
Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia


1
Department of Genetics and Molecular Biology, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Av. Instituto Politécnico Nacional 2508, San Pedro Zacatenco, Gustavo A. Madero, Mexico City 07360, Mexico
2
Nephrology Department, Hospital General Regional No. 1, Instituto Mexicano del Seguro Social, Av. Bosques de los Olivos No. 101, Comunidad La Goleta, Morelia 61301, Michoacán, Mexico
*
Author to whom correspondence should be addressed.
Hemato 2024, 5(1), 6-18; https://0-doi-org.brum.beds.ac.uk/10.3390/hemato5010002
Submission received: 20 November 2023
/
Revised: 13 December 2023
/
Accepted: 18 December 2023
/
Published: 26 December 2023
(This article belongs to the Section Leukemias)
Abstract
:Acute lymphoblastic leukemia (ALL), a remarkable cancer that mainly affects children, has seen commendable advances in its treatment. However, the occurrence of relapses after initial treatments poses a major threat and is one of the leading causes of cancer-related mortality in pediatric patients. To address this problem, innovative therapeutic approaches for ALL need to be continuously developed and refined. Synthetic lethality, an interaction between genes in which alteration of only one allows survival, but simultaneous alteration of both leads to inviability, is emerging as a promising therapeutic approach against ALL and other cancers. In this regard, the review aims to examine the documented cases of synthetic lethality in ALL reported to date (2023) and to elucidate the molecular mechanisms underlying this phenomenon. Furthermore, this review explores possible targets that have so far gone unnoticed, justifying their importance in this context.
1. Introduction
Leukemia constitutes a collective term for a diverse group of malignant disorders characterized by the excessive production of abnormal cells in hematopoietic tissues, such as bone marrow and the lymphatic system [1,2,3]. The classification of leukemias encompasses several types, which are distinguished mainly by the speed of growth—acute (fast) or chronic (slower)—and the type of cell originating, either myeloid (giving rise to erythrocytes, platelets, monocytes, neutrophils, basophils and eosinophils) or lymphoid (giving rise to T lymphocytes, B lymphocytes and NK cells) [4,5,6].
Leukemia is the most prevalent form of childhood cancer and accounts for approximately one-third of all pediatric cancer cases (children under 15 years of age) [7]. Within this demographic group, acute lymphoblastic leukemia (ALL) outnumbers acute myelogenous leukemia (AML) by five times, constituting about 78% of all childhood leukemia diagnoses [1,8]. Among pediatric hematological malignancies, B-acute lymphoblastic leukemia (B-ALL) ranks first, not only for being the most frequent, but also for being the leading cause of cancer-related childhood mortality [7]. Indeed, the incidence of B-ALL has shown a persistent increase over the years [9]. Meanwhile, T-cell-acute lymphoblastic leukemia (T-ALL) accounts for 10% to 15% of childhood ALL cases. Historically, the prognosis of children with T-ALL has been poor, but progressive improvement has been achieved through intensified therapeutic approaches [10,11].
The treatment protocol for ALL comprises three crucial phases: induction (or remission), consolidation (or intensification) and maintenance (or continuation) [2]. Remission induction therapy involves a combination of three drugs (a glucocorticoid such as prednisone or dexamethasone, vincristine and asparaginase) or, in selected cases, a full four-drug regimen (incorporating the aforementioned three plus an anthracycline). Administered for 4–6 weeks, this therapeutic protocol demonstrates impressive efficacy, achieving a remarkable complete remission (CR) rate in approximately 98% of pediatric patients [2,12]. The consolidation phase, the second stage of treatment, is carried out to prevent the re-emergence of leukemic cells. Therapeutic agents such as cyclophosphamide, cytarabine and mercaptopurine are used for this purpose [13]. Finally, maintenance therapy aims to maintain remission over a prolonged period, usually 2 to 3 years. This regimen involves daily administration of mercaptopurine and weekly doses of methotrexate, occasionally supplemented with vincristine and steroid pulses [2].
Notably, the high incidence of relapses (in children it is around 10%, while in adults it is close to 50%) after initial treatments represents a major challenge, constituting the main cause of mortality in pediatric patients diagnosed with ALL [14]. Indeed, this problem persists despite notable advances such as the introduction of multidrug chemotherapy regimens, allogeneic hematopoietic stem cell transplantation (HSCT) and the recent integration of CD19-targeted chimeric antigen receptor (CAR) T-cell immunotherapy [15,16]. Given its relevance, there is an inherent need to evolve and improve ALL treatment strategies by considering new and innovative approaches. In this regard, synthetic lethality has emerged as a promising therapeutic avenue against ALL and other cancers. A synthetic lethality interaction develops between two genes when alteration of only one allows survival, but simultaneous alteration of both causes loss of viability (Figure 1) [17]. With this goal in mind, the present review aims to showcase the synthetic lethality approaches described for the treatment of ALL, while explaining the intricate molecular mechanisms underlying these strategies. In addition, the review explores potential targets that have so far escaped scrutiny, offering a reasoned justification for their importance in this particular context.
2. Challenges in the Identification of Synthetic Lethal Interactions
Screening for synthetic lethal interactions poses three major challenges. First, by definition, these genetic interactions produce lethality, making mutant recovery and identification difficult. Second, many synthetic lethal interactions are condition-dependent and may not be conserved in all genetic contexts or in different cellular conditions. Third, synthetic lethal interactions are rare, requiring a large number of mutant gene pair combinations to be queried to identify synthetic lethal interactions [17]. For these reasons, most large-scale synthetic lethal interaction studies have been carried out in budding yeast or fission yeast, as technologies are available that facilitate the generation and high-throughput analysis of double mutants under defined laboratory conditions. Advances in the field of RNA interference (RNAi) and, more recently, CRISPR/Cas technology have made it possible to conduct large-scale, unbiased synthetic lethality screening assays directly in human cell cultures [18]. These technologies offer the potential to expand our understanding of synthetic lethality interactions in the context of human disease, including cancer. In addition, functional genomic screening, based on the genetic concept of synthetic lethality, offers a promising avenue for the identification of potential drug targets. This strategy is in line with the evolving landscape of personalized or precision medicine, where understanding the tumor-specific genetic context is paramount to develop personalized and impactful treatment strategies [19].
