
Composites of (C4F)n and (CF)n Synthesized by Uncatalyzed Fluorination of Graphite

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
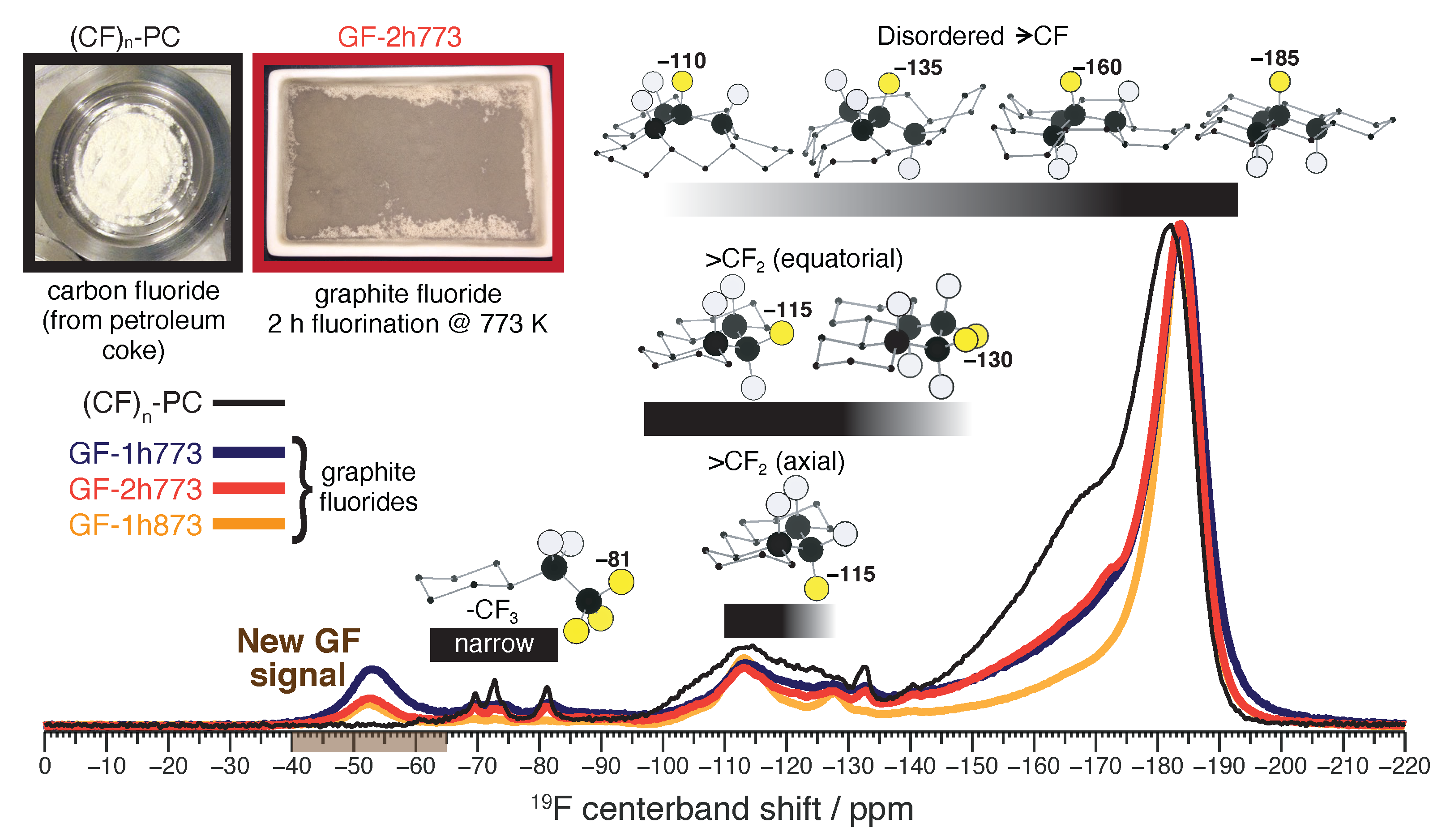
2.1. Fluorine-19 MAS NMR: A New Spectral Signature from Graphite Fluorides
2.2. Carbon-13 CP MAS NMR: Evidence for a Fluorine-Deficient Graphite Fluoride Composition
2.3. Two-Dimensional {19F}13C CP HETCOR CPMG NMR: Indication of Covalent C–F Bonds
2.4. Additional 19F NMR Characterization: Evidence for Distinct yet Well-Mixed Graphite Fluoride Phases
2.5. Stoichiometric Analysis of the New Graphite Fluoride Domain by Quantitative NMR
2.6. Calculation of NMR Chemical Shifts: Support for Layers with Single-Sided Fluorination
- The structure contains only “isolated” groups, i.e., whose three framework bonds are all to bare, sp hybridized framework carbon.
