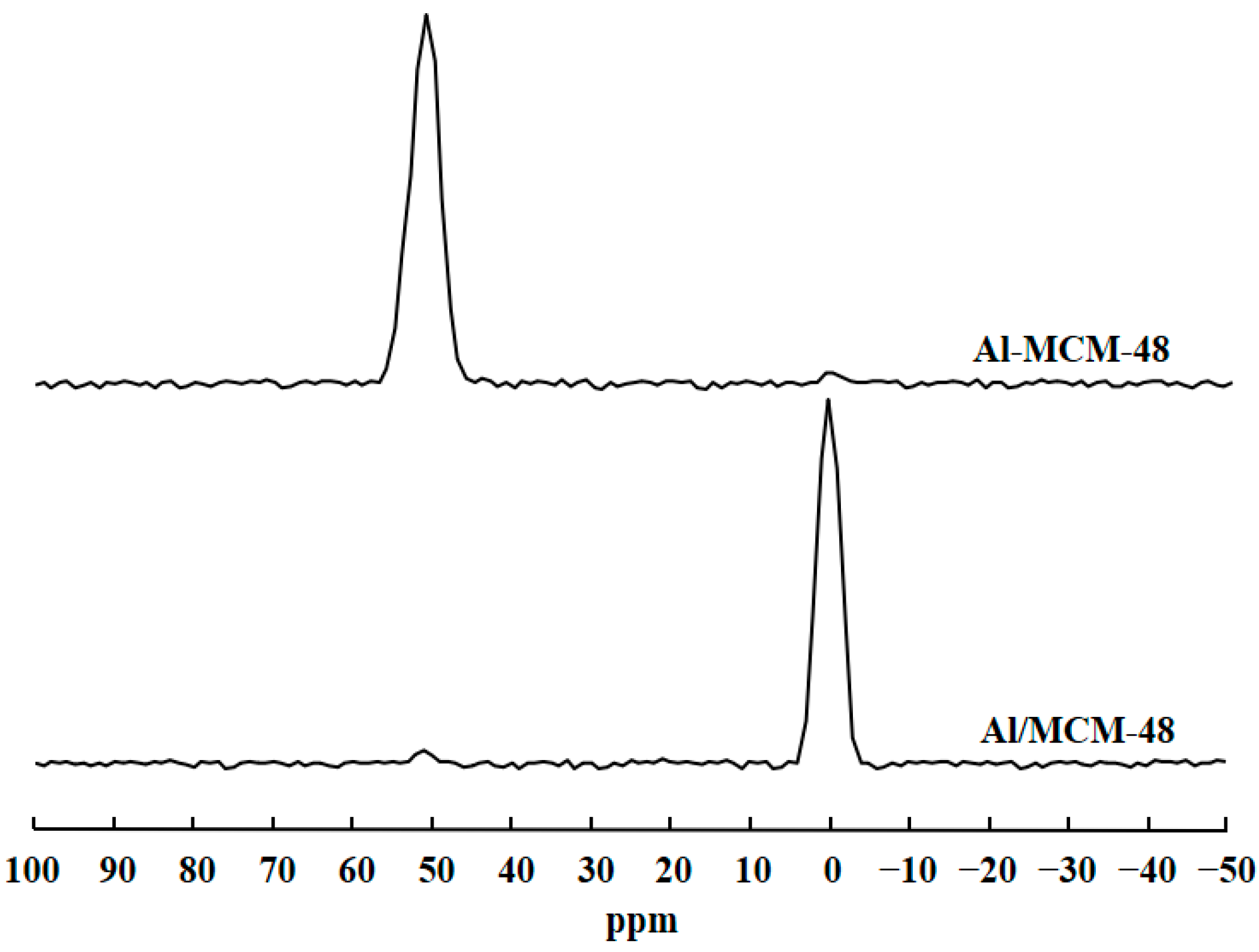
Figure 2 shows the 27Al MAS NMR spectra of Al-MCM-48 and Al/MCM-48 after calcination at 823 K for 4 h. Al-MCM-48 exhibited a strong signal at 51 ppm and a very weak signal at 0 ppm in the 27Al MAS NMR spectrum. On the other hand, Al/MCM-48 exhibited a strong signal at about 0 ppm and a very weak signal at 51 ppm in the 27Al MAS NMR spectrum. 27Al MAS NMR is a useful tool to probe the situation of Al3+ ions in Al-containing mesoporous silica materials [19]. The peak at about 51 ppm is assigned to tetrahedrally coordinated Al, and the peak at 0 ppm is assigned to octahedrally coordinated Al in the 27Al MAS NMR spectra. Hence, the signal at 51 ppm represented Al3+ ions entering the MCM-48 framework, and the signal at 0 ppm represented Al3+ ions existing in the extra-framework of MCM-48. The results of 27Al MAS NMR spectra proved that Al3+ ions in Al-MCM-48 existed in the mesoporous framework and that Al3+ ions in Al/MCM-48 existed in the extra-framework of MCM-48, which was consistent with the results of XRD patterns.
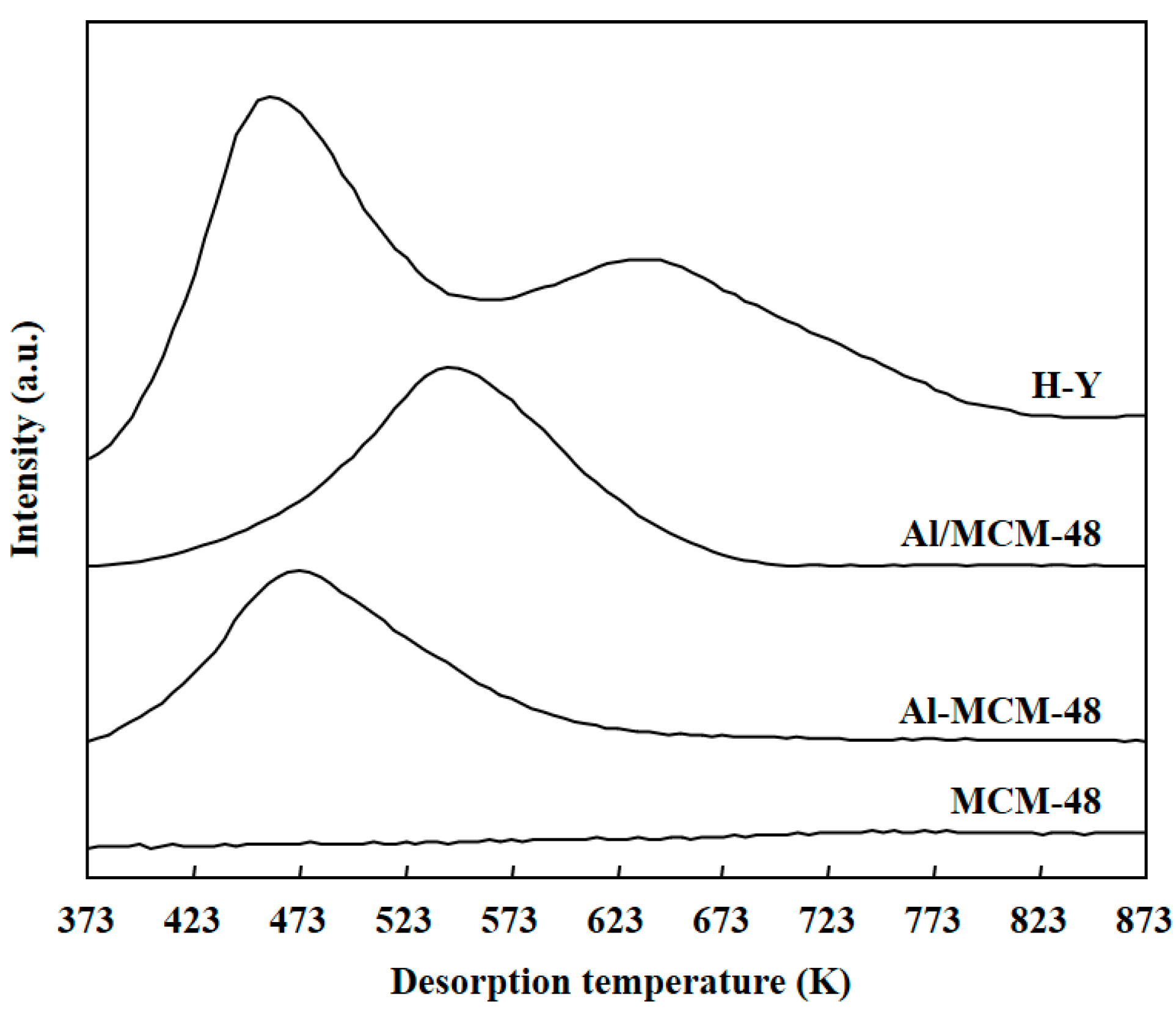
Figure 3 shows the profiles of NH3-TPD for various solid samples after calcination at 823 K for 4 h. NH3 molecules were absorbed on the surface of solid samples at 373 K in vacuum, and the temperature was then increased from 373 to 873 K to desorb NH3 molecules. The NH3 molecules that were absorbed on weak acid sites desorbed at low temperatures, and the NH3 molecules that were absorbed on strong acid sites desorbed at high temperatures. MCM-48 did not show any bands in the NH3-TPD profile, which implied that MCM-48 did not possess any solid acid sites. Al-MCM-48 and Al/MCM-48 showed bands with maximum at 473 and at 543 K in the NH3-TPD profiles, respectively. Hence, Al/MCM-48 possessed stronger solid acid sites than Al-MCM-48. As discussed above, Al3+ was located uniformly inside the framework of mesoporous silica in Al-MCM-48, but the Al3+ ions existed in the extra-framework in Al/MCM-48. It has been reported that the extra-framework of Al3+ ions have stronger acidity compared to the intra-framework of Al3+ ions in Al-containing MCM-41 catalysts [30]. In addition, the acid sites in Al/HMS were stronger than those in Al-HMS [19]. As for H-Y, it exhibited two bands at 473 and 643 K in the NH3-TPD profile, which indicates that H-Y had two types of solid acid sites on the surface. Because the acidity of a solid acid is mainly determined by the strongest solid acid sites on the surface, H-Y zeolite possessed the strongest sites among the various samples used in this study. As a result, the acidic strength of various solid samples was in the order of H-Y > Al/MCM-48 > Al-MCM-48 > MCM-48.