3. Synthetic Lethality Reports in ALL
Genomic studies in ALL samples have revealed a myriad of entities characterized by distinctive driver mutations, often with unique gene expression profiles. These findings have necessitated an expansion or revision of the 2016 World Health Organization (WHO) classification (Table 1) [20]. Recognizing these genomic complexities is of paramount importance, especially in the context of synthetic lethality, where the identification of specific genetic interactions offers the promise of targeted therapeutic interventions [18].
Currently, there are few reports studying synthetic lethality in the context of ALL. However, this emerging strategy holds great promise as it may allow for personalized treatments tailored to each patient’s unique genetic and molecular landscape. An illustrative example of this phenomenon is seen in ALL patients harboring the Philadelphia (Ph) chromosome [21,22]. The Ph chromosome is the most prevalent cytogenetic abnormality among adult patients diagnosed with ALL, manifesting in approximately 20% to 30% of all cases [23]. This aberration arises from a reciprocal translocation between the ABL1 oncogene on the long arm of chromosome 9 and a breakpoint cluster region (BCR) on the long arm of chromosome 22 (t(9;22)(q34.1;q11.2)/BCR::ABL1). This translocation results in the formation of a fusion gene, BCR-ABL1, which encodes an oncogenic protein with constitutively active tyrosine kinase activity [24]. Patients with Ph-positive (Ph+) ALL face a high risk of central nervous system (CNS) involvement and often experience an aggressive clinical course. Historically, this subgroup has shown inferior results compared to their Ph-negative (Ph-) counterparts, although this scenario has improved with the introduction of tyrosine kinase inhibitors [25]. Synthetic lethality has been reported in Ph+ B-ALL cell lines when combining a phosphoinositide-3-kinase (PI3K) inhibitor, such as taselisib, with olaparib, a poly(ADP-ribose) polymerase (PARP) inhibitor. This is because taselisib causes a reduction in breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) levels that affects homologous recombination (HR) repair, and olaparib inhibits base excision repair (BER). Both DNA repair pathways play a crucial role in preserving genomic stability. Thus, Ph+ ALL cells show increased vulnerability to genetic instability secondary to BCRA1 downregulation and BER inhibition, ultimately leading to apoptosis [21]. Similarly, a recent article reported synthetic lethality in relapsed ALL when mismatch repair (MMR) deficiency coincides with targeting of DNA polymerase β (POLB). MMR deficiency has been associated with thiopurine resistance and hypermutation in this disease. However, downregulation of POLB levels increased sensitivity to thiopurines in resistant cells by also altering the BER pathway. Meanwhile, the use of the POLB inhibitor oleanolic acid (OA) in combination with thiopurines showed a synergistic effect, inducing cell death in ALL cell lines, patient-derived xenograft (PDX) cells, as well as mouse xenograft models [26]. In an effort to improve the efficacy of Ph+ B-ALL treatment with a different approach, a study analyzed the combination of BCR-ABL1 inhibition using the tyrosine kinase inhibitor (TKI) nilotinib, together with the IRE1α RNase domain inhibitor MKC-8866. IRE1α assumes a critical role in the unfolded protein response (UPR) by performing messenger RNA splicing of the transcription factor XBP1 [27]. XBP1 targets encode proteins that contribute to improved folding and quality control of endoplasmic reticulum (ER) proteins [28]. This therapy fights the disease through specific and complementary mechanisms of action affecting UPR proteins, multiple inducers of apoptosis and negative regulators of the cell cycle. It is important to note that p38 mitogen-activated protein kinase (MAPK) activation, driven primarily by MKC-8866 activity, and Jun N-terminal kinase 1/2 (JNK1/2) activation, a consequence of nilotinib treatment, are key factors contributing to the success of combination therapy [22]. The UPR plays a crucial role in managing ER stress by halting protein translation, eliminating misfolded proteins and activating signaling pathways that enhance the synthesis of molecular chaperones involved in protein folding. If these targets are not reached within a defined time frame or if prolonged disruption occurs, the UPR triggers apoptosis [29].
One study proposed that administration of N-(3-oxododecanoyl)-L-homoserine lactone (3OC12-HSL) could induce synthetic lethality in refractory B-ALL when combined with chemotherapeutic drugs. The underlying mechanism for this phenomenon in B-ALL involves the overexpression of PON2, an arylesterase. PON2 plays a pivotal role by facilitating glucose uptake and energy production through the inhibition of the interaction between glucose transporter 1 (GLUT1) and its stomatin (STOM) inhibitor, effectively mitigating the energy crisis imposed by metabolic gatekeepers and promoting disease progression. In the context of synthetic lethality, PON2 rapidly hydrolyzes 3OC12-HSL, causing intracellular acidification. This process leads to the activation of caspase-3, triggering a cascade of events that ultimately results in cell death [30]. Particularly in B-ALL cells with elevated WEE1 levels, one study mentioned synthetic lethality resulting from the combination of the CHK1/CHK2 inhibitor PF-0477736 and the WEE1 inhibitor adavosertib (AZD1775) [31]. Both CHK1/2 and WEE1 are cell cycle checkpoint kinases, and their sustained inhibition leads to cell death [32]. Thus, the rationale for this drug regimen was to activate the S-phase checkpoint and induce DNA damage, thereby elevating replicative stress and causing the collapse of the replicative forks [31]. On the other hand, transcription factor 3 (TCF3)-hepatic leukemia factor (HLF)-positive B-ALL cell lines showed increased susceptibility to olaparib and veliparib. This susceptibility is attributed to the TCF3-HLF protein, which causes increased PARP activity and defective HR repair [33]. Notably, TCF3-HLF ALL represents an exceptionally aggressive subtype characterized by a remarkably poor outcome with existing standard treatment modalities, including chemotherapy and stem cell transplantation [34].