- All valence electrons are paired.
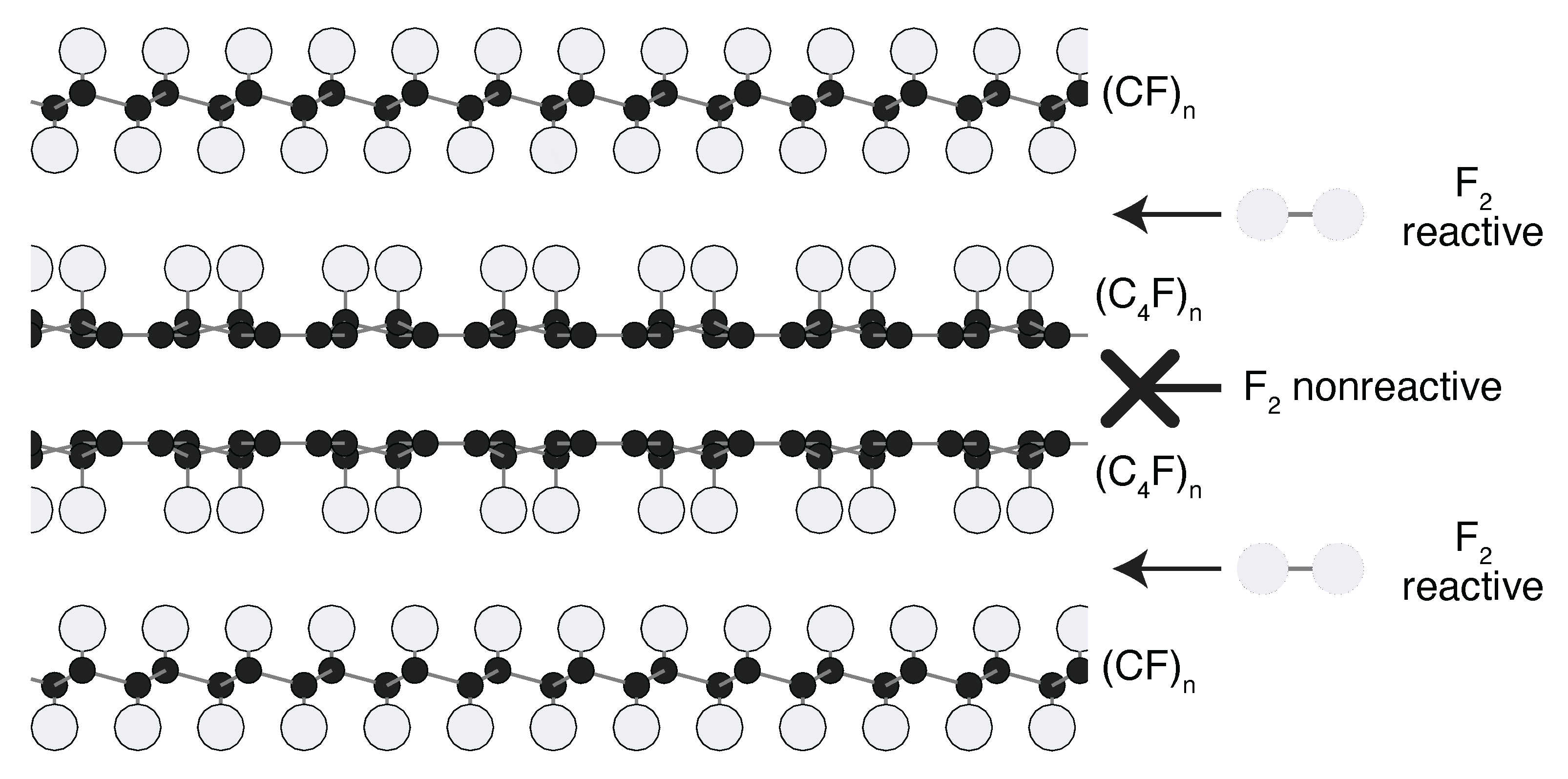
2.7. Structural Model: (CF)/(CF) Graphite Fluoride Composites
2.8. Electrochemical Properties of Cathodes Made from (C4F)/(CF) Graphite Fluoride Composites
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. Solid-State Nuclear Magnetic Resonance
4.3. Calculation of NMR Chemical Shifts
4.4. Electrochemistry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GF | graphite fluoride |
| F-GIC | fluorine graphite intercalation compound |
| HOPG | highly oriented pyrolytic graphite |
| MAS | magic-angle spinning |
| NMR | nuclear magnetic resonance |
| DFT | density functional theory |
| PC | petroleum coke |
| XRD | X-ray diffraction |
| CPMG | Carr-Purcell-Meiboom-Gill |
| CP | cross-polarization |
| HETCOR | heteronuclear correlation |
| EXSY | exchange spectroscopy |
| MRE | matched reconstructed echo |
| FID | free induction decay |
| S/N | signal-to-noise ratio |
| WISE | wideline separation |
| AFM | atomic force microscopy |
| SCCM | standard cubic centimeters per minute |
| TOP-aMAT | two-dimensional one-pulse adiabatic magic angle turning |
| SPINAL | small phase incremental alternation |
| PVDF | poly(vinylidene fluoride) |
References
- Ruff, O.; Bretschneider, O. Die Reaktionsprodukte der verschiedenen Kohlenstoffformen mit Fluor II (Kohlenstoff-monofluorid). Z. Anorg. Allg. Chem. 1934, 217, 1–18. [Google Scholar] [CrossRef]
- Lagow, R.J.; Badachhape, R.B.; Wood, J.L.; Margrave, J.L. Synthesis of superstoichiometric poly(carbon monofluoride). J. Am. Chem. Soc. 1974, 96, 2628–2629. [Google Scholar] [CrossRef]
- Kita, Y.; Watanabe, N.; Fujii, Y. Chemical composition and crystal structure of graphite fluoride. J. Am. Chem. Soc. 1979, 101, 3832–3841. [Google Scholar] [CrossRef]
- Touhara, H.; Kadono, K.; Fujii, Y.; Watanabe, N. On the Structure of Graphite Fluoride. Z. Anorg. Allg. Chem. 1987, 544, 7–20. [Google Scholar] [CrossRef]
- Rüdorff, W.; Rüdorff, G. Tetrakohlenstoffmonofluorid, eine neue Graphit-Fluor-Verbindung. Chem. Berichte 1947, 80, 417–423. [Google Scholar] [CrossRef]
- Nakajima, T.; Kawaguchi, M.; Watanabe, N. Ternary intercalation compound of graphite with aluminum fluoride and fluorine. Chem. Lett. 1981, 10, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Palchan, I.; Davidov, D.; Selig, H. Preparation and properties of new graphite–fluorine intercalation compounds. J. Chem. Soc. Chem. Commun. 1983, 657–658. [Google Scholar] [CrossRef]
