Metallosupramolecular Compounds Based on Copper(II/I) Chloride and Two Bis-Tetrazole Organosulfur Ligands


Abstract
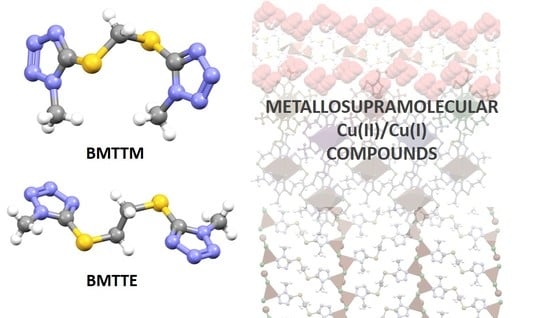
:
1. Introduction
2. Results and Discussion
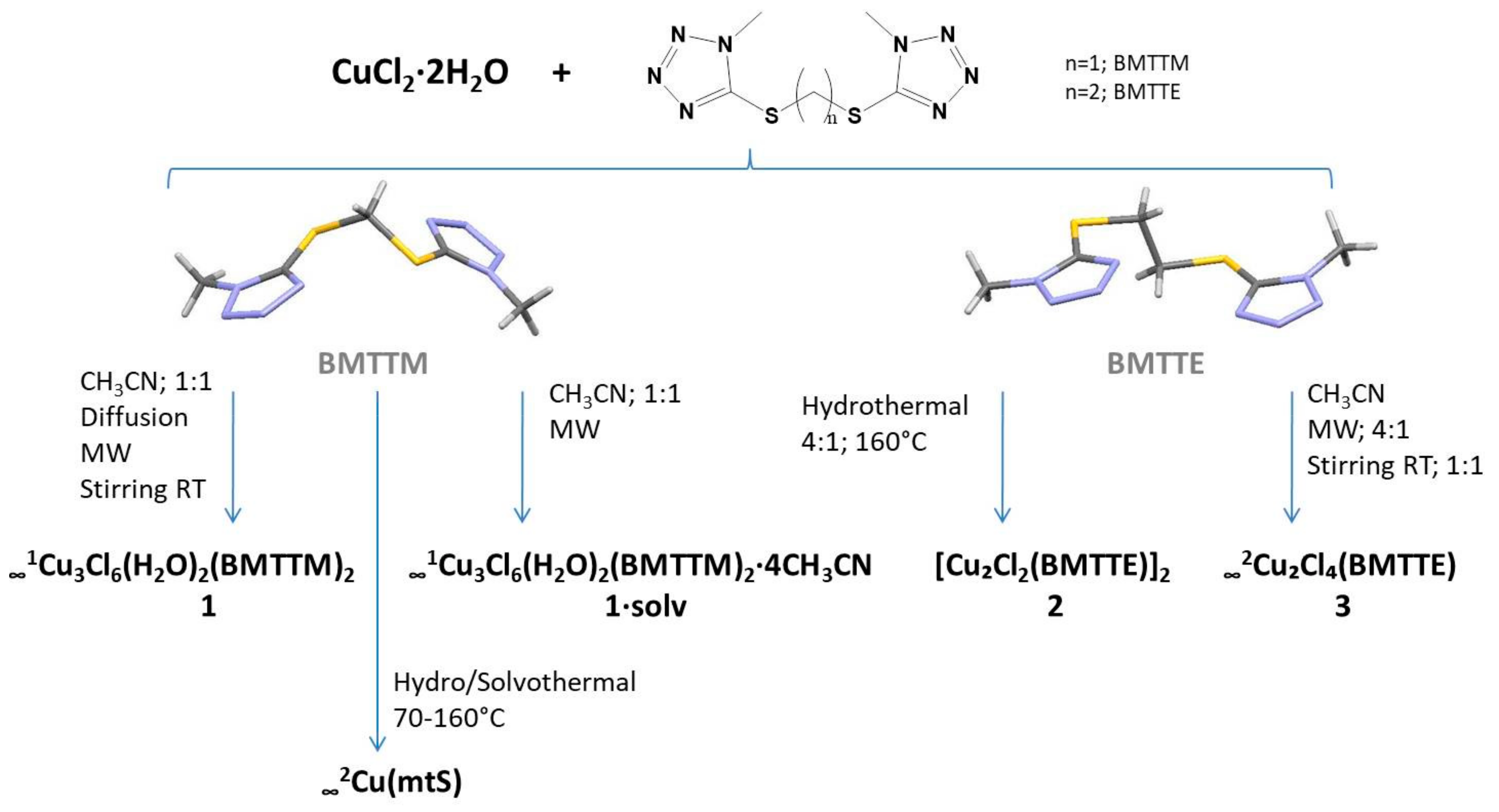
2.1. Synthesis of the Complexes
2.2. Structural Studies: General Features
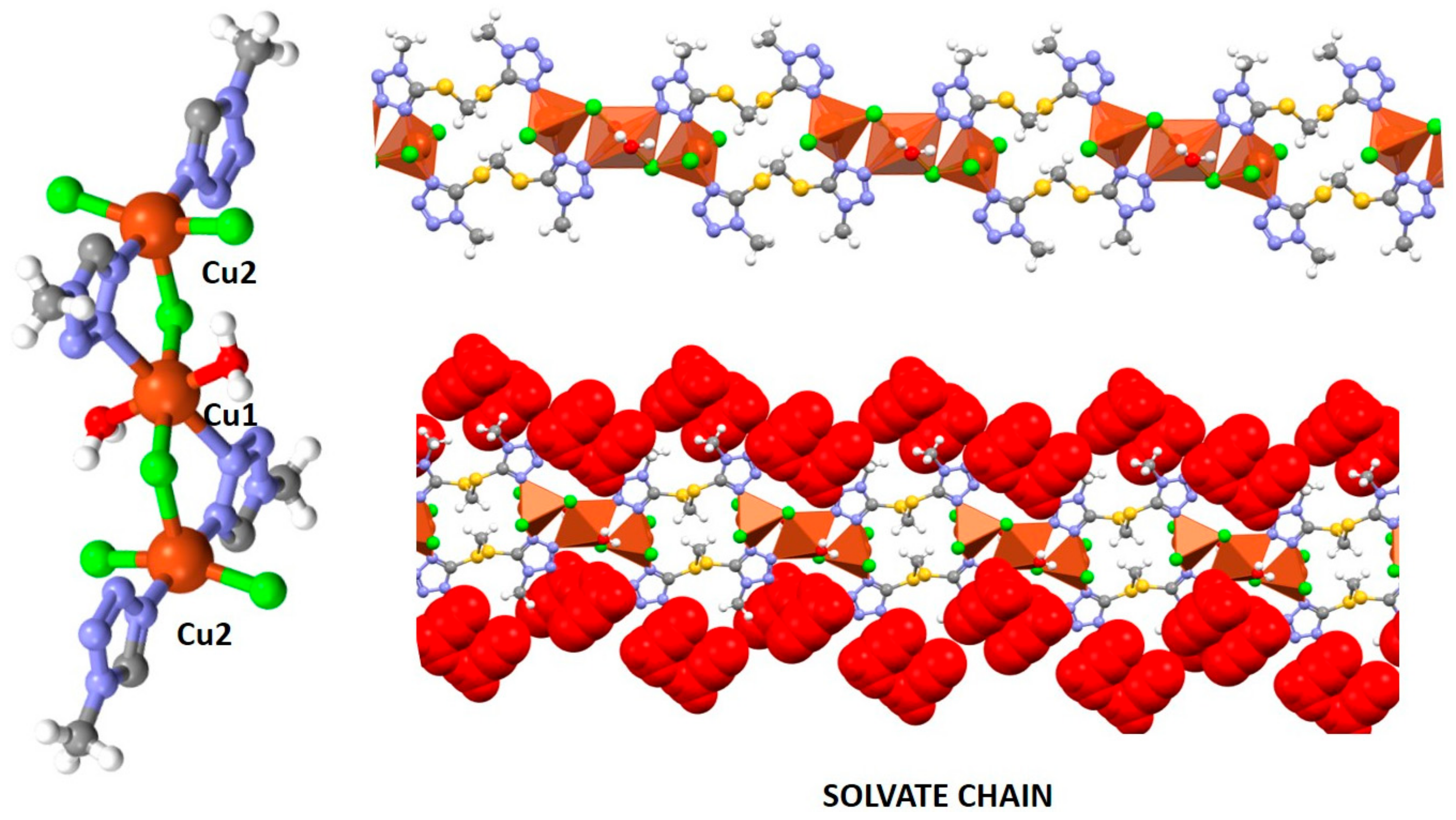
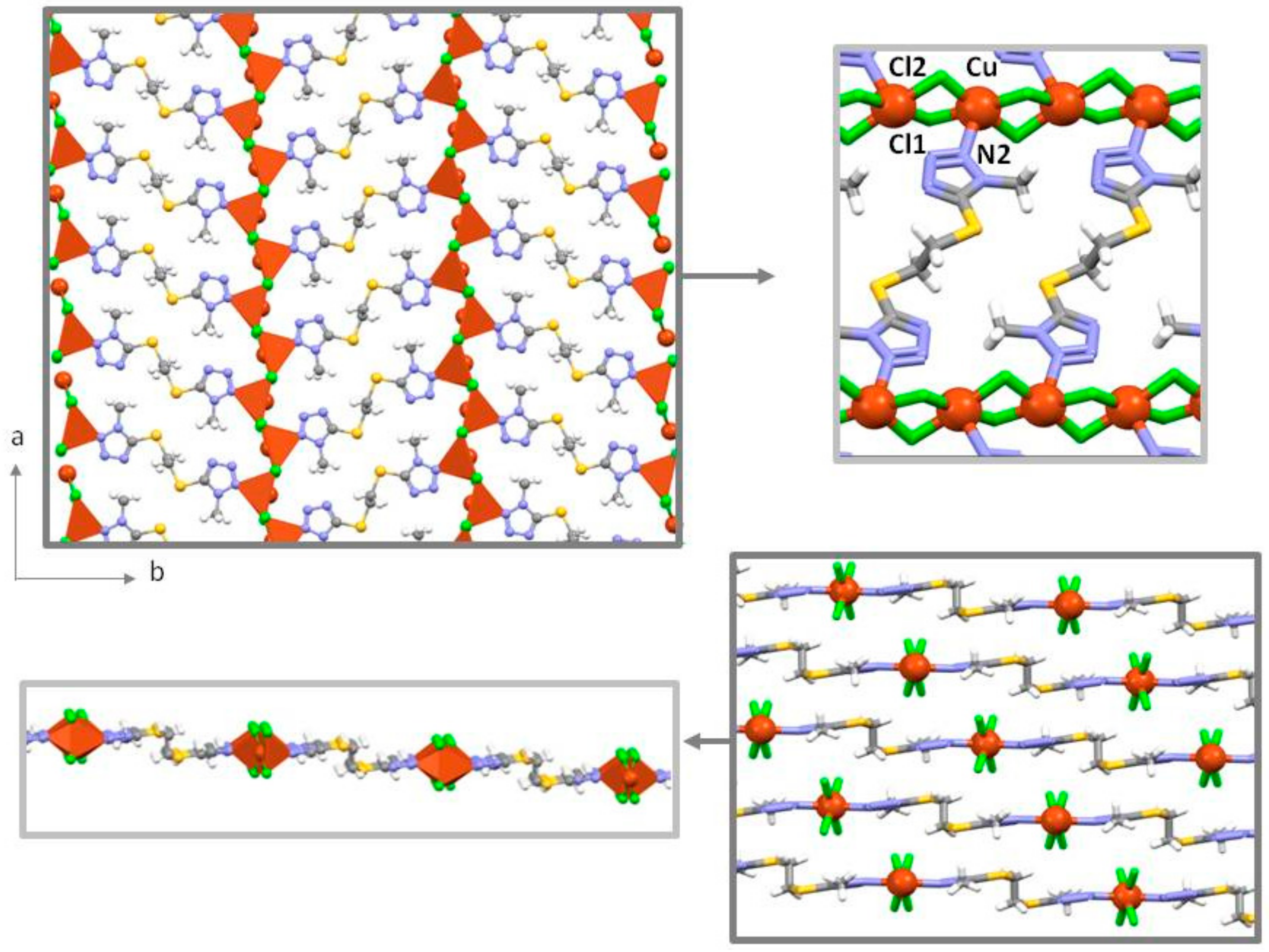
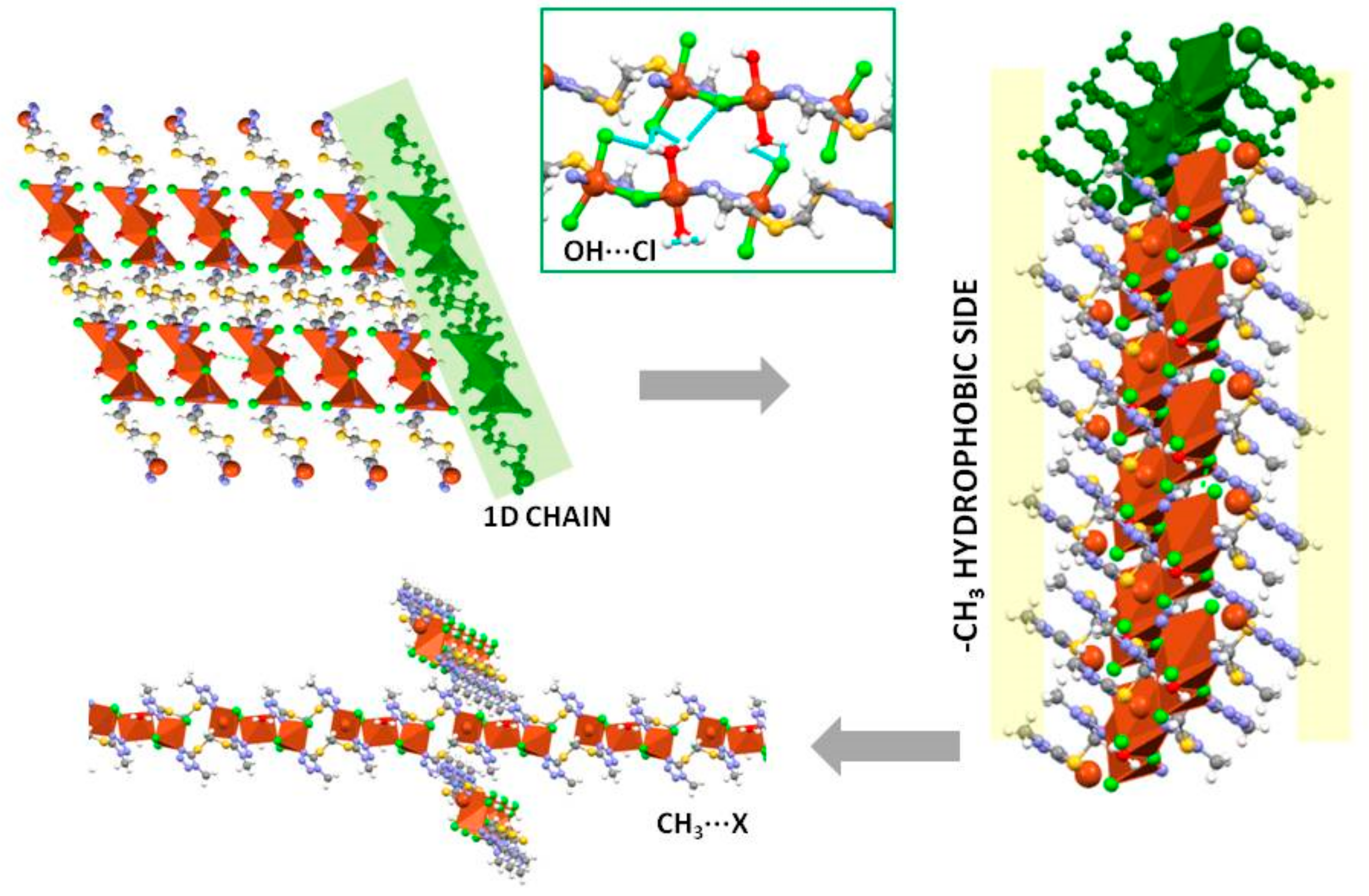
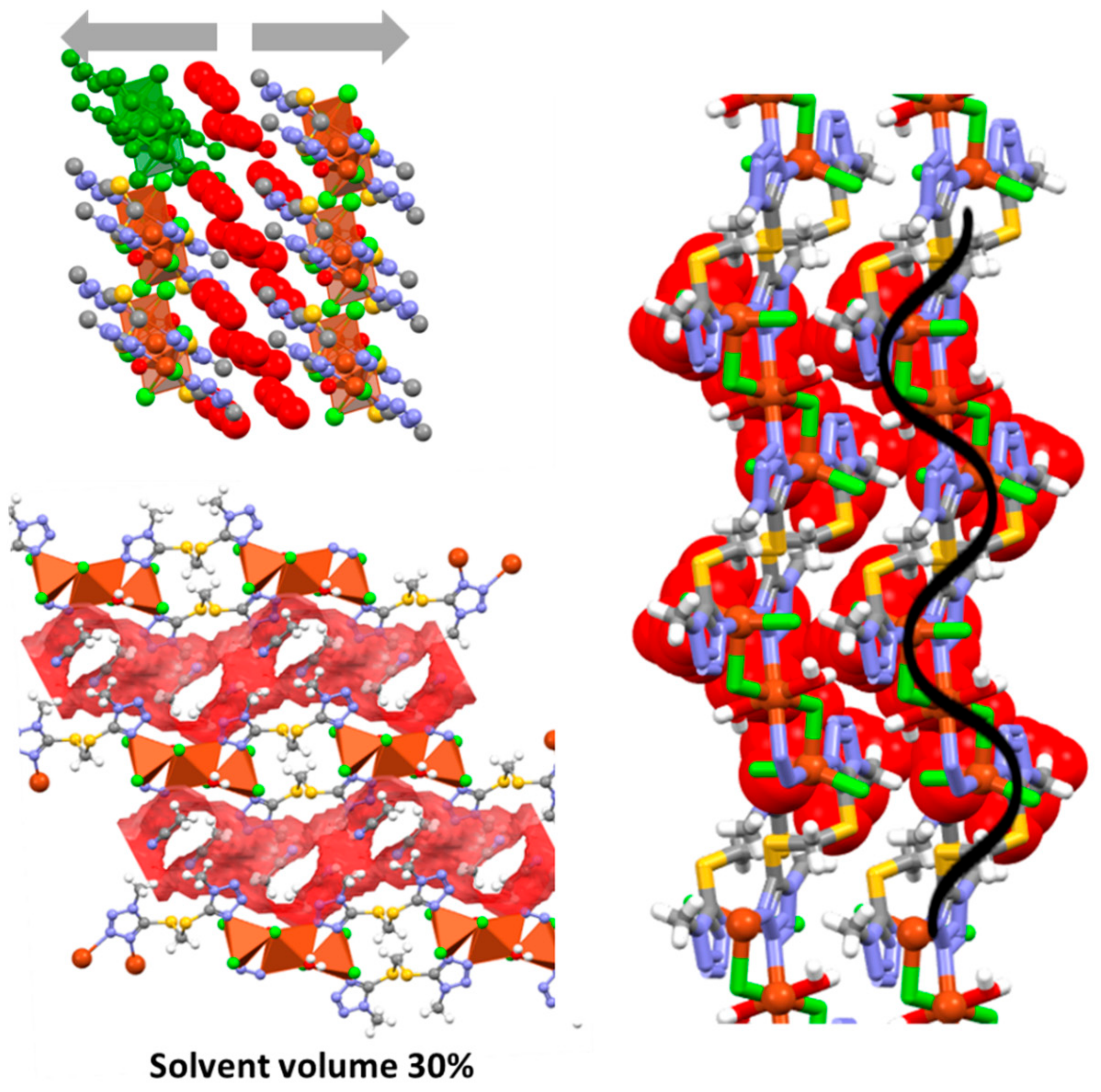
2.3. Crystal Structures of 1 and 1∙solv
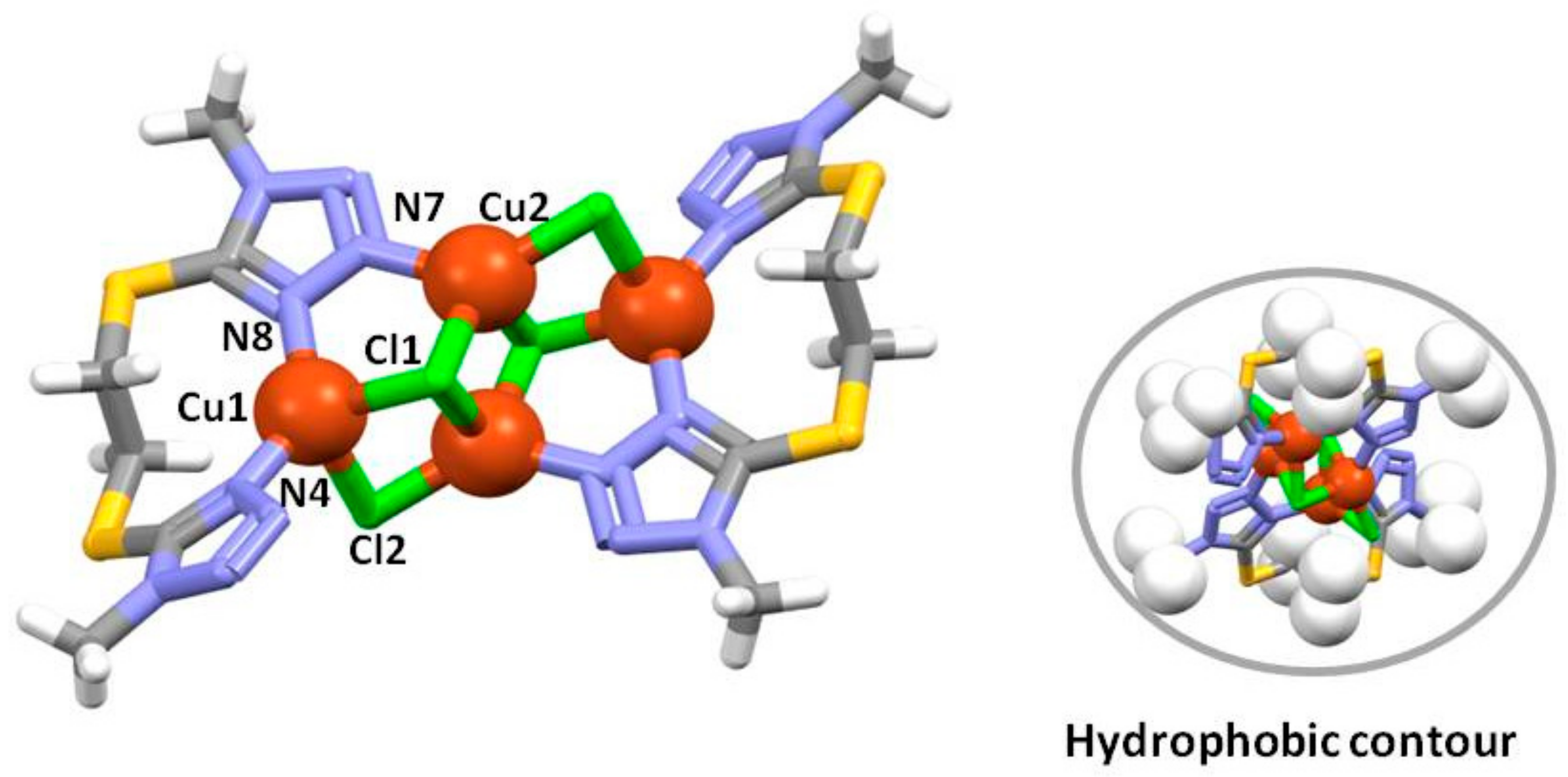
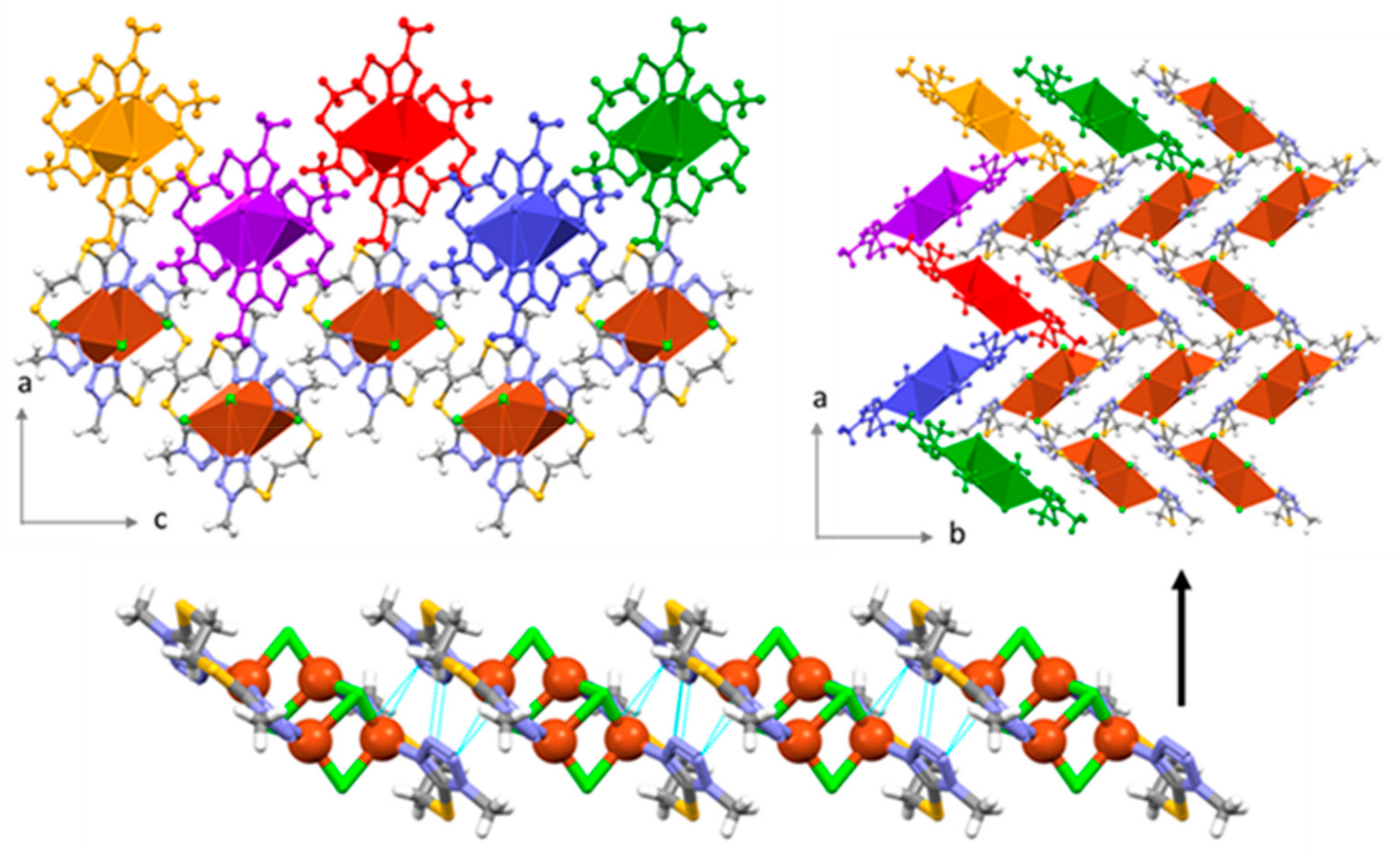
2.4. Crystal Structure of 2
2.5. Crystal Structure of 3
2.6. Supramolecular Arrangements
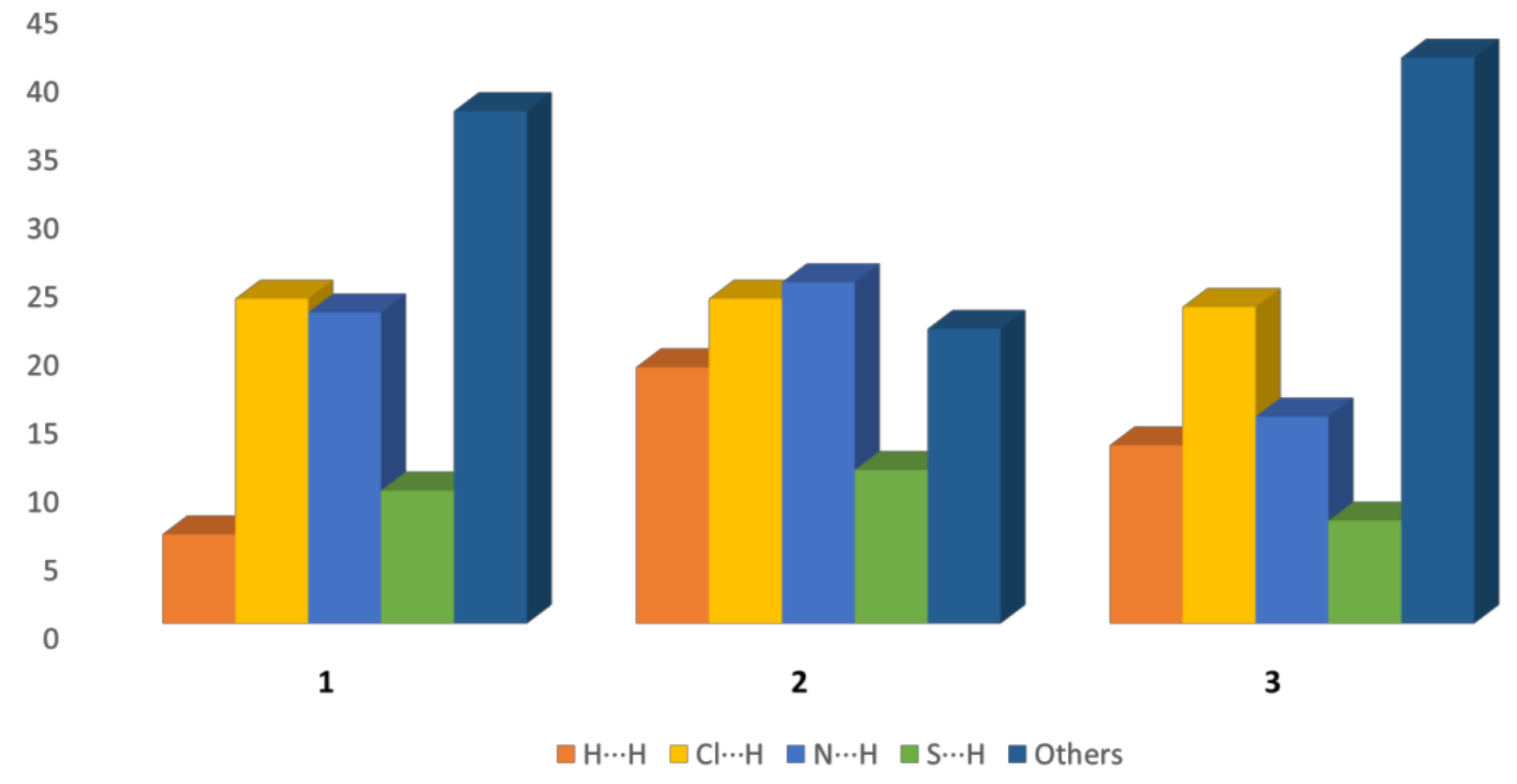
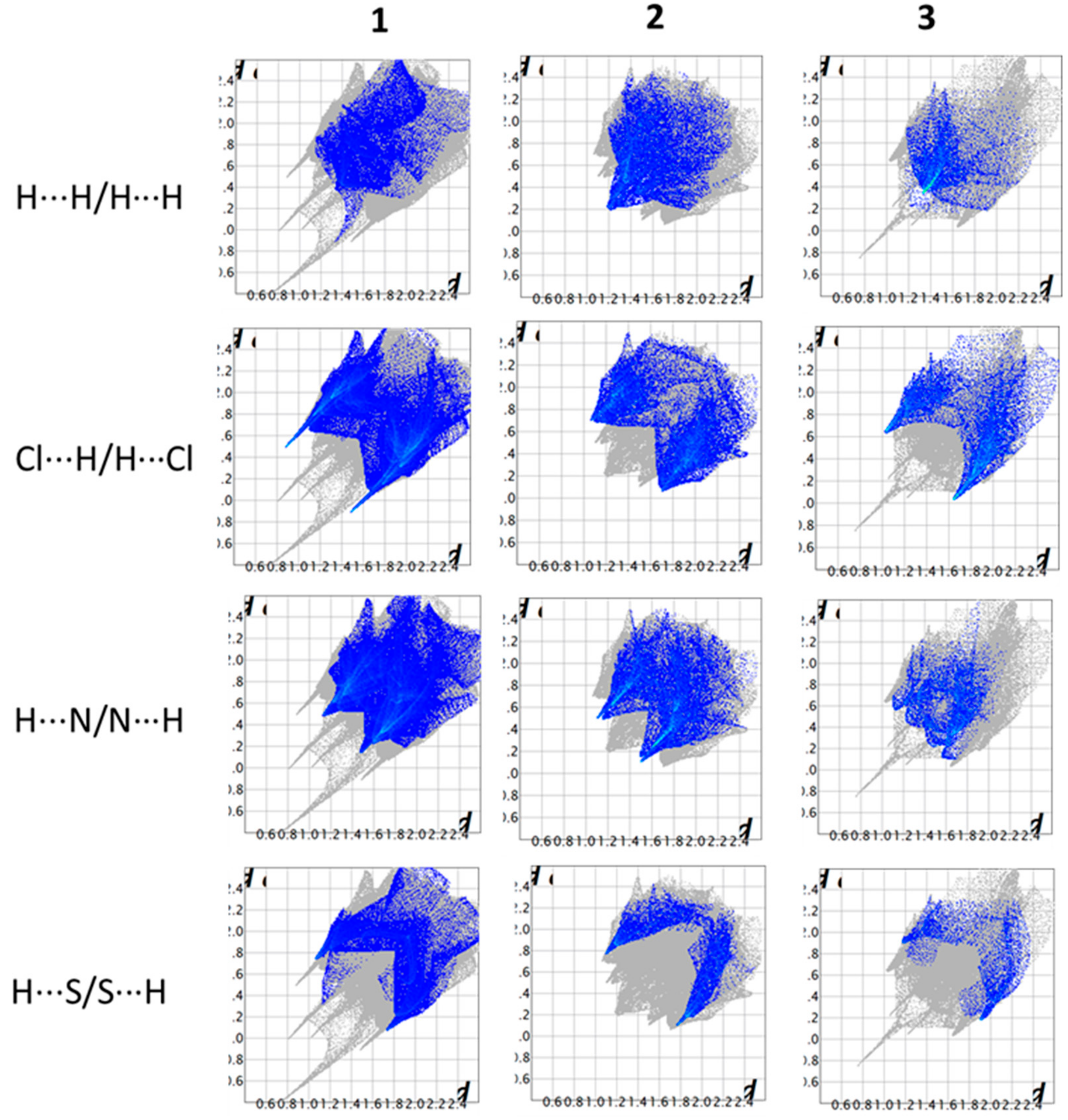
2.7. Hirshfeld Surface Study
2.8. Thermal Analysis
3. Conclusions
4. Experimental
4.1. Materials and Physical Measurements
4.2. Synthesis and Crystallization of the Complexes
4.2.1. ∞1Cu3Cl6(H2O)2(BMTTM)2 (1) and ∞1Cu3Cl6(H2O)2(BMTTM)2·4CH3CN (1∙solv)
4.2.2. [Cu2Cl2(BMTTE)]2 (2) and ∞2Cu2Cl4(BMTTE) (3)
Synthesis of [Cu2Cl2(BMTTE)]2 (2)
Synthesis of ∞2Cu2Cl4(BMTTE) (3)
4.3. X-ray Structure Determination
4.4. Hirshfeld Surface Study
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirao, T.; Kim, D.S.; Chi, X.; Lynch, V.M.; Ohara, K.; Park, J.S.; Yamaguchi, K.; Sessler, J.L. Control over multiple molecular states with directional changes driven by molecular recognition. Nat. Commun. 2018, 9, 823. [Google Scholar] [CrossRef]