Table 1 lists the physical properties of various Pt-supported catalysts. The BET surface areas and the pore sizes were calculated using the N2 adsorption–desorption isotherms. All MCM-48-based samples (Pt/MCM-48, Pt/Al-MCM-48, and Pt/Al/MCM-48) showed BET surface areas larger than 1000 m2/g. The pore sizes (calculated by the BHJ method) were 39.2, 37.9, and 38.0 Å for Pt/MCM-48, Pt/Al-MCM-48, and Pt/Al/MCM-48, respectively. The pore size slightly decreased after Al modification in MCM-48. The small Al3+ ion entered the framework of mesoporous silica in Al-MCM-48, which caused a decrease of pore size in Pt/Al-MCM-48. The wall of mesoporous silica in Al/MCM-48 became thicker after grafting Al3+ ion on the wall, which caused a decrease of pore size in Pt/Al/MCM-48. Pt-supported MCM-48 materials had very high BET surface area and large pore size compared to Pt/H-Y. The actual Pt loading measured by the ICP element analyses was almost consistent with the designed Pt loading (1 wt %) for each sample. The Pt particle size in each sample was calculated using the amount of CO adsorption and the actual Pt loading. The Pt particle sizes were 2.2, 2.3, 2.1, and 2.6 nm for Pt/MCM-48, Pt/Al-MCM-48, Pt/Al/MCM-48, and Pt/H-Y, respectively. All catalysts with 1 wt % Pt loading possessed well-dispersed Pt particles on the surfaces.
3.2. Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons
Table 2 shows the reaction results of polyethylene hydrocracking over various catalysts at 573 K for 4 h. Before the reaction, 4 MPa of H2 was charged into the autoclave reactor at room temperature. The blank (reaction without a catalyst) showed a very low total yield of C1–C21 (1.2%) and did not form any C9–C15 hydrocarbons. The C9–C15 hydrocarbons are the desired feedstock because the carbon numbers of hydrocarbons in current jet fuel are the same as those distributed in kerosene. Pt/MCM-48 (without solid acid site) showed a low total C1–C22 yield of 7.1% after reaction at 573 K for 4 h. On the other hand, both Pt/Al-MCM-48 and Pt/Al/MCM-48 showed high total C1–C22 yields and formed C9–C15 jet fuel range hydrocarbons as the main products after reaction at 573 K for 4 h. These results indicate that the solid acid sites are important for the hydrocracking of polyethylene over Pt-loaded catalysts. Pt/Al-MCM-48 formed a relatively large yield of C16–C21 heavy hydrocarbons (13.4%). Pt/HY showed the highest total yield of C1–C21 (99.6%) among various catalysts but formed C1–C8 light hydrocarbons as the main products. Pt/Al/MCM-48 showed the largest yield of C9–C15 jet fuel range hydrocarbons (85.9%) among various catalysts for the hydrocracking of polyethylene.