Turning attention to T-ALL, a comprehensive genetic screening in a zebrafish T-ALL model uncovered synthetic lethality between NU7026—an inhibitor targeting DNA-PK and PI3K—and pladienolide B—an inhibitor of mRNA splicing. Additionally, synthetic lethality was also observed when NU7026 was combined with thapsigargin, a noncompetitive inhibitor of sarco/endoplasmic reticulum Ca+2 ATPase (SERCA) [35]. Enhancer of zeste 2 polycomb repressive complex 2 (EZH2)-deficient T-ALL cells exhibit synthetic lethality upon exposure to MK8776, a CHK1 inhibitor. This phenomenon is due to the fact that EZH2 deficiency leads to MYCN-induced replication stress, which makes cells more dependent on CHK1 activity for replication fork integrity [36]. Synthetic lethality also occurred in chemoresistant NOTCH1-mutated T-ALL cells treated with IACS-010759, a potent small molecule inhibitor of complex I of the mitochondrial electron transport chain (ETC), together with L-asparaginase-based chemotherapy. This is because oncogenic activation of NOTCH1 is associated with an increased reliance on glutamine-driven oxidative phosphorylation to generate ATP. Thus, inhibition of oxidative phosphorylation combined with the glutaminase inhibitory activity of L-asparaginase effectively induces a cellular transcriptomic and metabolic catastrophe that prevents leukemia progression [37]. T-ALL cells overexpressing LMO2 show high sensitivity to olaparib. Mechanistically, this occurs because LMO2 inhibits the recruitment of BRCA1 to double-strand breaks (DSBs) by interacting with p53 binding protein 1 (53BP1) during repair [38]. The oncogenic KRAS G13D variant has been described to elevate the expression levels of components related to the alternative nonhomologous end-joining (alt-NHEJ) pathway in the T-ALL cell line CCRF-HSB2. Indeed, this variant results in a preference for the use of the highly error-prone alt-NHEJ pathway for DNA DSB repair. In this regard, action on DNA ligase 3 (LIG3a) or PARP1 inhibitors such as NU1025 specifically sensitizes KRAS-mutated cells to genotoxic anticancer drugs such as daunorubicin, cytarabine or etoposide (VP-16), offering a promising avenue for improving therapeutic responses in the context of KRAS-mutated cells [39]. Lastly, in preclinical studies performed with leukemia cell lines and primary leukemia samples, it was observed that TAL1-AKTE17K-positive T-ALL cells showed increased sensitivity to PI3K inhibitors, namely dactolisib (BEZ235) and MK2206. This increased sensitivity derives from the fact that, in the absence of AKT pathway activation, TAL1 exerts adverse effects by inducing positive regulation of proapoptotic genes. Likewise, in the context of TAL1-AKTE17K-positive cells, a marked elevation of susceptibility to PARP inhibitors such as olaparib was observed. This increased vulnerability may be attributed to their increased reliance on DNA repair genes that span multiple pathways, with a greater emphasis on the NER pathway. Thus, significant synergy was evident when olaparib was combined with dactolisib in TAL1-AKTE17K-positive cells [40]. A comprehensive compilation of all identified cases of synthetic lethality in ALL is presented in Table 2.
4. Exploring Synthetic Lethality Targets for ALL
The heterogeneity of ALL, characterized by diverse genetic mutations and altered gene expression, adds a layer of complexity to the identification of synthetic lethal pairs of broad applicability. The challenge is not only to identify these pairs but also to discern their relevance in a specific context, taking into account the various subtypes and individual variations within ALL patients. However, there are some avenues that merit further exploration in future research. For example, the combination of the mechanistic target of rapamycin kinase (mTOR) inhibitor sirolimus and methotrexate showed a synergistic effect on B-ALL cells, both in vitro and in vivo. Mice xenografted with ALL cells that underwent the dual treatment regimen achieved sustained and durable complete remission, in marked contrast to the outcome of single-agent treatments, which initially resulted in a partial response before eventually progressing. The observed effect can be attributed, at least partially, to the decrease in dihydrofolate reductase (DHFR) expression, a consequence of the sirolimus-induced decrease in cyclin D1 levels [41]. In the same line of research, sirolimus also potentiated the cytotoxicity of dexamethasone in some ALL cell lines and prolonged event-free survival in ALL xenograft models. These effects were attributed to induction of cell arrest, depolarization of the mitochondrial membrane, inactivation of the PI3K pathway and activation of caspases 3 and 9 [42]. Given these considerations, mTOR emerges as a viable candidate to determine its potential synthetic lethality in specific ALL contexts.
In Ph+ ALL patients, the concurrent administration of a cyclosporine with dasatinib has been observed to increase the peak plasma concentrations of dasatinib. These higher plasma concentrations are closely related to enhanced tyrosine kinase inhibition, which correlates with improved disease control in murine models [43]. The underlying rationale for this synergistic interaction involves the identification of a noncanonical WNT/Calcineurin/nuclear factor of activated T-cells (NFAT) signaling cascade, which acts as an escape route mechanism in BCR-ABL1-positive cells following exposure to TKIs [44]. In this sense, induction of synthetic lethality could be achieved by inhibition of this pathway using WNT inhibitors or NFAT inhibitors. In addition, this has the potential advantage of reducing the required doses of dasatinib and thereby mitigating the reported hematological toxicities [43]. Other potential targets in Ph+ ALL, which could be explored for their potential synthetic lethality in conjunction with ABL1 hyperactivity, were identified using a CRISPR-based genotoxic assay against DNA-damaging agents. Among these targets are the fucose-1-phosphate guanylyltransferase (FPGT)-cardiac troponin I-interacting kinase (TNNI3K), NADH:ubiquinone oxidoreductase subunit C1 (NDUFC1), β-1,3-galactosyltransferase 6 (B3GALT6), neuromedin U receptor 2 (NMUR2), AP2-associated kinase 1 (AAK1), zinc finger X-linked duplicated A (ZXDA), calpastatin (CAST), γ-aminobutyric acid type A receptor subunit γ2 (GABRG2), GFB-induced factor 2 (TGIF2), crystallin γS (CRYGS) and family with sequence similarity 105, member B (FAM105B) [45,46]. It is crucial to note that these predictions must undergo experimental validation to corroborate their significance, and it is imperative to thoroughly elucidate the molecular mechanisms involved.
The combination of dasatinib with dexamethasone showed remarkable pharmacological synergy, resulting in resensitization of glucocorticoid-resistant ALL cell lines and patient-derived xenograft samples. Furthermore, this drug combination had a significant impact on the expansion of T-ALL in immunocompromised mice. This response can be attributed, in part, to inhibition of the lymphocyte-specific protein tyrosine kinase (LCK) pathway, which ultimately leads to cell cycle arrest [47]. Furthermore, desatinib-mediated LCK inhibition, together with mTORC1 inhibition, has been documented to produce potent cell killing in T-ALL cells through downregulation of antiapoptotic myeloid cell leukemia 1 (MCL1) protein expression [48]. Thus, the possible synthetic lethal interaction between LCK and mTOR warrants further investigation in future studies.