- Thomas-Gipson, J.; Pérez-Aguirre, R.; Beobide, G.; Castillo, O.; Luque, A.; Pérez-Yañez, S.; Román, P. Unravelling the Growth of Supramolecular Metal–Organic Frameworks Based on Metal-Nucleobase Entities. Cryst. Growth Des. 2015, 15, 975–983. [Google Scholar] [CrossRef]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; John Wiley & Sons: London, UK, 2013. [Google Scholar]
- Lehn, J.-M. Supramolecular Chemistry; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Taylor, R.; Kennard, O. Crystallographic Evidence for the Existence of C−H⋯O, C−H⋯N, and C−H⋯Cl Hydrogen Bonds. J. Am. Chem. Soc. 1982, 104, 5063–5070. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steinter, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Brammer, L.; Bruton, E.A.; Sherwood, P. Understanding the Behavior of Halogens as Hydrogen Bond Acceptors. Cryst. Growth Des. 2001, 1, 277–290. [Google Scholar] [CrossRef]
- Thallapally, P.K.; Nangia, A. A Cambridge Structural Database analysis of the C–H⋯Cl interaction: C–H⋯Cl− and C–H⋯Cl–M often behave as hydrogen bonds but C–H⋯Cl–C is generally a van der Waals interaction. CrystEngComm 2001, 3, 114–119. [Google Scholar] [CrossRef]
- Popova, E.A.; Trifonov, R.E.; Ostrovskii, R.A. Tetrazoles for biomedicine. Russ. Chem. Rev. 2019, 88, 644–676. [Google Scholar] [CrossRef]
- Neochoritis, C.G.; Zhao, T.; Dömling, A. Tetrazoles via Multicomponent Reactions. Chem. Rev. 2019, 119, 1970–2042. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Hu, H.L.; Tian, A.X. Influence of Transition Metal Coordination Nature on the Assembly of Multinuclear Subunits in Polyoxometalates-Based Compounds. Cryst. Growth Des. 2010, 10, 4786–4794. [Google Scholar] [CrossRef]