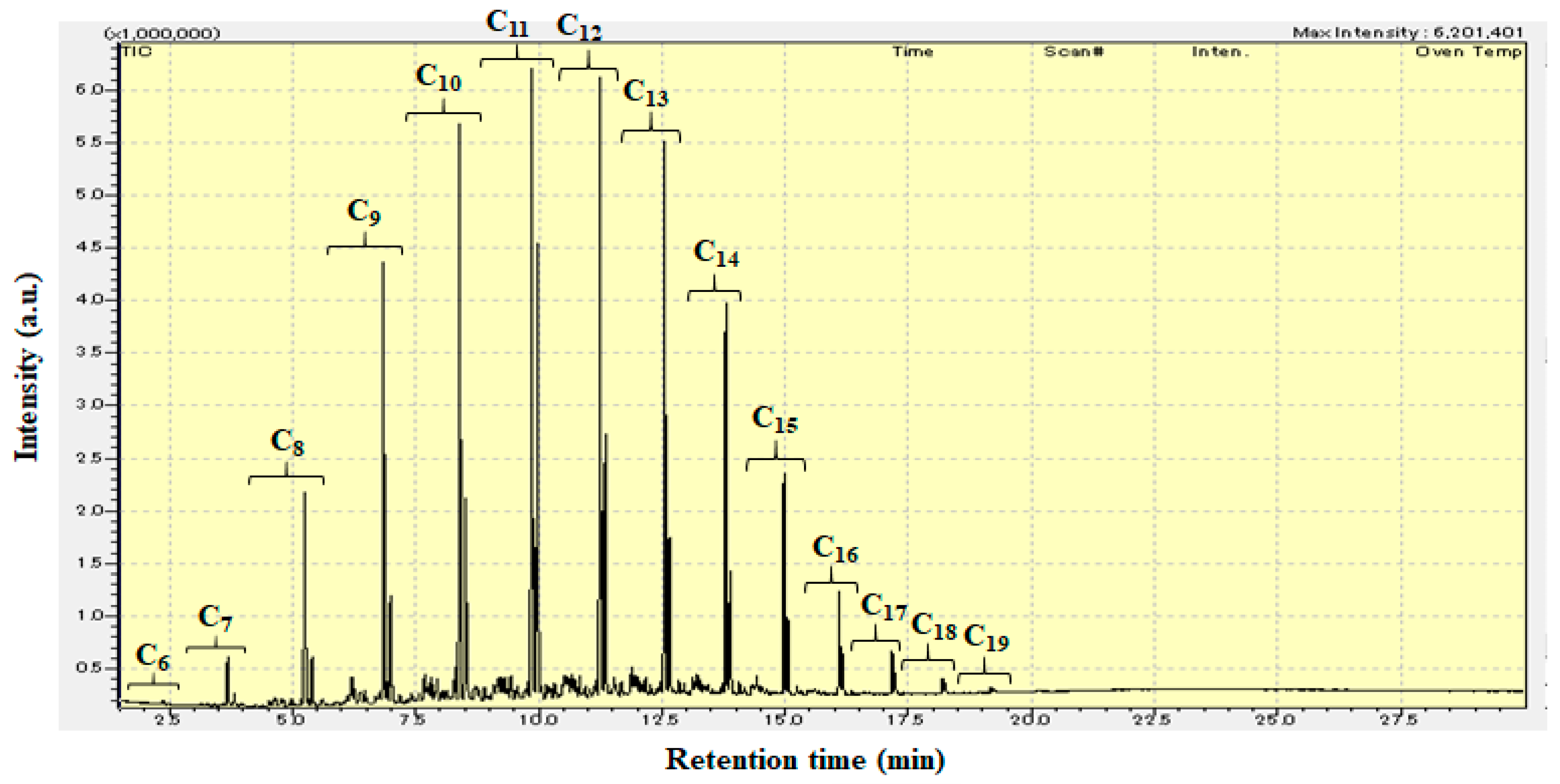
Figure 4 shows the GC-MS chromatogram (retention time: 1.5–30 min) of liquid products from the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K for 4 h. The liquid products obtained from the hydrocracking of polyethylene over Pt/Al/MCM-48 contained many hydrocarbons with carbon numbers ranging from 6 to 19. The C11 and C12 hydrocarbons were the strongest signals among the various signals in the GC-MS chromatogram. The C9–C15 jet fuel range hydrocarbons occupied a large percentage in the liquid products. Bioethylene can be obtained from the dehydration of bioethanol, and biopolyethylene can be obtained from the polymerization of bioethylene. Hence, these liquid products from the hydrocracking of polyethylene over Pt/Al/MCM-48 have the potential to be used as an alternative biojet fuel in current jet engines.
With reference to the NIST-11 database, the signals of alkenes and aromatic compounds were very weak and almost all signals were saturated hydrocarbons in the GC-MS chromatogram. The amount of alkenes was low because polyethylene was cracked under high hydrogen pressure (4 MPa H2 at room temperature). Moreover, MCM-48-based materials had larger pores compared to ZSM-5 zeolite, which caused the hydrocarbon products to be easily desorbed from the catalyst, and aromatic compounds were hardly formed during the reaction.
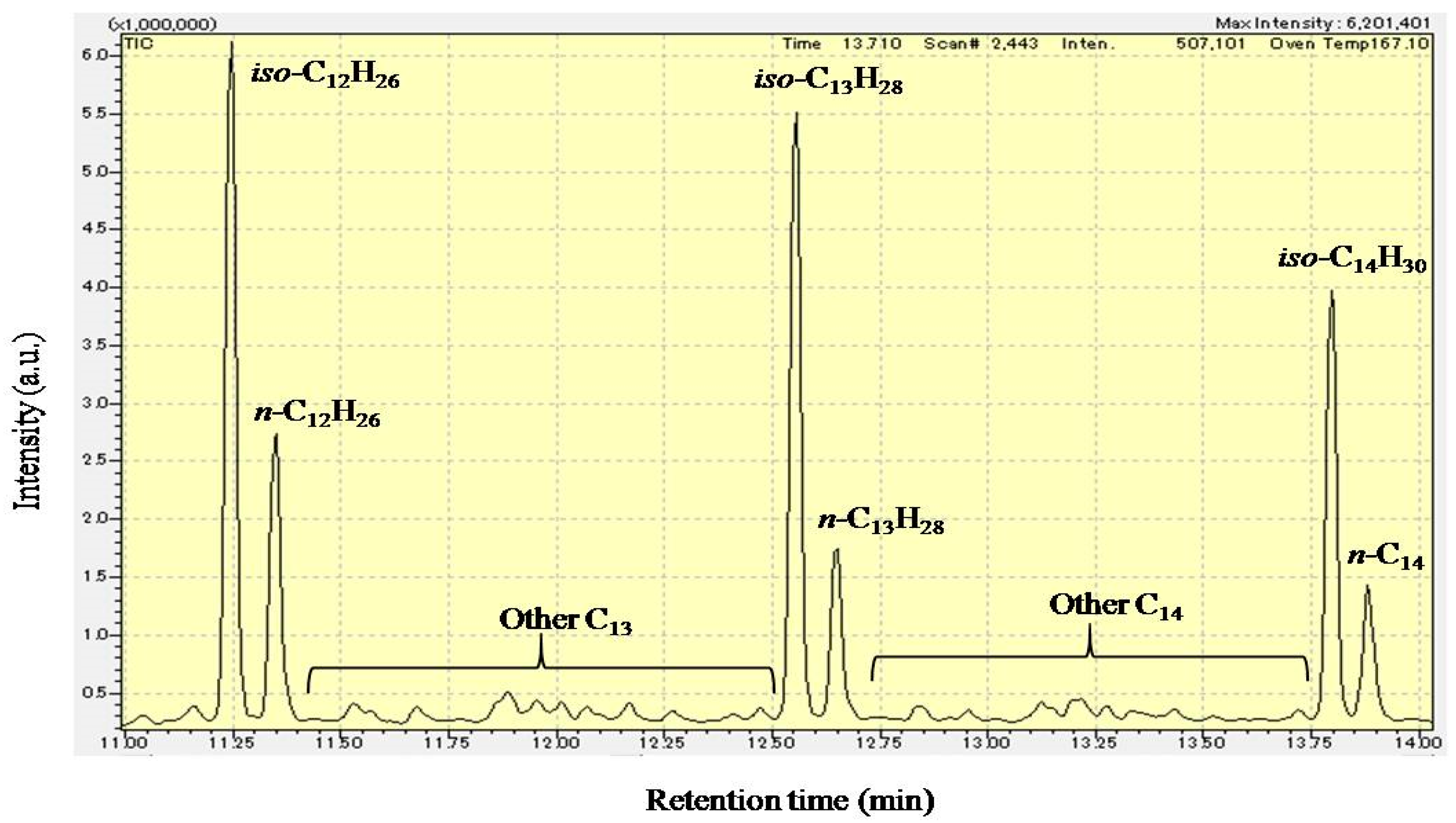
Figure 5 shows the GC-MS chromatogram (retention time: 11–14 min) of liquid products from the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K for 4 h. This liquid sample was the same as that analyzed in Figure 4. The positions of n-alkanes in the GC-MS chromatogram were determined using standard reagents. The strong signals before each n-alkane in the GC-MS chromatogram were the mixture of iso-alkanes with one branch carbon chain, in which 2-methyl isomers and 3-methyl isomers (formed from the isomerization of α-olefins) were the main components. The signals between iso-C13H28 and n-C12H26 were the other hydrocarbons with 13 carbons, such as the iso-alkane with two or more branch of carbon chains. Jet fuel has a carbon distribution of hydrocarbons similar to that of kerosene. However, jet fuel has a low pour point compared to kerosene because it is used in the cold sky. As shown in Figure 5, the amount of iso-alkanes was much larger than the amount of n-alkanes in the liquid products from the hydrocracking of polyethylene over Pt/Al/MCM-48. Because the freezing points of iso-alkanes are much lower than those of n-alkanes, the liquid products from the hydrocracking of biopolyethylene probably have a low freezing point to be used as an alternative biojet fuel in current jet engines.