Exposure to various chemotherapeutic drugs, such as vincristine, daunorubicin, etoposide, cytosine arabinoside, dexamethasone and asparaginase, followed by treatment with the FMS-related receptor tyrosine kinase 3 (FLT3) inhibitor CEP-701, demonstrated a synergistic killing effect on KMT2A-rearranged ALL cell lines (specifically, HB-1119 and SEM-K2 cells). Mechanistically, CEP-701 induces cell cycle arrest in the G1 phase and increases sensitivity to the action of chemotherapeutic drugs to subsequently activate proapoptotic signals [49]. Another study suggested that the use of inhibitors directed against FLT3 and TGFβ1 could prove to be a valuable strategy to eradicate minimal residual disease (MRD) in KMT2A-rearranged ALL [50]. Concurrently, MRX-2843, a small molecule that acts as a dual inhibitor of MERTK and FLT3 kinases, demonstrated strong synergy with vincristine to prevent proliferation of B-ALL and T-ALL cell lines. This effect was further corroborated in an orthotopic murine xenograft model designed specifically for thymus precursor T-ALL [51]. All these observations suggest that FLT3 could be a potential target for synthetic lethality in KMT2A-rearranged ALL. Furthermore, predictions indicate that FLT3 inhibition could potentially exhibit synthetic lethality when combined with silencing of proteinase 3 (PRTN3), protein tyrosine phosphatase receptor type K (PTPRK), eukaryotic translation initiation factor 1 (EIF1), Kelch domain-containing 7A (KLHDC7A), adrenoceptor α2A (ADRA2A), RAB5B, thrombospondin 3 (THBS3), transmembrane protein 214 (TMEM214), acyl-CoA dehydrogenase family member 9 (ACAD9), pantothenate kinase 4 (PANK4), cystatin 8 (CST8), and collagen type V α3 chain (COL5A3) [45,46]. Experimental research is vital to validate potential synthetic lethality and ensure its safety for implementation in future studies.
Recent studies have revealed a remarkable upregulation of PARP enzymes in T-ALL patients, which intricately influences the expression and post-transcriptional modification of the TET1 gene [52]. The increased expression of TET1 in T-ALL cells establishes an increased susceptibility to olaparib. This increased sensitivity derives from the ability of olaparib to counteract TET1 activity, a crucial mechanism that is biologically significant [53]. Thus, combining PARP inhibitors with TET1 silencing or employing its inhibition by small molecules such as NSC-370284 may pave the way to synthetic lethality. This strategic approach holds promise for synergistically targeting key pathways, potentially improving therapeutic outcomes in a synergistic manner.
Combination therapy of ABT-737, a B-cell lymphoma 2 (BCL-2) inhibitor, and N-(4-hydroxyphenyl)retinamide, a retinoid receptor activator, synergistically induced B-ALL cell death. This effect was due to phosphorylation of MCL-1 via JNK activation and facilitated by the production of reactive oxygen species (ROS). In addition, the combined treatment caused depolarization of the mitochondrial membrane and activation of caspases 3, 8 and 9 [54]. In another study, it was observed that the combination of ABT-737 with the multi-TKI sunitinib showed synergy in the elimination of KMT2A-rearranged ALL cells. This could suggest synthetic lethality between BCL-2 inhibition and receptor tyrosine kinase (RTK) signaling [55].
Proteasome inhibitors, such as bortezomib, exhibit highly cytotoxic effects on NOTCH1-overactivated T-ALL cells. Mechanistically, bortezomib represses transcription of NOTCH1 and downstream effectors, such as HES1, GATA3, RUNX3 and nuclear factor κB (NF-κB) (p65 and p50). This occurs through downregulation of the central transactivator Sp1 and its dissociation from the NOTCH1 promoter [56]. Similar effects were observed with withaferin A, which induces inhibition of protein translation mediated by eIF2A phosphorylation [57]. In this context, co-inhibition of NOTCH1, together with proteasome inhibition, could potentially engender a synthetic lethality interaction within NOTCH1-overactivated T-ALL cells. Further research is needed to thoroughly validate and elucidate the mechanistic intricacies of this potential interaction. Other targets predicted to exhibit synthetic lethality along with NOTCH1 inhibition include casein β (CSN2), keratin 31 (KRT31), matrix metallopeptidase 2 (MMP2), zinc finger protein 83 (ZNF83), aminolevulinate dehydratase (ALAD), cell division cycle 45 (CDC45), FRAT regulator of WNT signaling pathway 1 (FRAT1), methyltransferase-like 25 (METTL25), aspartylglucosaminidase (AGA), carboxypeptidase Z (CPZ), keratin-associated protein 15-1 (KRTAP15-1) and RAB40C [45,46]. It is imperative to note that these predictions must undergo rigorous experimental confirmation to thoroughly assess their applicability and safety.
Studies indicate that simultaneous action against receptor tyrosine kinase-like orphan receptor 1 (ROR1) and inhibition of B-cell receptor (BR) signaling is more effective in eliminating ROR1-positive B-ALL cells, suggesting a synthetic lethality interaction between the ROR1 and BR pathway [58]. Inhibition of ROR1 with KAN0441571C has also shown synergy with the BCL2 inhibitor venetoclax [59].
5. Conclusions and Future Directions
Synthetic lethality is gaining attention as a promising therapeutic avenue for ALL and other cancers. The concept revolves around exploiting specific vulnerabilities of cancer cells without affecting normal cells, thus offering a targeted therapeutic strategy. Furthermore, since these treatments are more personalized, they should have limited adverse effects and require lower doses of drugs to be effective. Despite this, its practical application faces significant challenges that require careful consideration. The intricate landscape of genetic interactions and the unique molecular profile of ALL present complexities in identifying robust synthetic lethal pairs. Moreover, translating these findings into effective and safe clinical interventions requires a thorough understanding of the dynamic interplay within the cellular machinery. For these reasons, unfortunately only one synthetic lethal interaction—that between PARP and BRCA1 and/or BRCA2—has successfully moved from discovery to clinical approval. Specifically in the ALL context, it is noteworthy that most synthetic lethality findings, so far, have been identified only in cell lines, with a limited number in murine models. For this reason, the ALL treatment landscape lacks ongoing clinical trials dedicated to exploring synthetic lethality strategies. In this sense, to change this trend, it is imperative to broaden the scope of research and address the associated challenges through interdisciplinary collaboration.