- Wang, X.; Hu, H.; Tian, A.; Lin, H.; Li, J. Application of Tetrazole-Functionalized Thioethers with Different Spacer Lengths in the Self-Assembly of Polyoxometalate-Based Hybrid Compounds. Inorg. Chem. 2010, 49, 10299–10306. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Liu, G.; Hu, H.; Tian, A. Polyoxometalates-Directed Assembly of Inorganic-Organic Hybrid Compounds with Copper Multinuclear Nano-Cluster Based on Flexible Double Tetrazole-Based Thioether. J. Clust. Sci. 2011, 22, 211–223. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Q.; Tian, A.; Hu, H.; Liu, G. Assembly of a Series of Keggin-Based Multi- and Mono-Nuclear Structures by Tuning the Bis (Tetrazole)-Functionalized Thioether Ligands. Inorg. Chim. Acta 2012, 384, 62–68. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, S.Z. Crystal structure of poly[(μ4-1-methyl-1Htetrazole-5-thiolato-κ3S:S:N:N’) copper(I)], C2H3CuN4S. Kristallogr. NCS 2016, 231, 791–792. [Google Scholar]
- Li, C.-R.; Chen, T.; Xia, Z.-Q. Bis[(1-methyl-1H-tetrazol-5-yl)sulfanyl]-ethane. Acta Crystallogr. 2011, E67, o769. [Google Scholar] [CrossRef]
- Argibay-Otero, S.; Gómez-Paz, O.; Carballo, R. A Monoclinic Polymorph of 1,2-Bis[(1-Methyl-1H -Tetrazol-5-yl)Sulfanyl]ethane (BMTTE). Acta Crystallog. 2017, E73, 1523–1525. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Shearer, J.; Catalano, V.J. Subtle Modulation of Cu4X4L2 Phosphine Cluster Cores Leads to Changes in Luminescence. Inorg. Chem. 2015, 54, 6245–6256. [Google Scholar] [CrossRef] [PubMed]
- Robin, A.Y.; Fromm, K.M. Coordination polymer networks with O- and N-donors: What they are, why and how they are made. Coord. Chem. Rev. 2006, 250, 2127–2157. [Google Scholar] [CrossRef]