- Hamwi, A.; Daoud, M.; Cousseins, J. Graphite Fluorides Prepared at Room-Temperature 1. Synthesis and Characterization. Synth. Met. 1988, 26, 89–98, WOS:A1988Q485700010. [Google Scholar] [CrossRef]
- Vaknin, D.; Palchan, I.; Davidov, D.; Selig, H.; Moses, D. Resistivity and E.S.R. studies of graphite HOPG/fluorine intercalation compounds. Synth. Metals 1986, 16, 349–365. [Google Scholar] [CrossRef]
- Sato, Y.; Shiraishi, S.; Mazej, Z.; Hagiwara, R.; Ito, Y. Direct conversion mechanism of fluorine–GIC into poly(carbon monofluoride), (CF)n. Carbon 2003, 41, 1971–1977. [Google Scholar] [CrossRef]
- Dubois, M.; Guérin, K.; Pinheiro, J.P.; Fawal, Z.; Masin, F.; Hamwi, A. NMR and EPR studies of room temperature highly fluorinated graphite heat-treated under fluorine atmosphere. Carbon 2004, 42, 1931–1940. [Google Scholar] [CrossRef]
- Rüdorff, W.; Rüdorff, G. Zur Konstitution des Kohlenstoff-Monofluorids. Z. Anorg. Chem 1947, 253, 281–296. [Google Scholar] [CrossRef]
- Mallouk, T.; Bartlett, N. Reversible intercalation of graphite by fluorine: A new bifluoride, C12HF2, and graphite fluorides, CxF (5 > x > 2). J. Chem. Soc. Chem. Commun. 1983, 103–105. [Google Scholar] [CrossRef]
- Mallouk, T.; Hawkins, B.L.; Conrad, M.P.; Zilm, K.; Maciel, G.E.; Bartlett, N. Raman, Infrared and n.m.r. Studies of the Graphite Hydrofluorides CxF1-δ(HF)δ (2 ≤ x ≤ 5). Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Sci. 1985, 314, 179–187. [Google Scholar]
- Nakajima, T.; Ino, T.; Watanabe, N.; Takenaka, H. Preparation, structure, and electrical conductivity of fluorine-graphite intercalation compound. Carbon 1988, 26, 397–401. [Google Scholar] [CrossRef]
- Nakajima, T.; Molinier, M.; Motoyama, M. Structure of fluorine-graphite intercalation compounds. Carbon 1991, 29, 429–437. [Google Scholar] [CrossRef]
- Nakajima, T.; Tamura, T. The c-axis structures of stage 1 and 2 fluorine-graphite intercalation compounds prepared from highly oriented pyrolytic grahite and natural graphite flake. Synth. Met. 1995, 73, 63–67. [Google Scholar] [CrossRef]
- Oshida, K.; Endo, M.; Nakajima, T.; Vittorio, S.L.d.; Dresselhaus, M.S.; Dresselhaus, G. Image analysis of TEM pictures of fluorine-intercalated graphite fibers. J. Mater. Res. 1993, 8, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Di Vittorio, S.L.; Dresselhaus, M.S.; Dresselhaus, G. A model for disorder in fluorine-intercalated graphite. J. Mater. Res. 1993, 8, 1578–1585. [Google Scholar] [CrossRef] [Green Version]
- Panich, A.M. Nuclear magnetic resonance study of fluorine-graphite intercalation compounds and graphite fluorides. Synth. Met. 1999, 100, 169–185, WOS:000080386100001. [Google Scholar] [CrossRef]