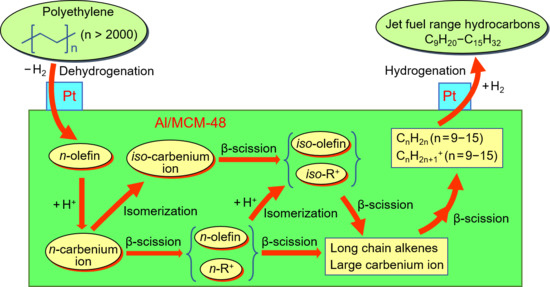
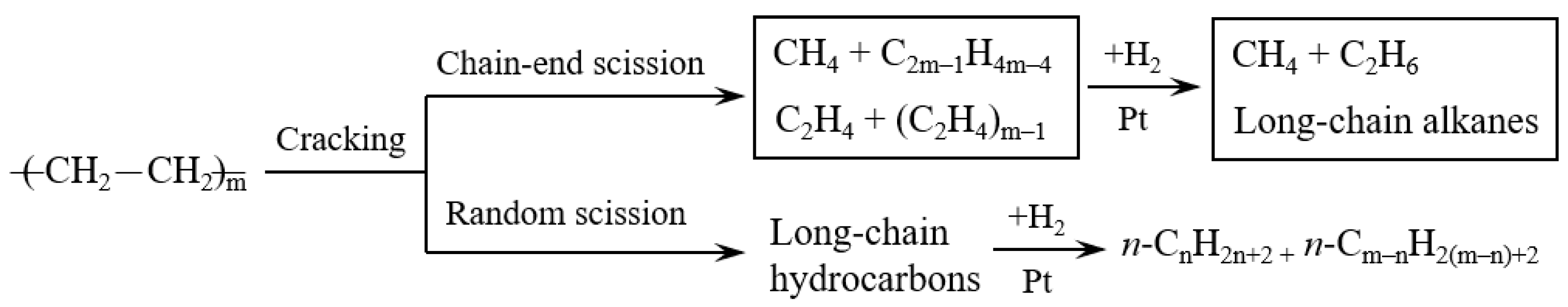
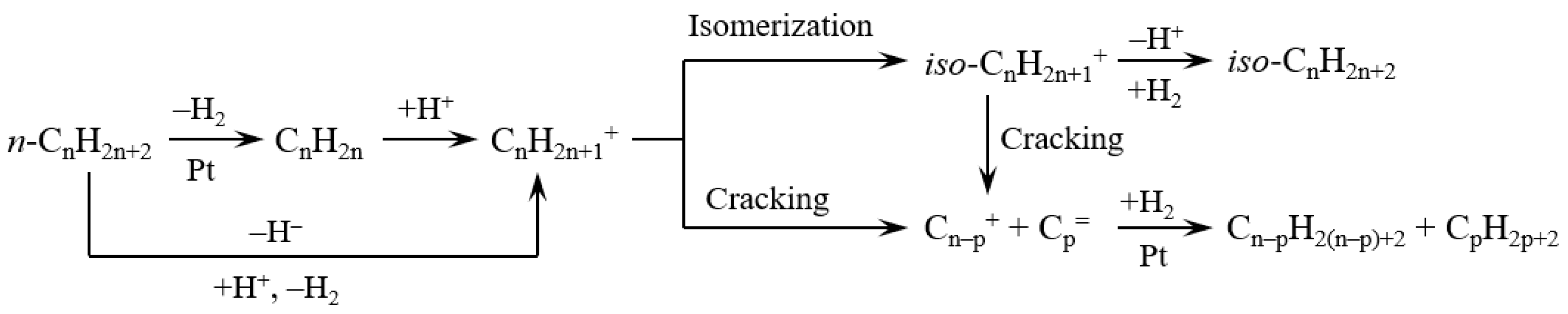
Figure 6 exhibits the scission types of carbon chain in the hydrocracking of polyethylene. The polyethylene molecules can be cracked through chain-end scission and random scission during the reaction. The chain-end scission of polyethylene forms CH4, C2H6, and long-chain hydrocarbons. The formed long-chain hydrocarbons subsequently undergo a further cracking by chain-end scission. Hence, the chain-end scission forms a large amount of gas hydrocarbons. On the other hand, random scission of polyethylene easily forms long-chain hydrocarbons rather than gas hydrocarbons (CH4 and C2H6). From the view of thermodynamics in random scission, the C–C bonds in the middle of long-chain hydrocarbons are easily cracked compared to the chain-end C–C bonds. The nature of the catalyst has a great influence in the scission types of carbon chain. As shown in Table 2, Pt/MCM-48 without any acid sites formed a large yield of light hydrocarbons by the chain-end scission. Pt promoted polyethylene hydrogenolysis, which caused chain-end scission on Pt/MCM-48. By introducing Al3+ ions to the acid sites, Pt/Al-MCM-48 and Pt/Al/MCM-48 showed low yields of light hydrocarbons and large yields of C9–C15 hydrocarbons from the hydrocracking of polyethylene. The random scission was accelerated by introducing solid acid sites to the catalysts in the hydrocracking of polyethylene.
Figure 7 exhibits the reaction pathways in the hydrocracking of long-chain hydrocarbons over catalysts containing Pt and solid acids. Carbenium ion is a key intermediate for the hydrocracking of long-chain hydrocarbons because it undergoes β-scission to achieve the hydrocracking of hydrocarbons. The carbenium ion formed in the middle of the carbon chain is more stable than that formed in the end of the carbon chain. This is the reason why the yields of light hydrocarbons were suppressed over catalysts containing solid acid sites (as shown in Table 2).
The acid sites are necessary for the formation of carbenium ions from alkanes. Carbenium ions could be formed on solid acids by hydride abstraction of n-alkanes or by protonation to form carbonium ion followed by transformation to carbenium ion and hydrogen. On the other hand, carbenium ions formed by dehydrogenation of n-alkanes to n-alkenes and subsequent protonation on catalysts simultaneously had Pt and solid acids. As for the formation of carbenium ion, the protonation of an alkene molecule is much faster than hydride abstraction or the protonation of an alkane molecule. Hence, the addition of Pt in the solid acids greatly increased the speed of carbenium ion formation in the reaction system [31,32,33,34].
The isomerization of n-alkanes is a competitive reaction with respect to cracking because of the common carbenium ion intermediate. All n-alkanes in the catalytic system (including reactants and products) undergo isomerization in parallel with cracking. Hence, the amount of iso-alkanes was larger than the amount of n-alkanes in the liquid product from the hydrocracking of polyethylene over Pt/Al/MCM-48 (As shown in Figure 5).