- Xue, K.; Chai, W.X.; Wu, Y.W.; Ling, C.; Song, L. Syntheses, Structures and Properties of Two Coordination Polymers Built Upon Copper(I, II) Halide Clusters and a New Thiodiazole Ligand. J. Clust. Sci. 2014, 25, 1005–1017. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedij, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua [1,7-bis(N-methylbenzimidazol-2′yl)-2,6-dithiaheptane] copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Amoedo, A.; Carballo, R.; García-Martínez, E.; Lago, A.B.; Vázquez López, E.M. Molecular metallocycles, acyclic metallodimers and 2D coordination polymers containing the twisted ligand bis(pyrimidin-2-ylthio)methane. Dalton Trans. 2010, 39, 2385–2394. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Ren, Z.; Lang, J. Unique Deca- and Tetranuclear Halocuprate(I) Clusters of a Clamplike Ligand: Isolation, Structure, and Luminescence Properties. Eur. J. Inorg. Chem. 2014, 5, 824–830. [Google Scholar] [CrossRef]
- Kwon, H.; Lee, E. Static and dynamic coordination behaviours of copper(I) ions in hexa(2-pyridyl)benzene ligand systems. Dalton Trans. 2018, 47, 8448–8455. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.K.; Hoffmann, R. Copper(I)-copper(I) interactions. Bonding relationships in d10-d10 systems. Inorg. Chem. 1978, 17, 2187–2189. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural Variation in Copper(I) Complexes with Pyridylmethylamide Ligands: Structural Analysis with a New Four-Coordinate Geometry Index, τ(4). Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Kitaigorodskii, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Lago, A.B.; Carballo, R.; Rodríguez-Hermida, S.; Vázquez-López, E.M. Copper(II) Acetate/Bis(4-pyridylthio)methane System: Synthesis, Structural Diversity, and Single-Crystal to Single-Crystal Transformation. Cryst. Growth Des. 2014, 14, 3096–3109. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Xia, Z.-Q.; Chen, A.-P.; Gao, S.-L. Bis[(1-methyl-1H-tetrazol-5-yl)sulfanyl]-methane. Acta Crystallogr. 2011, E67, o999. [Google Scholar] [CrossRef] [Green Version]
- Ardon, M.; Hayes, P.D.; Hogarth, G. Microwave-Assisted Reflux in Organometallic Chemistry: Synthesis and Structural Determination of Molybdenum Carbonyl Complexes. An Intermediate-Level Organometallic-Inorganic Experiment. J. Chem. Educ. 2002, 79, 1249–1251. [Google Scholar] [CrossRef]
- SMART, Version 5.054; Bruker AXS: Madison, WI, USA, 1997.
- SAINT, Version 8.38A; Bruker AXS: Madison, WI, USA, 2017.
- Sheldrick, G.M. SADABS, Version 2016/2; Program for Absorption Corrections; Göttingen University: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef]
- Rodriguez-Carvajal, J. Fullprof: A Program for Rietveld Refinement and Pattern Matching Analysis. In Proceedings of the Satellite Meeting on Powder Diffraction of the XV Congress of the IUCr, Toulouse, France, 19–28 July 1990; p. 127. [Google Scholar]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer, Version 3.1; University of Western Australia: Crawley, Australia, 2012. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Dimensionality | Coordination Mode of Ligand | Inorganic Units | Coordination Geometry |
|---|---|---|---|
| ∞1Cu3Cl6(H2O)2(BMTTM)2 (1) 1D ∞1Cu3Cl6(H2O)2(BMTTM)2·4CH3CN (1∙solv) 1D |  |  |  |
| [Cu2Cl2(BMTTE)]2 (2) 0D, tetramer |  |  |  |
| ∞2Cu2Cl4(BMTTE) (3) 2D |  |  |  |