- Mitkin, V.N. Types of Inorganic Fluorocarbon Polymer Materials and Structure–Property Correlation Problems. J. Struct. Chem. 2003, 44, 82–115. [Google Scholar] [CrossRef]
- Sato, Y.; Itoh, K.; Hagiwara, R.; Fukunaga, T.; Ito, Y. On the so-called “semi-ionic” C-F bond character in fluorine-GIC. Carbon 2004, 42, 3243–3249. [Google Scholar] [CrossRef]
- Bettinger, H.F.; Kudin, K.N.; Scuseria, G.E. Structural Models of Fluorine-Graphite Intercalation Compounds from Density Functional Theory. J. Phys. Chem. A 2004, 108, 3016–3018. [Google Scholar] [CrossRef]
- Hamwi, A. Fluorine reactivity with graphite and fullerenes. fluoride derivatives and some practical electrochemical applications. J. Phys. Chem. Solids 1996, 57, 677–688. [Google Scholar] [CrossRef]
- Watanabe, N. Two types of graphite fluorides, (CF)n and (C2F)n, and discharge characteristics and mechanisms of electrodes of (CF)n and (C2F)n in lithium batteries. Solid State Ionics 1980, 1, 87–110. [Google Scholar] [CrossRef]
- Walder, B.J.; Alam, T.M. Modes of Disorder in Poly(carbon monofluoride). J. Am. Chem. Soc. 2021, 143, 11714–11733. [Google Scholar] [CrossRef]
- Leung, K.; Schorr, N.B.; Mayer, M.; Lambert, T.N.; Meng, Y.S.; Harrison, K.L. Edge-Propagation Discharge Mechanism in CFx Batteries—A First-Principles and Experimental Study. Chem. Mater. 2021, 33, 1760–1770. [Google Scholar] [CrossRef]
- Sayahpour, B.; Hirsh, H.; Bai, S.; Schorr, N.B.; Lambert, T.N.; Mayer, M.; Bao, W.; Cheng, D.; Zhang, M.; Leung, K.; et al. Revisiting Discharge Mechanism of CFx as a High Energy Density Cathode Material for Lithium Primary Battery. Adv. Energy Mater. 2022, 12, 2103196. [Google Scholar] [CrossRef]
- Krawietz, T.R.; Haw, J.F. Characterization of poly(carbon monofluoride) by 19F and 19F to 13C cross polarization MAS NMR spectroscopy. Chem. Commun. 1998, 2151–2152. [Google Scholar] [CrossRef]
- Panich, A.M.; Nakajima, T.; Vieth, H.M.; Privalov, A.F.; Goren, S.D. Nuclear magnetic resonance study of fluorine-graphite intercalation compounds. J. Phys. Condens. Matter 1998, 10, 7633–7642. [Google Scholar] [CrossRef]
- Dubois, M.; Guérin, K.; Hamwi, A.; Vinogradov, A. Nature of C–F Bonds in Fluorinated Carbons. In New Fluorinated Carbons: Fundamentals and Applications; Progress in Fluorine Science; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2, Chapter 9; pp. 215–243. [Google Scholar] [CrossRef]
- Takashima, M.; Watanabe, N. Formation and Structure of Crystalline Graphite Fluoride. Nippon Kagaku Kaishi 1975, 432–436. [Google Scholar] [CrossRef]