In the hydrocracking of large hydrocarbons, the relative reactivity of hydrocracking greatly increases with increasing carbon number in the carbon chain [35,36]. For example, the speed of n-C17H36 hydrocracking is 2.4 times faster than that of n-C16H34 hydrocracking and 4.0 times faster than that of n-C15H32 hydrocracking over catalysts containing metal and solid acid [36]. Hence, a long-chain hydrocarbon is easily cracked, while it is difficult to crack a short-carbon-chain hydrocarbon in a reaction system [35]. In addition, the acidic strength of solid acid is crucial for the hydrocracking of large hydrocarbons [31,32,33,34]. A weak acid cannot crack a short-carbon-chain hydrocarbon, while a strong acid can crack a short-carbon-chain hydrocarbon with the same reaction conditions.
As shown in Table 2, Pt/Al-MCM-48 formed a relatively large yield of C16–C21 heavy alkanes because the solid acidity of Al-MCM-48 was too weak. Pt/H-Y formed a large yield of C1–C8 light hydrocarbons because the acidity of Pt/H-Y was too strong. The products underwent further cracking to form light hydrocarbons (<C9) on the catalysts containing Pt and strong solid acids [37]. Pt/Al/MCM-48 showed the highest yield of C9–C15 hydrocarbons among the various catalysts, indicating that Al/MCM-48 had proper acidic strength for the hydrocracking of polyethylene to jet fuel range hydrocarbons.
Figure 8 shows the effect of Pt loading in Pt/Al/MCM-48 for the hydrocracking of polyethylene at 573 K for 4 h. Al/MCM-48 (without Pt) showed a low total yield of C1–C21 hydrocarbons (16.3%) and a low yield of C9–C15 jet fuel range hydrocarbons (10.4%). Both the total yield of C1–C21 hydrocarbons and the yield of C9–C15 hydrocarbons greatly increased by introducing 0.2 wt % Pt into Al/MCM-48. As discussed above, the hydrocracking of long-chain hydrocarbons over Pt-promoted solid acid resulted in a bifunctional mechanism, in which the Pt site achieved dehydrogenation and hydrogenation, and the acid site achieved carbenium intermediate formation [31,32,33,34]. The carbenium ion was formed much faster from an alkene molecule (on bifunctional catalysts containing Pt and solid acid) than from an alkane molecule (on solid acids without Pt). Introducing Pt to Al/MCM-48 greatly increased the total yield of C1–C21 hydrocarbons because more carbenium intermediates could be formed in the presence of Pt. As shown in Figure 8, the total yield of C1–C21 hydrocarbons greatly increased with increasing Pt loading when the Pt loading was less than 1.0 wt %, and almost kept constant when the Pt loading was larger than 1 wt % in Pt/Al/MCM-48. This implies that the hydrogenation/dehydrogenation on Pt sites was the limiting step when the Pt loading was less than 1 wt %, and the formation of carbenium intermediates (for hydrocracking and isomerization) on solid acid sites was the limiting step when the Pt loading was larger than 1 wt % in Pt/Al/MCM-48.
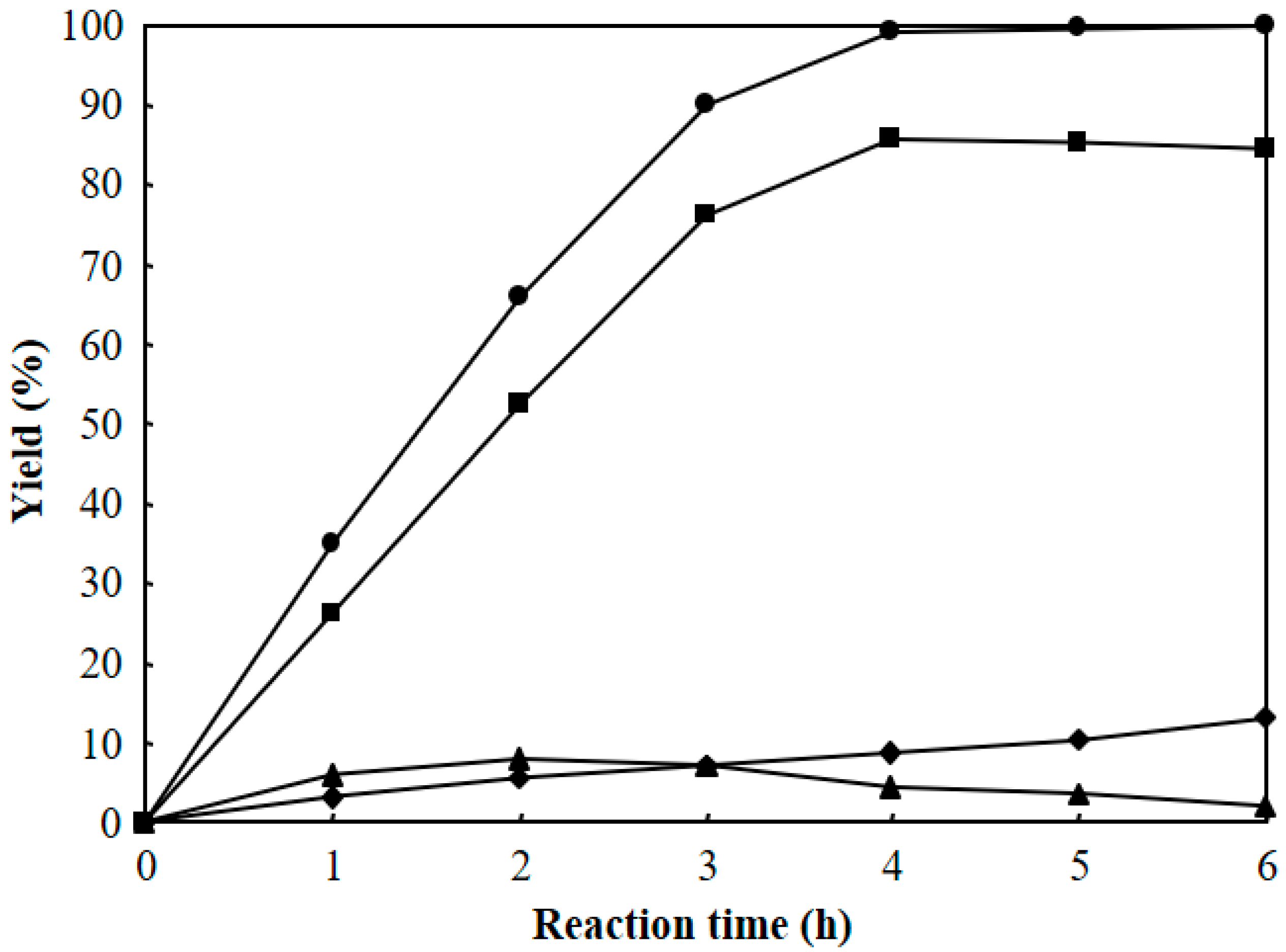
Figure 9 shows the effect of reaction time on the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K. The same amount of polyethylene (10 g) and the same amount of 1 wt % Pt/Al/MCM-48 catalyst (1 g) were added to six autoclave reactors. Then, 4 MPa H2 was charged to the autoclaves at room temperature. The autoclaves were then heated to 573 K with stirring for the reaction. The reaction at 573 K was finished for each autoclave reactor in each one hour, and the products were analyzed to determine the yields of various hydrocarbons at that reaction time. The total yield of C1–C21 hydrocarbons increased by prolonging the reaction time and reached 99.3% after reaction at 573 K for 4 h. Hence, a reaction time of 4 h was enough for converting solid polyethylene to liquid hydrocarbons over Pt/Al/MCM-48 at 573 K. The yield of C1–C8 light hydrocarbons increased by prolonging the reaction time from 1 to 6 h. The yield of C16–C21 heavy hydrocarbons increased by prolonging the reaction time from 1 to 2 h and then decreased with further prolonging of the reaction time. A long reaction time caused further cracking of the C16–C21 products. The yield of C9–C15 jet fuel range hydrocarbons increased by prolonging the reaction time from 1 to 4 h and then slightly decreased when the reaction time was longer than 4 h.