| Atoms | 1 | 1∙solv | Atoms | 1 | 1∙solv |
|---|---|---|---|---|---|
| Cu1–Cl1 | 2.5477(12) | 2.5119(4) | N4–Cu1–N8#1 | 171.83(15) | 174.19(4) |
| Cu1–Cl2 | 2.2895(11) | 2.2660(4) | N8#1–Cu1–Cl1 | 93.88(11) | 93.94(3) |
| Cu1–Cl3 | 2.2744(12) | 2.2927(3) | N8#1–Cu1–Cl2 | 90.83(11) | 90.23(3) |
| Cu2–N3 | 2.561(4) | 2.4137(11) | N8#1–Cu1–Cl3 | 90.22(11) | 87.76(3) |
| Cu1–N4 | 2.038(4) | 2.0500(11) | Cl1#2–Cu2–Cl1 | 180.00(5) | 180.000(14) |
| Cu1–N8#1 | 2.052(4) | 2.0346(11) | Cl1–Cu2–N3 | 83.54(9) | 85.98(3) |
| Cu2–Cl1 | 2.2981(10) | 2.2716(3) | Cl1–Cu2–N3#2 | 96.46(9) | 94.02(3) |
| Cu2–Cl1#2 | 2.2980(10) | 2.2716(3) | N3#2–Cu2–N3 | 180.0 | 180.0 |
| Cu2–O1 | 1.959(3) | 2.0140(9) | O1–Cu2–Cl1 | 90.18(10) | 88.06(3) |
| Cl2–Cu1–Cl1 | 97.16(4) | 102.660(12) | O1–Cu2–Cl1#2 | 89.82(10) | 91.94(3) |
| Cl3–Cu1–Cl1 | 105.98(4) | 100.253(12) | O1–Cu2–N3 | 89.96(14) | 93.80(4) |
| Cl3–Cu1–Cl2 | 156.72(4) | 157.082(13) | O1#2–Cu2–N3 | 90.03(13) | 86.20(4) |
| N4–Cu1–Cl1 | 94.23(11) | 91.57(3) | O1–Cu2–O1#2 | 180.0 | 180.00(5) |
| N4–Cu1–Cl2 | 87.23(11) | 90.35(3) | Cu2–Cl1–Cu1 | 107.19(4) | 106.772(12) |
| N4–Cu1–Cl3 | 88.45(11) | 89.45(3) |
| Atoms | 2 | 3 | Atoms | 2 |
|---|---|---|---|---|
| Cu1–Cu2#1 | 2.9556(3) | N4–Cu1–Cu2#1 | 124.81(4) | |
| Cu1–Cl1 | 2.4039(4) | 2.3132(4) | N4–Cu1–Cl1 | 106.22(4) |
| Cu1–Cl1#2 | 2.3163(4) | N4–Cu1–Cl2#1 | 96.70(4) | |
| Cu1–Cl2 | 2.2987(4) | N4–Cu1–N8 | 137.74(6) | |
| Cu1–Cl2#1 | 2.4794(4) | 2.2979(4) | N8–Cu1–Cu2#1 | 96.92(4) |
| Cu1–N2 | 2.3981(13) | N8–Cu1–Cl1 | 100.74(4) | |
| Cu1–N4 | 2.0097(14) | N8–Cu1–Cl2#1 | 108.89(4) | |
| Cu1–N8 | 2.0268(14) | Cu2#1–Cu2–Cu1#1 | 72.140(10) | |