- Hagaman, E.W.; Murray, D.K.; Del Cul, G.D. Solid State 13C and 19F NMR Characterization of Fluorinated Charcoal. Energy Fuels 1998, 12, 399–408. [Google Scholar] [CrossRef]
- Dubois, M.; Giraudet, J.; Guérin, K.; Hamwi, A.; Fawal, Z.; Pirotte, P.; Masin, F. EPR and Solid-State NMR Studies of Poly(dicarbon monofluoride) (C2F)n. J. Phys. Chem. B 2006, 110, 11800–11808. [Google Scholar] [CrossRef] [PubMed]
- Chamssedine, F.; Dubois, M.; Guérin, K.; Giraudet, J.; Masin, F.; Ivanov, D.A.; Vidal, L.; Yazami, R.; Hamwi, A. Reactivity of Carbon Nanofibers with Fluorine Gas. Chem. Mater. 2007, 19, 161–172. [Google Scholar] [CrossRef]
- Giraudet, J.; Dubois, M.; Guérin, K.; Delabarre, C.; Hamwi, A.; Masin, F. Solid-State NMR Study of the Post-Fluorination of (C2.5F)n Fluorine–GIC. J. Phys. Chem. B 2007, 111, 14143–14151. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, B.J.; Boender, G.J.; de Groot, H.J.M. High Magnetic Field for Enhanced Proton Resolution in High-Speed CP/MAS Heteronuclear 1H–13C Dipolar-Correlation Spectroscopy. J. Magn. Reson. Ser. A 1996, 120, 274–277. [Google Scholar] [CrossRef]
- Giraudet, J.; Dubois, M.; Guérin, K.; Delabarre, C.; Pirotte, P.; Hamwi, A.; Masin, F. Heteronuclear dipolar recoupling using Hartmann–Hahn cross polarization: A probe for 19F–13C distance determination of fluorinated carbon materials. Solid State Nucl. Magn. Reson. 2007, 31, 131–140. [Google Scholar] [CrossRef]
- Ahmad, Y.; Dubois, M.; Guérin, K.; Hamwi, A.; Fawal, Z.; Kharitonov, A.P.; Generalov, A.V.; Klyushin, A.Y.; Simonov, K.A.; Vinogradov, N.A.; et al. NMR and NEXAFS Study of Various Graphite Fluorides. J. Phys. Chem. C 2013, 117, 13564–13572. [Google Scholar] [CrossRef]
- Vyalikh, A.; Bulusheva, L.G.; Chekhova, G.N.; Pinakov, D.V.; Okotrub, A.V.; Scheler, U. Fluorine Patterning in Room-Temperature Fluorinated Graphite Determined by Solid-State NMR and DFT. J. Phys. Chem. C 2013, 117, 7940–7948. [Google Scholar] [CrossRef]
- Murakami, M.; Matsumoto, K.; Hagiwara, R.; Matsuo, Y. 13C/19F high-resolution solid-state NMR studies on layered carbon-fluorine compounds. Carbon 2018, 138, 179–187. [Google Scholar] [CrossRef]
- Chen, X.; Dubois, M.; Radescu, S.; Rawal, A.; Zhao, C. Liquid-phase exfoliation of F-diamane-like nanosheets. Carbon 2021, 175, 124–130. [Google Scholar] [CrossRef]
- Wardell, G.E.; McBrierty, V.J.; Douglass, D.C. Application of NMR to the study of molecular motion in SBS copolymers. J. Appl. Phys. 1974, 45, 3441–3446. [Google Scholar] [CrossRef]