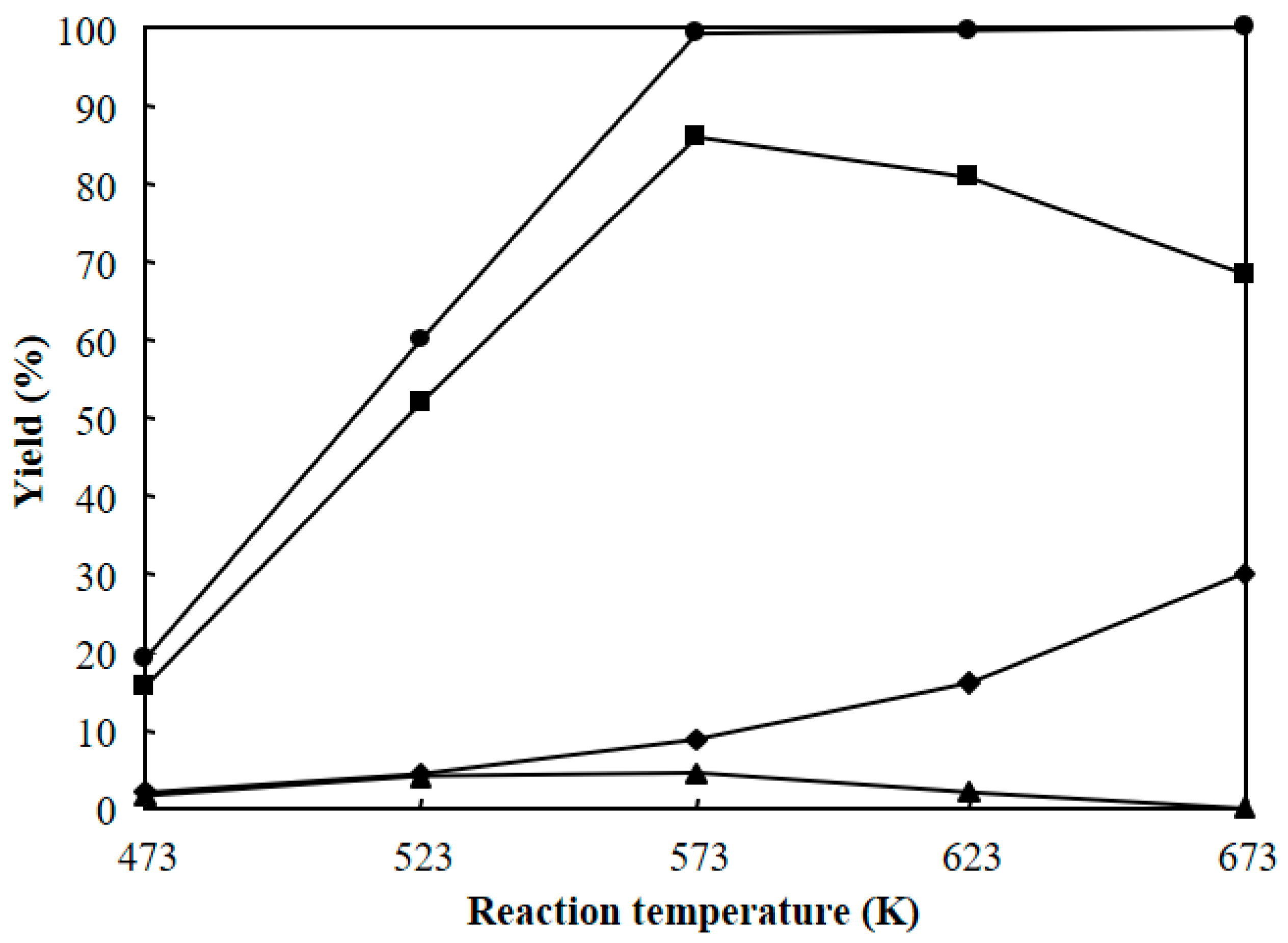
Figure 10 shows the dependence of reaction temperature on the hydrocracking of polyethylene over Pt/Al/MCM-48 for 4 h. The total yield of C1–C21 hydrocarbons was low (19.3%) when the reaction was carried out at a low temperature of 473 K for 4 h. The total yield of C1–C21 hydrocarbons greatly increased with increasing reaction temperature and showed a high value of 99.3% when the reaction was carried out at 573 K for 4 h. Further, reaction at a reaction temperature of 623 K for 4 h showed a high yield of C1–C21 hydrocarbons approaching 100%, but the yield of C1–C8 light hydrocarbons greatly increased due to the further cracking of C9–C15 and C16–C21 products. As a result, the reaction at 573 K for 4 h obtained the highest yield of C9–C15 jet fuel range hydrocarbons among the various reaction temperatures in the hydrocracking of polyethylene over Pt/Al/MCM-48.
Table 3 shows the reusability of Pt/Al/MCM-48 in the hydrocracking of polyethylene at 573 K for 4 h. The used solid catalyst was obtained by filtering out the liquid product in the slurry that was taken from the autoclave after reaction at 573 K for 4 h. The obtained used solid catalyst and 10 g polyethylene were put in a clean autoclave. Then, 4 MPa H2 was introduced to the reactor at room temperature. The reactor was heated to 573 K and held at 573 K for 4 h for the second reaction cycle. The total yield of C1–C21 hydrocarbons and the yield of C9–C15 jet fuel range hydrocarbons did not decrease after reaction for four cycles. The hydrocracking of polyethylene was carried out under high H2 pressure, which ensured that Pt did not oxidize during the reaction. In addition, the BET surface area of Pt/Al/MCM-48 did not decrease, and the size of Pt particles in Pt/Al/MCM-48 did not increase after reaction for four cycles. These results proved that the mesoporous structure did not collapse and the Pt particles did not sinter when the reaction was carried out at a medium temperature of 573 K over Pt/Al/MCM-48. In addition, the catalytic performance of the used catalyst was maintained after reaction for four cycles, indicating that the active components (Pt and Al3+) in Pt/Al/MCM-48 did not leach into the liquid during the reaction.