The WHO classification for ALL is primarily a genome-based classification of entities that has been updated based on extensive research conducted over the past decade. In the context of this manuscript, the meticulously identified distinct driver mutations deserve consideration for their potential to induce synthetic lethality when combined with inhibition of other genes. This inhibition can be achieved through the use of inhibitory drugs, by causing metabolic changes, or by genetic modifications employing techniques such as CRISPR-Cas9, among others. Overall, the translation of synthetic lethality into therapeutic strategies is aided by the synthesis of genetic interaction data from model organisms, tumor genomes and human cell lines. Advances in next-generation sequencing technologies are facilitating the identification of numerous ALL-specific mutations and alterations in gene expression, providing potential targets for a synthetic lethality approach. High-throughput analyses, encompassing genomic, transcriptomic, proteomic and metabolomic data, could play a crucial role in delineating synthetic lethality targets and unraveling the underlying mechanisms. Given the inherent complexity of this molecular network, machine learning and artificial intelligence emerge as valuable tools for discerning synthetic lethality targets in ALL patients. The design and development of small molecules capable of modulating the activity of key enzymes becomes more relevant, especially if their activity can develop synthetic lethality in conjunction with prevalent ALL mutations.
While current research predominantly emphasizes certain subtypes of ALL, it may be prudent to explore synthetic lethal interactions within each of the subtypes outlined in the 2016 World Health Organization (WHO) ALL classification. This is particularly relevant for those subtypes that are associated with poor clinical prognosis for patients.
Author Contributions
Conceptualization, F.A.L.-R.; investigation, F.A.L.-R. and V.C.-V.; resources, F.A.L.-R.; data curation, F.A.L.-R.; writing—original draft preparation, F.A.L.-R.; writing—review and editing, F.A.L.-R. and V.C.-V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.Á.; Cortes-Penagos, C. Acute Myeloid Leukemia-Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Stem Cell Res. 2017, 11, 328–339. [Google Scholar]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Liu, W.; Schiöth, H.B. Interaction between Environmental Pollutants and Cancer Drug Efficacy: Bisphenol A, Bisphenol A Diglycidyl Ether and Perfluorooctanoic Acid Reduce Vincristine Cytotoxicity in Acute Lymphoblastic Leukemia Cells. J. Appl. Toxicol. 2023, 43, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V. FLT3–ITD and Its Current Role in Acute Myeloid Leukaemia. Med. Oncol. 2017, 34, 114. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; de Smith, A.J.; Vergara-Lluri, M.; Muskens, I.S.; McKean-Cowdin, R.; Kogan, S.; Brynes, R.; Wiemels, J.L. Trends in Acute Lymphoblastic Leukemia Incidence in the United States by Race/Ethnicity from 2000 to 2016. Am. J. Epidemiol. 2021, 190, 519–527. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Kudłak, B.; Liu, W.; Williams, M.J.; Schiöth, H.B. The Potential Interaction of Environmental Pollutants and Circadian Rhythm Regulations that may Cause Leukemia. Crit. Rev. Environ. Sci. Technol. 2022, 52, 4094–4112. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved Survival for Children and Adolescents with Acute Lymphoblastic Leukemia Between 1990 and 2005: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef]
- Pui, C.-H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Manabe, A. Treatment and Biology of Pediatric Acute Lymphoblastic Leukemia. Pediatr. Int. 2018, 60, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Estlin, E.J.; Yule, S.M.; Lowis, S.P. Consolidation Therapy for Childhood Acute Lymphoblastic Leukaemia: Clinical and Cellular Pharmacology of Cytosine Arabinoside, Epipodophyllotoxins and Cyclophosphamide. Cancer Treat. Rev. 2001, 27, 339–350. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Jabbour, E.; Advani, A. Recent Advances in Managing Acute Lymphoblastic Leukemia. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Robison, L.L.; Look, A.T. Acute Lymphoblastic Leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of Acute Lymphoblastic Leukemia/Lymphoma. Virchows Arch. 2023, 482, 11–26. [Google Scholar] [CrossRef]