- Walder, B.J.; Alam, T.M. Quantification of Uncoupled Spin Domains in Spin-Abundant Disordered Solids. Int. J. Mol. Sci. 2020, 21, 3938. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.A. EDF Statistics for Goodness of Fit and Some Comparisons. J. Am. Stat. Assoc. 1974, 69, 730–737. [Google Scholar] [CrossRef]
- Marsaglia, G.; Marsaglia, J. Evaluating the Anderson-Darling Distribution. J. Stat. Softw. 2004, 9, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rimsza, J.M.; Walder, B.J.; Alam, T.M. Influence of Polymorphs and Local Defect Structures on NMR Parameters of Graphite Fluorides. J. Phys. Chem. C 2021, 125, 2699–2712. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Ivanovskii, A.L. Layers and tubes of fluorographene C4F: Stability, structural and electronic properties from DFTB calculations. Chem. Phys. Lett. 2013, 576, 44–48. [Google Scholar] [CrossRef]
- Bulusheva, L.G.; Okotrub, A.V.; Yudanov, N.F. Atomic arrangement and electronic structure of graphite fluoride C2F. Phys. Low-Dimens. Struct. 2002, 7–8, 1–14. [Google Scholar]
- Pelikán, P.; Noga, J.; Biskupič, S. On the structural and electronic properties of poly(dicarbon monofluoride): Solid-state semi-empirical INDO study. J. Solid State Chem. 2003, 174, 233–240. [Google Scholar] [CrossRef]
- Ewels, C.P.; Van Lier, G.; Charlier, J.C.; Heggie, M.I.; Briddon, P.R. Pattern Formation on Carbon Nanotube Surfaces. Phys. Rev. Lett. 2006, 96, 216103. [Google Scholar] [CrossRef] [Green Version]
- Ohana, I.; Palchan, I.; Yacoby, Y.; Davidov, D.; Selig, H. Electronic charge transfer in stage-2 fluorine-intercalated graphite compounds. Phys. Rev. B 1988, 38, 12627–12632. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, A.K.; Margrave, J.L. Kinetics of the Reactions of Elemental Fluorine. IV. Fluorination of Graphite. J. Phys. Chem. 1965, 69, 2772–2775. [Google Scholar] [CrossRef]
- Lam, P.; Yazami, R. Physical characteristics and rate performance of (CFx)n (0.33 < x < 0.66) in lithium batteries. J. Power Sources 2006, 153, 354–359. [Google Scholar] [CrossRef]
- Giraudet, J.; Delabarre, C.; Guérin, K.; Dubois, M.; Masin, F.; Hamwi, A. Comparative performances for primary lithium batteries of some covalent and semi-covalent graphite fluorides. J. Power Sources 2006, 158, 1365–1372. [Google Scholar] [CrossRef]
- Yazami, R.; Hamwi, A.; Guérin, K.; Ozawa, Y.; Dubois, M.; Giraudet, J.; Masin, F. Fluorinated carbon nanofibres for high energy and high power densities primary lithium batteries. Electrochem. Commun. 2007, 9, 1850–1855. [Google Scholar] [CrossRef]
- Şahin, H.; Topsakal, M.; Ciraci, S. Structures of fluorinated graphene and their signatures. Phys. Rev. B 2011, 83, 115432. [Google Scholar] [CrossRef] [Green Version]