4. Conclusions
Catalysts containing Pt- and Al-modified MCM-48 showed high yields of C9–C15 jet fuel range hydrocarbons from the hydrocracking of polyethylene at 573 K in autoclave reactors. Al/MCM-48 synthesized by grafting Al3+ ions on the surface of calcined MCM-48 acted as an excellent support for Pt in the hydrocracking of polyethylene to C9–C15 jet fuel range hydrocarbons because Al/MCM-48 had property surface acidity. Solid acid sites achieved formation of carbenium ion intermediates to promote random scission of polyethylene. Pt sites achieved dehydrogenation/hydrogenation of long-carbon-chain hydrocarbons to accelerate the formation of carbenium ion in the presence of acid sites. The yield of C9–C15 jet fuel range hydrocarbons increased from 10.4 to 85.9% with increasing Pt loading from 0 to 1 wt % and almost did not change when the Pt loading ranged from 1 to 2 wt % in Pt/Al/MCM-48. The highest value of C9–C15 yield was obtained at a reaction temperature of 573 K and a reaction time of 4 h over Pt/Al/MCM-48. Prolonging the reaction time or increasing the reaction temperature improved the total yield of C1–C21 hydrocarbons but decreased yields of C9–C15 jet fuel range hydrocarbons due to further cracking. The Pt/Al/MCM-48 catalyst could be reused by a simple filtration method, and the catalytic performance did not decrease after reaction for four cycles.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 1044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesaro, A.; Belgiorno, V. Combined biogas and bioethanol production: Opportunities and challenge for industrial application. Energies 2015, 8, 8121–8144. [Google Scholar] [CrossRef]
- Thanh, L.T.; Okitsu, K.; Boi, L.V.; Maeda, Y. Catalytic technologies for biodiesel fuel production and utilization of glycerol: A review. Catalysts 2012, 2, 191–222. [Google Scholar] [CrossRef] [Green Version]
- Hanaoka, T.; Liu, Y.; Matsunaga, K.; Miyazawa, T.; Hirata, S.; Sakanishi, K. Bench-scale production of liquid fuel from woody biomass via gasification. Fuel Process. Technol. 2010, 91, 859–865. [Google Scholar] [CrossRef]
- Liu, Y.; Hanaoka, T.; Miyazawa, T.; Murata, K.; Okabe, K.; Sakanishi, K. Fischer-Tropsch synthesis in slurry-phase reactors over Mn- and Zr-modified Co/SiO2 catalysts. Fuel Process. Technol. 2009, 90, 901–908. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyas, R.; Murata, K.; Minowa, T.; Sakanishi, K. Production of bio-hydrogenated diesel by hydrotreatment of high-acid-value waste cooking oil over ruthenium catalyst supported on Al-polyoxocation-pillared pontmorillonite. Catalysts 2012, 2, 171–190. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyas, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of vegetable oils to produce bio-hydrogenated diesel and liquefied petroleum gas fuel over catalysts containing sulfided Ni-Mo and solid Acids. Energy Fuels 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Hydrocracking of algae oil to aviation fuel-ranged hydrocarbons over NiMo-supported catalysts. Catal. Today 2019, 332, 115–121. [Google Scholar] [CrossRef]
- Liu, Y. Catalytic Deoxygenation of hexadecyl palmitate as a model compound of Euglena oil in H2 and N2 atmospheres. Catalysis 2017, 7, 333. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Staples, M.D.; Malina, R.; Tyner, W.E. Stochastic teno-economic analysis of alcohol-to-jet fuel production. Biotechnol. Biofuels 2017, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Catalytic ethylene oligomerization over Ni/Al-HMS: A key step in conversion of bio-ethanol to higher olefins. Catalysis 2018, 8, 537. [Google Scholar] [CrossRef] [Green Version]
- Andrei, R.D.; Popa, M.I.; Fajula, F.; Hulea, V. Hetergeneous oligomerization of ethylene over highly active and stable Ni-AlSBA-15 mesoporous catalysts. J. Catal. 2015, 323, 76–84. [Google Scholar] [CrossRef]
- Pan, Z.; Xue, X.; Zhang, C.; Wang, D.; Xie, Y.; Zhang, R. Evaluation of process parameters on high-density polyethylene hydro-liquefaction products. J. Anal. Appl. Pyrolysis 2018, 136, 146–152. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Zhu, L.; Zhu, X.; Qian, M.; Yadavalli, G.; Yan, D.; Wu, J.; Chen, S. Optimizing carbon efficiency of jet fuel range alkanes from cellulose co-fed with polyethylene via catalytically combined processes. Bioresour. Technol. 2016, 214, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Duan, D.; Lei, H.; Villota, E.; Ruan, R. Jet fuel production from waste plastics via catalytic pyrolysis with activated carbons. Appl. Energy 2019, 251, 113337. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H. Synthesis of high-density jet fuel from plastics via catalytically integral processes. RCS Adv. 2016, 6, 6154–6163. [Google Scholar] [CrossRef]
- Tao, L.; Markham, J.N.; Haq, Z.; Biddy, M.J. Techno-economic analysis for upgrading the bio-mass-derived ethanol-to-jet blendstocks. Green Chem. 2017, 19, 1082–1101. [Google Scholar] [CrossRef]
- Han, J.; Tao, L.; Wang, M. Well-to-wake analysis of ethanol-to-jet and sugar-to jet pathways. Biotechnol. Biofuels 2017, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Murata, K.; Inaba, M.; Mimura, N. Syntheses of Ti- and Al-containing hexagonal mesoporous silicas for gas-phase epoxidation of propylene by molecular oxygen. Appl. Catal. A Gen. 2006, 309, 91–105. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.C. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Romero, A.A.R.; Alba, M.D.; Zhou, W.; Klinoeski, J. Synthesis and characterization of the mesoporous silicate molecular sieve MCM-48. J. Phys. Chem. B 1997, 101, 5294–5300. [Google Scholar] [CrossRef]
- Kim, H.; Choi, S.J.; Kim, J.M.; Jeon, J.K.; Park, S.H.; Jung, S.C.; Kim, S.C.; Park, Y.K. Catalytic copyrolysis of particle board and polypropylene over Al-MCM-48. Mater. Res. Bull. 2016, 82, 61–66. [Google Scholar] [CrossRef]
- Park, Y.K.; Lee, H.W.; Lee, J.Y.; Kim, Y.M. The use of high density polyethylene (HDPE) as a co-feeding feedstock on the catalytic pyrolysis of yellow poplar over Al-MCM-48 and Al-MSU-F. J. Anal. Appl. Pyrolysis 2018, 135, 390–396. [Google Scholar] [CrossRef]
- Romero, A.; Nieto-Marquez, A.; Essayem, N.; Alonso, E.; Pinel, C. Improving conversion of D-glucose into short-chain alkanes over Ru/MCM-48 based catalysts. Microporous Mesoporous Mater. 2019, 286, 25–35. [Google Scholar] [CrossRef]
- Zhang, W.; Pinnavaia, T.J. Transition metal substituted derivatives of cubic MCM-48 mesoporous molecular sieves. Catal. Lett. 1996, 38, 261–265. [Google Scholar] [CrossRef]
- Chen, F.; Huang, L.; Yang, X.; Wang, Z. Synthesis of Al-substitute MCM-41 and MCM-48 solid acids with mixed cationic-anionic surfactants as templates. Mater. Lett. 2013, 109, 299–301. [Google Scholar] [CrossRef]
- Morey, M.S.; Stucky, G.D.; Schwarz, S.; Froba, M. Isomorphic substitution and postsynthesis incorporation of zirconium into MCM-48 mesoporous silica. J. Phys. Chem. B 1999, 103, 2037–2041. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Okabe, K.; Hanaoka, T.; Sakanishi, K. Synthesis of Zr-grafted SBA-15 as an Effective Support for Cobalt Catalyst in Fischer-Tropsch Synthesis. Chem. Lett. 2008, 37, 984–985. [Google Scholar] [CrossRef]
- Komai, S.; Yazawa, Y.; Satsuma, A.; Hattori, T. Determination of metal dispersion of Pt/CeO2 catalyst by CO-pulse method. J. Jpn. Pet. Inst. 2005, 48, 173–177. [Google Scholar] [CrossRef]
- Park, K.C.; Yim, D.J.; Ihm, S.K. Characteristics of Al-MCM-41 supported Pt catalysts:effect of Al distribution in Al-MCM-41 on its catalyticactivity in naphthalene hydrogenation. Catal. Today 2002, 74, 281–290. [Google Scholar] [CrossRef]
- Liu, Y.; Misono, M. Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid. Materials 2009, 2, 2319–2336. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Sakanishi, K. Hydroisomerization-cracking of gasoline distillate from Fischer–Tropsch synthesis over bifunctional catalysts containing Pt and heteropolyacids. Fuel 2011, 90, 3056–3065. [Google Scholar] [CrossRef]
- Liu, Y.; Koyano, G.; Misono, M. Hydroisomerization of n-hexane and n-heptane over platinum-promoted Cs2.5H0.5PW12O40 (Cs2.5) studied in comparison with several other solid acids. Top. Catal. 2000, 11, 239–246. [Google Scholar] [CrossRef]
- Liu, Y.; Na, K.; Misono, M. Skeletal isomerization of n-pentane over Pt-promoted cesium hydrogen salts of 12-tungstophosphoric acid. J. Mol. Catal. A Chem. 1999, 141, 145–153. [Google Scholar] [CrossRef]
- Fernandes, F.A.N.; Teles, U.M. Modeling and optimization of Fischer-Tropsch products hydrocracking. Fuel Process. Technol. 2007, 88, 207–214. [Google Scholar] [CrossRef]
- Bouchy, C.; Hastoy, G.; Guillon, E.; Martens, J.A. Fischer-Tropsch waxes upgrading via hydrocracking and selective hydroisomerization. Oil Gas Sci. Technol. 2009, 64, 91–112. [Google Scholar]
- Liu, Y.; Murata, K.; Okabe, K.; Inaba, M.; Takahara, I.; Hanaoka, T.; Sakanishi, K. Selective hydrocracking of Fischer-Tropsch waxes to high-quality diesel fuel over Pt-promoted polyoxocation-pillared montmorillonites. Top. Catal. 2009, 52, 597–608. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction (XRD) patterns of various samples after calcination at 823 K for 4 h.