- Hiroki, H.; Akahane, K.; Inukai, T.; Morio, T.; Takagi, M. Synergistic Effect of Combined PI3 Kinase Inhibitor and PARP Inhibitor Treatment on BCR/ABL1-Positive Acute Lymphoblastic Leukemia Cells. Int. J. Hematol. 2023, 117, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brümmendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Targeting of BCR-ABL1 and IRE1α Induces Synthetic Lethality in Philadelphia-Positive Acute Lymphoblastic Leukemia. Carcinogenesis 2021, 42, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Liu-Dumlao, T.; Kantarjian, H.; Thomas, D.A.; O’Brien, S.; Ravandi, F. Philadelphia-Positive Acute Lymphoblastic Leukemia: Current Treatment Options. Curr. Oncol. Rep. 2012, 14, 387–394. [Google Scholar] [CrossRef]
- Geary, C.G. The Story of Chronic Myeloid Leukaemia. Br. J. Haematol. 2000, 110, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Vitale, A.; Guarini, A.; Chiaretti, S.; Foà, R. The Changing Scene of Adult Acute Lymphoblastic Leukemia. Curr. Opin. Oncol. 2006, 18, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.-Y.; Yang, D.-P.; Tang, C.; Fang, H.-S.; Sun, H.-Y.; Xiang, Y.-N.; Li, X.-M.; Yang, F.; Xia, R.-X.; Fan, F.; et al. Targeting DNA Polymerase β Elicits Synthetic Lethality with Mismatch Repair Deficiency in Acute Lymphoblastic Leukemia. Leukemia 2023, 37, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Pan, L.; Hong, C.; Chan, L.N.; Xiao, G.; Malvi, P.; Robinson, M.E.; Geng, H.; Reddy, S.T.; Lee, J.; Khairnar, V.; et al. PON2 Subverts Metabolic Gatekeeper Functions in B Cells to Promote Leukemogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2016553118. [Google Scholar] [CrossRef]
- Di Rorà, A.G.L.; Bocconcelli, M.; Ferrari, A.; Terragna, C.; Bruno, S.; Imbrogno, E.; Beeharry, N.; Robustelli, V.; Ghetti, M.; Napolitano, R.; et al. Synergism through WEE1 and CHK1 Inhibition in Acute Lymphoblastic Leukemia. Cancers 2019, 11, 1654. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fidalgo, J.A. Cell Proliferation Inhibitors and Apoptosis Promoters. Eur. J. Cancer Suppl. 2020, 15, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Takai, S.; Kamiya, T.; Inukai, T.; Sugita, K.; Ohyashiki, K.; Delia, D.; Masutani, M.; Mizutani, S.; Takagi, M. Poly (ADP-Ribose) Polymerase Inhibitors Selectively Induce Cytotoxicity in TCF3-HLF–Positive Leukemic Cells. Cancer Lett. 2017, 386, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and Drug Profiling of Fatal TCF3-HLF−positive Acute Lymphoblastic Leukemia Identifies Recurrent Mutation Patterns and Therapeutic Options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, C.P.; Guerri, L.; Lawir, D.-F.; Mateos, F.; Iconomou, M.; Iwanami, N.; Soza-Ried, C.; Sikora, K.; Siamishi, I.; Giorgetti, O.; et al. Genetic Landscape of T Cells Identifies Synthetic Lethality for T-ALL. Commun. Biol. 2021, 4, 1201. [Google Scholar] [CrossRef]
- León, T.E.; Rapoz-D’Silva, T.; Bertoli, C.; Rahman, S.; Magnussen, M.; Philip, B.; Farah, N.; Richardson, S.E.; Ahrabi, S.; Guerra-Assunção, J.A.; et al. EZH2 -Deficient T-Cell Acute Lymphoblastic Leukemia Is Sensitized to CHK1 Inhibition through Enhanced Replication Stress. Cancer Discov. 2020, 10, 998–1017. [Google Scholar] [CrossRef]
- Baran, N.; Lodi, A.; Dhungana, Y.; Herbrich, S.; Collins, M.; Sweeney, S.; Pandey, R.; Skwarska, A.; Patel, S.; Tremblay, M.; et al. Inhibition of Mitochondrial Complex I Reverses NOTCH1-Driven Metabolic Reprogramming in T-Cell Acute Lymphoblastic Leukemia. Nat. Commun. 2022, 13, 2801. [Google Scholar] [CrossRef]
- Parvin, S.; Ramirez-Labrada, A.; Aumann, S.; Lu, X.; Weich, N.; Santiago, G.; Cortizas, E.M.; Sharabi, E.; Zhang, Y.; Sanchez-Garcia, I.; et al. LMO2 Confers Synthetic Lethality to PARP Inhibition in DLBCL. Cancer Cell 2019, 36, 237–249.e6. [Google Scholar] [CrossRef]
- Hähnel, P.S.; Enders, B.; Sasca, D.; Roos, W.P.; Kaina, B.; Bullinger, L.; Theobald, M.; Kindler, T. Targeting Components of the Alternative NHEJ Pathway Sensitizes KRAS Mutant Leukemic Cells to Chemotherapy. Blood 2014, 123, 2355–2366. [Google Scholar] [CrossRef]
- Thielemans, N.; Demeyer, S.; Mentens, N.; Gielen, O.; Provost, S.; Cools, J. TAL1 Cooperates with PI3K/AKT Pathway Activation in T-Cell Acute Lymphoblastic Leukemia. Haematologica 2022, 107, 2304–2317. [Google Scholar] [CrossRef]