- Han, S.S.; Yu, T.H.; Merinov, B.V.; van Duin, A.C.T.; Yazami, R.; Goddard, W.A. Unraveling Structural Models of Graphite Fluorides by Density Functional Theory Calculations. Chem. Mater. 2010, 22, 2142–2154. [Google Scholar] [CrossRef] [Green Version]
- Asanov, I.P.; Bulusheva, L.G.; Dubois, M.; Yudanov, N.F.; Alexeev, A.V.; Makarova, T.L.; Okotrub, A.V. Graphene nanochains and nanoislands in the layers of room-temperature fluorinated graphite. Carbon 2013, 59, 518–529. [Google Scholar] [CrossRef]
- Clément, R.J.; Pell, A.J.; Middlemiss, D.S.; Strobridge, F.C.; Miller, J.K.; Whittingham, M.S.; Emsley, L.; Grey, C.P.; Pintacuda, G. Spin-Transfer Pathways in Paramagnetic Lithium Transition-Metal Phosphates from Combined Broadband Isotropic Solid-State MAS NMR Spectroscopy and DFT Calculations. J. Am. Chem. Soc. 2012, 134, 17178–17185. [Google Scholar] [CrossRef]
- Avalos, C.E.; Walder, B.J.; Viger-Gravel, J.; Magrez, A.; Emsley, L. Chemical exchange at the ferroelectric phase transition of lead germanate revealed by solid state 207Pb nuclear magnetic resonance. Phys. Chem. Chem. Phys. 2019, 21, 1100–1109. [Google Scholar] [CrossRef]
- An, K.H.; Heo, J.G.; Jeon, K.G.; Bae, D.J.; Jo, C.; Yang, C.W.; Park, C.Y.; Lee, Y.H.; Lee, Y.S.; Chung, Y.S. X-ray photoemission spectroscopy study of fluorinated single-walled carbon nanotubes. Appl. Phys. Lett. 2002, 80, 4235–4237. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Cho, T.H.; Lee, B.K.; Rho, J.S.; An, K.H.; Lee, Y.H. Surface properties of fluorinated single-walled carbon nanotubes. J. Fluor. Chem. 2003, 120, 99–104. [Google Scholar] [CrossRef]
- Robinson, J.T.; Burgess, J.S.; Junkermeier, C.E.; Badescu, S.C.; Reinecke, T.L.; Perkins, F.K.; Zalalutdniov, M.K.; Baldwin, J.W.; Culbertson, J.C.; Sheehan, P.E.; et al. Properties of Fluorinated Graphene Films. Nano Lett. 2010, 10, 3001–3005. [Google Scholar] [CrossRef]
- Bulusheva, L.; Okotrub, A. Electronic Structure of Fluorinated Graphene. In New Fluorinated Carbons: Fundamentals and Applications; Progress in Fluorine Science; Elsevier: Amsterdam, Netherlands, 2017; Volume 2, Chapter 8; pp. 177–213. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, S.; Wang, C.; Hui, Q. Fluorine adsorption on the graphene films: From metal to insulator. Comput. Mater. Sci. 2015, 97, 14–19. [Google Scholar] [CrossRef]
- Zhou, S.; Sherpa, S.D.; Hess, D.W.; Bongiorno, A. Chemical Bonding of Partially Fluorinated Graphene. J. Phys. Chem. C 2014, 118, 26402–26408. [Google Scholar] [CrossRef]