| Cu2–Cu2#1 | 2.7651(5) | Cl1–Cu2–Cu1#1 | 111.431(14) | |
| Cu2–Cl1 | 2.3556(5) | Cl1–Cu2– Cl1#1 | 113.629(13) | |
| Cu2–Cl1#1 | 2.6728(5) | Cl1#1–Cu2– Cl1#1 | 50.261(10) | |
| Cu2–Cl2 | 2.2330(4) | Cl1#1–Cu2–Cu2#1 | 51.302(12) | |
| Cu2–N7 | 2.0013(14) | Cl1–Cu2–Cu2#1 | 62.326(13) | |
| Cl1–Cu1–Cu2#1 | 58.759(12) | Cl2–Cu2–Cu1#1 | 54.982(12) | |
| Cl2#1–Cu1–Cu2#1 | 47.525(11) | Cl2–Cu2–Cu2#1 | 121.487(16) | |
| Cl1–Cu1–Cl1#2 | 176.292(10) | Cl2–Cu2–Cl1 | 113.376(17) | |
| Cl2–Cu1–Cl1 | 93.746(16) | Cl2–Cu2–Cl1#1 | 100.991(16) | |
| Cl2#1–Cu1–Cl1 | 102.006(16) | 85.599(15) | N7–Cu2–Cu1#1 | 137.43(4) |
| Cl2#1–Cu1–Cl1#2 | 94.895(15) | N7–Cu2–Cu2#1 | 99.24(4) | |
| Cl2–Cu1–Cl1#2 | 85.511(15) | N7–Cu2–Cl2 | 135.88(4) | |
| Cl2#1–Cu1–Cl2 | 176.067(11) | N7–Cu2–Cl1 | 99.50(4) | |
| Cl1–Cu1–N2 | 96.33(3) | N7–Cu2–Cl1#1 | 91.18(4) | |
| Cl1#2–Cu1–N2 | 87.29(3) | Cu2–Cl2–Cu1#1 | 77.492(15) | |
| Cl2–Cu1–N2 | 88.39(3) | Cu1–Cl1–Cu2#1 | 70.981(13) | |
| Cl2#1–Cu1–N2 | 95.53(3) | Cu2–Cl1–Cu1 | 90.204(15) | |
| Cu1#2–Cu1–N2 | 90.896(14) | Cu2–Cl1–Cu2#1 | 66.371(14) | |
| Cu1–Cl1–Cu1#1 | 90.072(14) |
| Structure | D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) |
|---|---|---|---|---|---|
| 1 | O1–HA…Cl3#1 | 0.84(2) | 2.82(6) | 3.250(4) | 114(5) |
| O1–HA…Cl1#1 | 0.84(2) | 2.93(4) | 3.685(3) | 152(6) | |
| C12–H12C…Cl2#4 | 0.98 | 2.49 | 3.418(5) | 158.6 | |
| C12–H12B…N7#3 | 0.98 | 2.86 | 3.207(6) | 102.0 | |
| C12–H12A…N6#2 | 0.98 | 2.68 | 3.327(6) | 124.0 | |
| C12–H12A…S2#1 | 0.98 | 2.90 | 3.717(5) | 141.2 | |
| 1∙solv | O1–H1C…Cl1#6 | 0.89 | 2.99 | 3.6713(10) | 135.4 |
| O1–H1C…Cl2#6 | 0.89 | 2.72 | 3.4096(10) | 135.1 | |