- Teachey, D.T.; Sheen, C.; Hall, J.; Ryan, T.; Brown, V.I.; Fish, J.; Reid, G.S.D.; Seif, A.E.; Norris, R.; Chang, Y.J.; et al. mTOR Inhibitors Are Synergistic with Methotrexate: An Effective Combination to Treat Acute Lymphoblastic Leukemia. Blood 2008, 112, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ryu, Y.-K.; Chen, T.Z.; Hall, C.P.; Webster, D.R.; Kang, M.H. Synergistic Activity of Rapamycin and Dexamethasone in Vitro and in Vivo in Acute Lymphoblastic Leukemia via Cell-Cycle Arrest and Apoptosis. Leuk. Res. 2012, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.A.; Klawitter, J.; Gregory, M.A.; Zaberezhnyy, V.; Baturin, D.; Pollyea, D.A.; Takebe, N.; Christians, U.; Gore, L.; DeGregori, J.; et al. Inhibition of Calcineurin Combined with Dasatinib Has Direct and Indirect Anti-Leukemia Effects against BCR-ABL1 + Leukemia. Am. J. Hematol. 2014, 89, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT Signaling Maintains Survival of Ph+ Leukemia Cells upon Inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.F.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying Chemogenetic Interactions from CRISPR Screens with DrugZ. Genome Med. 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Beckett, M.C.; Blair, H.J.; Tirtakusuma, R.; Nakjang, S.; Enshaei, A.; Halsey, C.; Vormoor, J.; Heidenreich, O.; Krippner-Heidenreich, A.; et al. Phase II-Like Murine Trial Identifies Synergy between Dexamethasone and Dasatinib in T-Cell Acute Lymphoblastic Leukemia. Haematologica 2020, 106, 1056–1066. [Google Scholar] [CrossRef]
- Laukkanen, S.; Veloso, A.; Yan, C.; Oksa, L.; Alpert, E.J.; Do, D.; Hyvärinen, N.; McCarthy, K.; Adhikari, A.; Yang, Q.; et al. Therapeutic Targeting of LCK Tyrosine Kinase and mTOR Signaling in T-Cell Acute Lymphoblastic Leukemia. Blood 2022, 140, 1891–1906. [Google Scholar] [CrossRef]
- Brown, P.; Levis, M.; McIntyre, E.; Griesemer, M.; Small, D. Combinations of the FLT3 Inhibitor CEP-701 and Chemotherapy Synergistically Kill Infant and Childhood MLL-Rearranged ALL Cells in a Sequence-Dependent Manner. Leukemia 2006, 20, 1368–1376. [Google Scholar] [CrossRef]
- Tamai, M.; Furuichi, Y.; Kasai, S.; Ando, N.; Harama, D.; Goi, K.; Inukai, T.; Kagami, K.; Abe, M.; Ichikawa, H.; et al. TGFβ1 Synergizes with FLT3 Ligand to Induce Chemoresistant Quiescence in Acute Lymphoblastic Leukemia with MLL Gene Rearrangements. Leuk. Res. 2017, 61, 68–76. [Google Scholar] [CrossRef]
- Kelvin, J.M.; Chimenti, M.L.; Zhang, D.Y.; Williams, E.K.; Moore, S.G.; Humber, G.M.; Baxter, T.A.; Birnbaum, L.A.; Qui, M.; Zecca, H.; et al. Development of Constitutively Synergistic Nanoformulations to Enhance Chemosensitivity in T-Cell Leukemia. J. Control Release 2023, 361, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP Proteins in Acute Leukemia: DNA Damage Response Inhibition and Therapeutic Strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; te Kronnie, G.; Meyer, L.-H.; Mohr, F.; et al. TET1 Promotes Growth of T-Cell Acute Lymphoblastic Leukemia and Can Be Antagonized via PARP Inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Wan, Z.; Kang, Y.H.; Sposto, R.; Reynolds, C.P. Mechanism of Synergy of N-(4-Hydroxyphenyl)Retinamide and ABT-737 in Acute Lymphoblastic Leukemia Cell Lines: Mcl-1 Inactivation. JNCI J. Natl. Cancer Inst. 2008, 100, 580–595. [Google Scholar] [CrossRef] [PubMed]
- Jayanthan, A.; Incoronato, A.; Singh, A.; Blackmore, C.; Bernoux, D.; Lewis, V.; Stam, R.; Whitlock, J.A.; Narendran, A. Cytotoxicity, Drug Combinability, and Biological Correlates of ABT-737 against Acute Lymphoblastic Leukemia Cells with MLL Rearrangement. Pediatr. Blood Cancer 2011, 56, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Koyama, D.; Kikuchi, J.; Hiraoka, N.; Wada, T.; Kurosawa, H.; Chiba, S.; Furukawa, Y. Proteasome Inhibitors Exert Cytotoxicity and Increase Chemosensitivity via Transcriptional Repression of Notch1 in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2014, 28, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, M.; Ambesi-Impiombato, A.; Qin, Y.; Herranz, D.; Bansal, M.; Girardi, T.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; De Keersmaecker, K.; et al. Synergistic Antileukemic Therapies in NOTCH1-Induced T-ALL. Proc. Natl. Acad. Sci. USA 2017, 114, 2006–2011. [Google Scholar] [CrossRef]
- Karvonen, H.; Niininen, W.; Murumägi, A.; Ungureanu, D. Targeting ROR1 Identifies New Treatment Strategies in Hematological Cancers. Biochem. Soc. Trans. 2017, 45, 457–464. [Google Scholar] [CrossRef]
- Wang, W.Z.; Shilo, K.; Amann, J.M.; Shulman, A.; Hojjat-Farsangi, M.; Mellstedt, H.; Schultz, J.; Croce, C.M.; Carbone, D.P. Predicting ROR1/BCL2 Combination Targeted Therapy of Small Cell Carcinoma of the Lung. Cell Death Dis. 2021, 12, 577. [Google Scholar] [CrossRef]
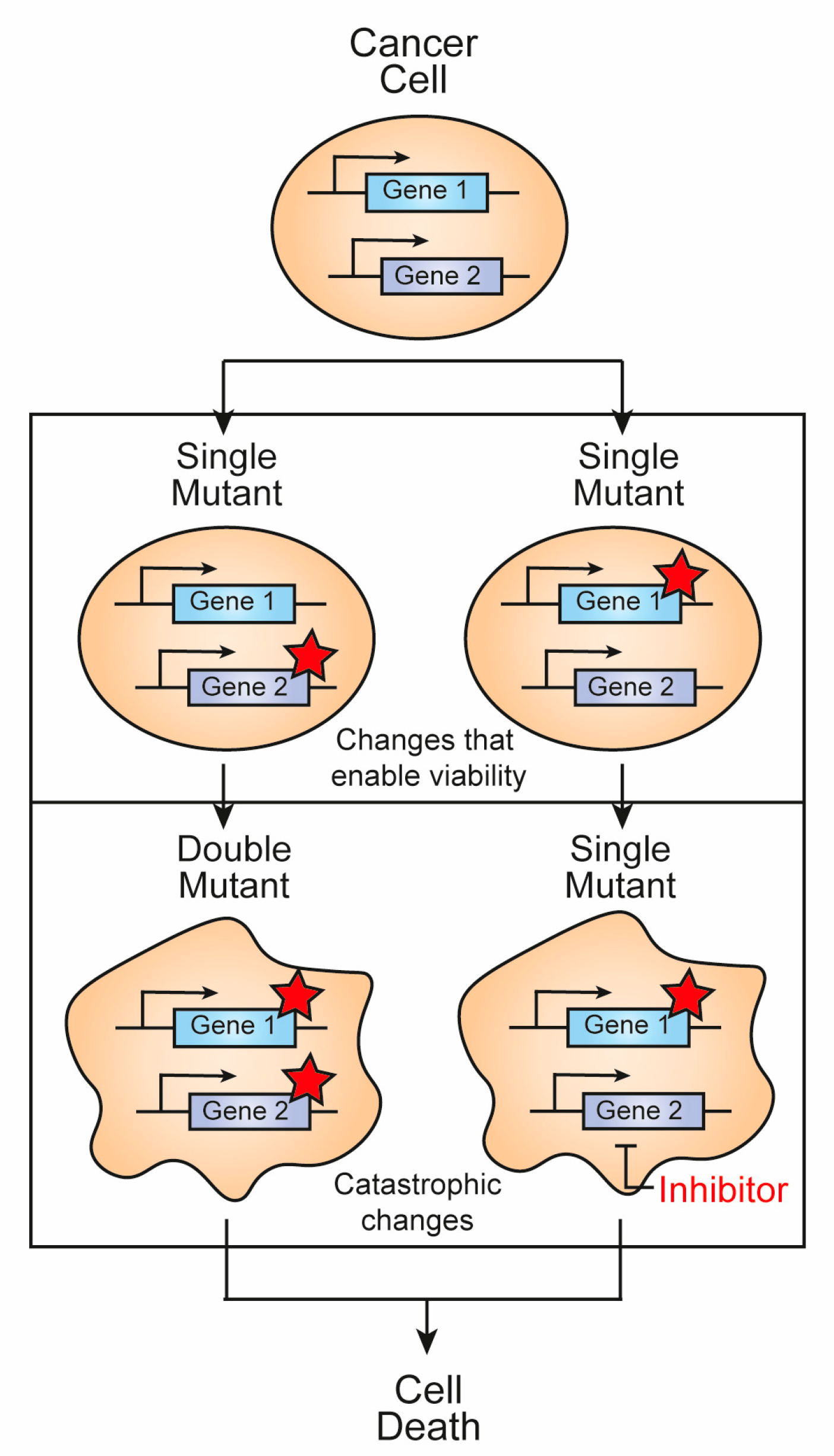
Figure 1.