Figure 2.
27Al MAS NMR spectra of Al-modified MCM-48 materials after calcination at 823 K for 4 h.

Figure 3.
Profiles of ammonia temperature-programmed desorption (NH3-TPD) for various solid samples after calcination at 823 K for 4 h.
Figure 3.
Profiles of ammonia temperature-programmed desorption (NH3-TPD) for various solid samples after calcination at 823 K for 4 h.

Figure 4.
GC-MS chromatogram (retention time: 1.5–30 min) of liquid products from the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K for 4 h.
Figure 4.
GC-MS chromatogram (retention time: 1.5–30 min) of liquid products from the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K for 4 h.

Figure 5.
GC-MS chromatogram (retention time: 11–14 min) of liquid products from the hydrocracking of hydrocracking over Pt/Al/MCM-48 at 573 K for 4 h.
Figure 5.
GC-MS chromatogram (retention time: 11–14 min) of liquid products from the hydrocracking of hydrocracking over Pt/Al/MCM-48 at 573 K for 4 h.

Figure 6.
Scission types of carbon chain in the hydrocracking of polyethylene.

Figure 7.
Reaction pathways in the hydrocracking of long-chain hydrocarbons over catalysts containing Pt and solid acids.
Figure 7.
Reaction pathways in the hydrocracking of long-chain hydrocarbons over catalysts containing Pt and solid acids.

Figure 8.
Effect of Pt loading in Pt/Al/MCM-48 for the hydrocracking of polyethylene at 573 K for 4 h. (●): total yield of C1–C21, (♦): C1–C8 yield, (■): C9–C15 yield, (▲): C16>–C21 yield.
Figure 8.
Effect of Pt loading in Pt/Al/MCM-48 for the hydrocracking of polyethylene at 573 K for 4 h. (●): total yield of C1–C21, (♦): C1–C8 yield, (■): C9–C15 yield, (▲): C16>–C21 yield.

Figure 9.
Effect of reaction time on the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K. (●): total yield of C1–C21, (♦): C1–C8 yield, (■): C9–C15 yield, (▲): C16–C21 yield.
Figure 9.
Effect of reaction time on the hydrocracking of polyethylene over Pt/Al/MCM-48 at 573 K. (●): total yield of C1–C21, (♦): C1–C8 yield, (■): C9–C15 yield, (▲): C16–C21 yield.

Figure 10.
Dependence of reaction temperature on the hydrocracking of polyethylene over Pt/Al/MCM-48 for 4 h. (●): total yield of C1–C21, (♦): C1–C8 yield, (■): C9–C15 yield, (▲): C16–C21 yield.
Figure 10.
Dependence of reaction temperature on the hydrocracking of polyethylene over Pt/Al/MCM-48 for 4 h. (●): total yield of C1–C21, (♦): C1–C8 yield, (■): C9–C15 yield, (▲): C16–C21 yield.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Physical properties of various Pt-supported catalysts.
| Catalyst 1 | BET Surface Area (m2 g−1) | Pore Size (Å) | Pt particle Size (nm) |
|---|---|---|---|
| Pt/MCM-48 | 1040 | 39.2 | 2.2 |
| Pt/Al-MCM-48 | 1062 | 37.9 | 2.3 |
| Pt/Al/MCM-48 | 1023 | 38.0 | 2.1 |
| Pt/H-Y | 246 | 7.5 | 2.6 |
1 Pt loading: 1 wt %.
Table 2.
Reaction results of polyethylene hydrocracking over various catalysts at 573 K for 4 h 1.
| Catalyst | Total Yield of C1–C21 (%) | C1–C8 Yield (%) | C9–C15 Yield (%) | C16–C21 Yield (%) |
|---|---|---|---|---|
| Blank | 1.2 | 1.2 | 0 | 0 |
| Pt/MCM-48 | 7.1 | 6.2 | 0.9 | 0 |
| Pt/Al-MCM-48 | 97.8 | 7.3 | 77.1 | 13.4 |
| Pt/Al/MCM-48 | 99.3 | 8.9 | 85.9 | 4.5 |
| Pt/H-Y | 99.6 | 80.7 | 18.9 | 0 |
1 Catalyst amount: 1 g; Pt loading: 1 wt %; polyethylene amount: 10 g.
Table 3.
Reusability of Pt/Al/MCM-48 in the hydrocracking of polyethylene at 573 K for 4 h 1.
| Reaction Cycle | Total Yield of C1–C21 (%) | C1–C8 Yield (%) | C9–C15 Yield (%) | C16–C21 Yield (%) |
|---|---|---|---|---|
| 1 | 99.3 | 8.9 | 85.9 | 4.5 |
| 2 | 99.2 | 8.8 | 85.8 | 4.6 |
| 3 | 99.1 | 8.8 | 85.8 | 4.5 |
| 4 | 99.2 | 8.9 | 85.9 | 4.4 |
1 Catalyst amount: 1 g; Pt loading: 1 wt %; polyethylene amount: 10 g.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, Y. Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48. Reactions 2020, 1, 195-209. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020014
AMA Style
Liu Y. Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48. Reactions. 2020; 1(2):195-209. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020014
Chicago/Turabian StyleLiu, Yanyong. 2020. "Hydrocracking of Polyethylene to Jet Fuel Range Hydrocarbons over Bifunctional Catalysts Containing Pt- and Al-Modified MCM-48" Reactions 1, no. 2: 195-209. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020014