| C12–H12C…N41#7 | 0.98 | 2.95 | 3.221(2) | 96.9 | |
| C12–H12B…N31#4 | 0.98 | 2.68 | 3.650(2) | 170.0 | |
| C22–H22C…Cl3#5 | 0.98 | 2.95 | 3.5901(15) | 123.9 | |
| C1–H1B…Cl2#3 | 0.99 | 2.95 | 3.5478(13) | 119.9 | |
| C1–H1B…S2#2 | 0.99 | 2.95 | 3.5065(13) | 116.3 | |
| C31–H31A…N6#5 | 0.98 | 2.64 | 3.559(2) | 156.5 | |
| C31–H31B…N7#1 | 0.98 | 2.59 | 3.473(2) | 150.1 | |
| C31–H31C…O1#7 | 0.98 | 2.71 | 3.536(2) | 141.7 | |
| C41–H12A…Cl1#6 | 0.98 | 2.98 | 3.3693(15) | 104.8 | |
| 2 | C2–H2B…Cl2#1 | 0.99 | 2.89 | 3.3201(17) | 107.2 |
| C1–H1B…Cl2#1 | 0.99 | 2.84 | 3.4698(17) | 122.1 | |
| C12–H12C…Cl2#3 | 0.98 | 2.87 | 3.4037(19) | 115.0 | |
| C22–H22C…Cl1#4 | 0.98 | 3.05 | 3.4498(17) | 106.1 | |
| C12–H12B…S2#2 | 0.98 | 2.95 | 3.7522(18) | 139.5 | |
| 3 | C1–H1A⋯Cl2#1 | 0.99 | 2.78 | 3.6987(16) | 153.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Paz, O.; Carballo, R.; Lago, A.B.; Vázquez-López, E.M. Metallosupramolecular Compounds Based on Copper(II/I) Chloride and Two Bis-Tetrazole Organosulfur Ligands. Chemistry 2020, 2, 33-49. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010005
Gómez-Paz O, Carballo R, Lago AB, Vázquez-López EM. Metallosupramolecular Compounds Based on Copper(II/I) Chloride and Two Bis-Tetrazole Organosulfur Ligands. Chemistry. 2020; 2(1):33-49. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010005
Chicago/Turabian StyleGómez-Paz, Olaya, Rosa Carballo, Ana Belén Lago, and Ezequiel M. Vázquez-López. 2020. "Metallosupramolecular Compounds Based on Copper(II/I) Chloride and Two Bis-Tetrazole Organosulfur Ligands" Chemistry 2, no. 1: 33-49. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010005