Concept of synthetic lethality. Loss or inhibition of either of the protein products of the 1 or 2 genes alone is viable. However, mutation or pharmacological inhibition of the protein product of gene 2 in cells with a mutation of gene 1 results in synthetic lethality. Red stars indicate deleterious mutations.
Figure 1.
Concept of synthetic lethality. Loss or inhibition of either of the protein products of the 1 or 2 genes alone is viable. However, mutation or pharmacological inhibition of the protein product of gene 2 in cells with a mutation of gene 1 results in synthetic lethality. Red stars indicate deleterious mutations.


{kind=link}
Table 1.
The International Consensus Classification of ALL (WHO).
| B-Acute Lymphoblastic Leukemia (B-ALL) |
|---|
| B-ALL with recurrent genetic abnormalities B-ALL with t(9;22)(q34.1;q11.2)/BCR::ABL1 with lymphoid only involvement with multilineage involvement B-ALL with t(v;11q23.3)/KMT2A-rearranged B-ALL with t(12;21)(p13.2;q22.1)/ETV6::RUNX1 B-ALL hyperdiploid B-ALL low hypodiploid B-ALL near haploid B-ALL with t(5;14)(q31.1;q32.3)/IL3::IGH B-ALL with t(1;19)(q23.3;p13.3)/TCF3::PBX1 B-ALL BCR::ABL1-like, ABL-1 class-rearranged B-ALL BCR::ABL1-like, JAK-STAT-activated B-ALL BCR::ABL1-like, NOS B-ALL with iAMP21 B-ALL with MYC rearrangement B-ALL with DUX4 rearrangement B-ALL with MEF2D rearrangement B-ALL with ZNF384 rearrangement B-ALL with NUTM1 rearrangement B-ALL with HLF rearrangement B-ALL with UBTF::ATXN7L3/PAN3,CDX2 (“CDX2/UBTF”) B-ALL with IKZF1 N159Y B-ALL with PAX5 P80R Provisional entities B-ALL ETV6::RUNX1-like B-ALL with PAX5 alteration B-ALL with mutated ZEB2 (p.H1038R)/IGH::CEBPE B-ALL ZNF384 rearranged-like B-ALL KMT2A rearranged-like T-acute lymphoblastic leukemia (T-ALL) Early T-cell precursor ALL, BCL11B-activated Early T-cell precursor ALL, NOS T-ALL, NOS Provisional entities T-ALL with BCL11B-activated T-ALL with TAL1/2-R T-ALL with TLX1-R T-ALL with TLX3-R T-ALL with HOXA T-ALL with LMO1/2-R T-ALL with NKX2-R T-ALL with SPI1-R T-ALL with BHLH, other Natural killer (NK) cell ALL |
NOS: No other specification.
Table 2.
Identified cases of synthetic lethality in ALL cell lines.
| ALL Subtype | Genes Involved | Observations | Reference |
|---|---|---|---|
| Ph+ ALL | PI3K PARP genes | Disrupts HR and BER repair pathways, promoting genomic instability and triggering apoptosis. | [21] |
| Ph+ ALL | POLB MMR genes | Impacts BER repair, increasing sensitivity to thiopurines. | [26] |
| Ph+ ALL | BCR-ABL1 IRE1α RNase | Negatively influences UPR and promotes apoptosis inducers and negative cell cycle regulators. | [22] |
| B-ALL | PON2 GLUT1 | PON2 hydrolyzes 3OC12-HSL, leading to intracellular acidification, and triggers caspase-3-mediated apoptosis. | [30] |
| B-ALL | WEE1 CHK1/2 | Triggers S-phase checkpoint activation and induces DNA damage that increases replicative stress and causes replication fork collapse. | [31] |
| TCF3-HLF B-ALL | TCF3-HLF PARP genes | TCF3-HLF protein increases PARP activity and defective HR repair | [33] |
| EZH2-deficient T-ALL | EZH2 CHK1 | EZH2 deficiency induces MYCN-driven replication stress, increasing CHK1 dependence for replication fork integrity. | [36] |
| NOTCH1-mutated T-ALL | NOTCH1 ETC genes | Metabolic and transcriptomic catastrophe occurs due to increased reliance on glutamine-driven oxidative phosphorylation. | [37] |
| T-ALL | LMO2 PARP1 | LMO2 inhibits the recruitment of BRCA1 to DSBs | [38] |
| KRAS-mutated T-ALL | KRAS-mutated PARP1 | KRAS mutated variant potentiates alt-NHEJ, increasing sensitivity to DSBs. | [39] |
| TAL1-AKTE17K-positive T-ALL | TAL1-AKTE17K PARP genes | TAL1-AKTE17K-positive T-ALL has an increased dependence on DNA repair genes. | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lagunas-Rangel, F.A.; Chávez-Valencia, V. Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia. Hemato 2024, 5, 6-18. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato5010002
AMA Style
Lagunas-Rangel FA, Chávez-Valencia V. Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia. Hemato. 2024; 5(1):6-18. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato5010002
Chicago/Turabian StyleLagunas-Rangel, Francisco Alejandro, and Venice Chávez-Valencia. 2024. "Synthetic Lethality Approaches in Acute Lymphoblastic Leukemia" Hemato 5, no. 1: 6-18. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato5010002