- Pischedda, V.; Radescu, S.; Dubois, M.; Cavallari, C.; Batisse, N.; Balima, F. Fluorine-graphite intercalation compound (C4F)n at high pressure: Experimental and theoretical study. Carbon 2018, 127, 384–391. [Google Scholar] [CrossRef]
- Cavallari, C.; Brunelli, M.; Radescu, S.; Dubois, M.; Batisse, N.; Vaughan, G.B.M.; Fischer, H.E.; Pischedda, V. Structural and electronic changes in graphite fluorides as a function of fluorination rate: An XRS, PDF and DFT study. Carbon 2019, 147, 1–8. [Google Scholar] [CrossRef]
- Cavallari, C.; Radescu, S.; Dubois, M.; Batisse, N.; Diaf, H.; Pischedda, V. Tuning C–F Bonding of Graphite Fluoride by Applying High Pressure: Experimental and Theoretical Study. J. Phys. Chem. C 2020, 124, 24747–24755. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Larsen, F.H.; Farnan, I. 29Si and 17O (Q)CPMG-MAS solid-state NMR experiments as an optimum approach for half-integer nuclei having long T1 relaxation times. Chem. Phys. Lett. 2002, 357, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D.J.; Vosegaard, T.; Massiot, D.; Grandinetti, P.J. Core Scientific Dataset Model: A lightweight and portable model and file format for multi-dimensional scientific data. PLoS ONE 2020, 15, e0225953. [Google Scholar] [CrossRef] [PubMed]
- PhySy Ltd. RMN 2.0.1. 2021. Available online: https://www.physyapps.com/rmn (accessed on 1 March 2022).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | f/mol% | /mol% | /mol% | /mol% |
|---|---|---|---|---|
| GF-1h773 | ||||
| GF-2h773 | ||||
| GF-1h873 |
| Sample | g/mol% | /mol% | /mol% | /mol% | /mol% |
|---|---|---|---|---|---|
| GF-1h773 | |||||
| GF-2h773 | |||||
| GF-1h873 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walder, B.J.; Schorr, N.B.; Brunke, L.B.; Siegal, M.P.; Alam, T.M.; Fritzsching, K.J.; Lambert, T.N. Composites of (C4F)n and (CF)n Synthesized by Uncatalyzed Fluorination of Graphite. Solids 2022, 3, 237-257. https://0-doi-org.brum.beds.ac.uk/10.3390/solids3020017
Walder BJ, Schorr NB, Brunke LB, Siegal MP, Alam TM, Fritzsching KJ, Lambert TN. Composites of (C4F)n and (CF)n Synthesized by Uncatalyzed Fluorination of Graphite. Solids. 2022; 3(2):237-257. https://0-doi-org.brum.beds.ac.uk/10.3390/solids3020017
Chicago/Turabian StyleWalder, Brennan J., Noah B. Schorr, Lyle B. Brunke, Michael P. Siegal, Todd M. Alam, Keith J. Fritzsching, and Timothy N. Lambert. 2022. "Composites of (C4F)n and (CF)n Synthesized by Uncatalyzed Fluorination of Graphite" Solids 3, no. 2: 237-257. https://0-doi-org.brum.beds.ac.uk/10.3390/solids3020017