
Advances in Escherichia coli-Based Therapeutic Protein Expression: Mammalian Conversion, Continuous Manufacturing, and Cell-Free Production


Abstract
:
1. Introduction
2. Background
3. Advantages
- It is the most well-understood expression system. The genome of Escherichia coli strain K-12 MG1655, which is the most studied and best-characterized strain, has been fully sequenced and annotated. It was first completely sequenced in 1997, and the annotation and analysis have been continually updated since then as our understanding of genomics and the biology of E. coli has advanced [9]. This knowledge base is critical in its utilization as a robust expression system.
- Numerous prokaryotic genes [10] are expressed in operons [11], where a solitary promoter leads to the synthesis of multiple proteins from a single mRNA molecule, which has a ribosome binding site (RBS) preceding the beginning AUG codon of each protein. This enables the simultaneous production [12] of subunits that assemble into complexes or the simultaneous expression of auxiliary components that may be necessary for the protein to attain its native shape.
- Simpler scale-up compared to eukaryotic systems, including mechanical cell disruption, which is less variable than eukaryotic cells, requires gentler lysis to preserve more fragile organelles and structures [16].
- Avoiding virus contamination risk. Proteins synthesized in mammalian cell lines, the host cells possess multiple copies of endogenous retrovirus-like sequences, which subsequently generate retrovirus-like particles (RVLPs) together with the target protein. While RVLPs are commonly regarded as dysfunctional, certain instances have demonstrated their ability to infect cell lines that are not of rodent origin. Exogenous viral contamination resulting from raw materials or persons is also possible; however, such concerns are not relevant in the context of E. coli-based expression systems [17].
- Low-cost growth medium, fast cellular proliferation, uncomplicated fermentation procedures, no viral contaminants in the final product, and high product yields [18].
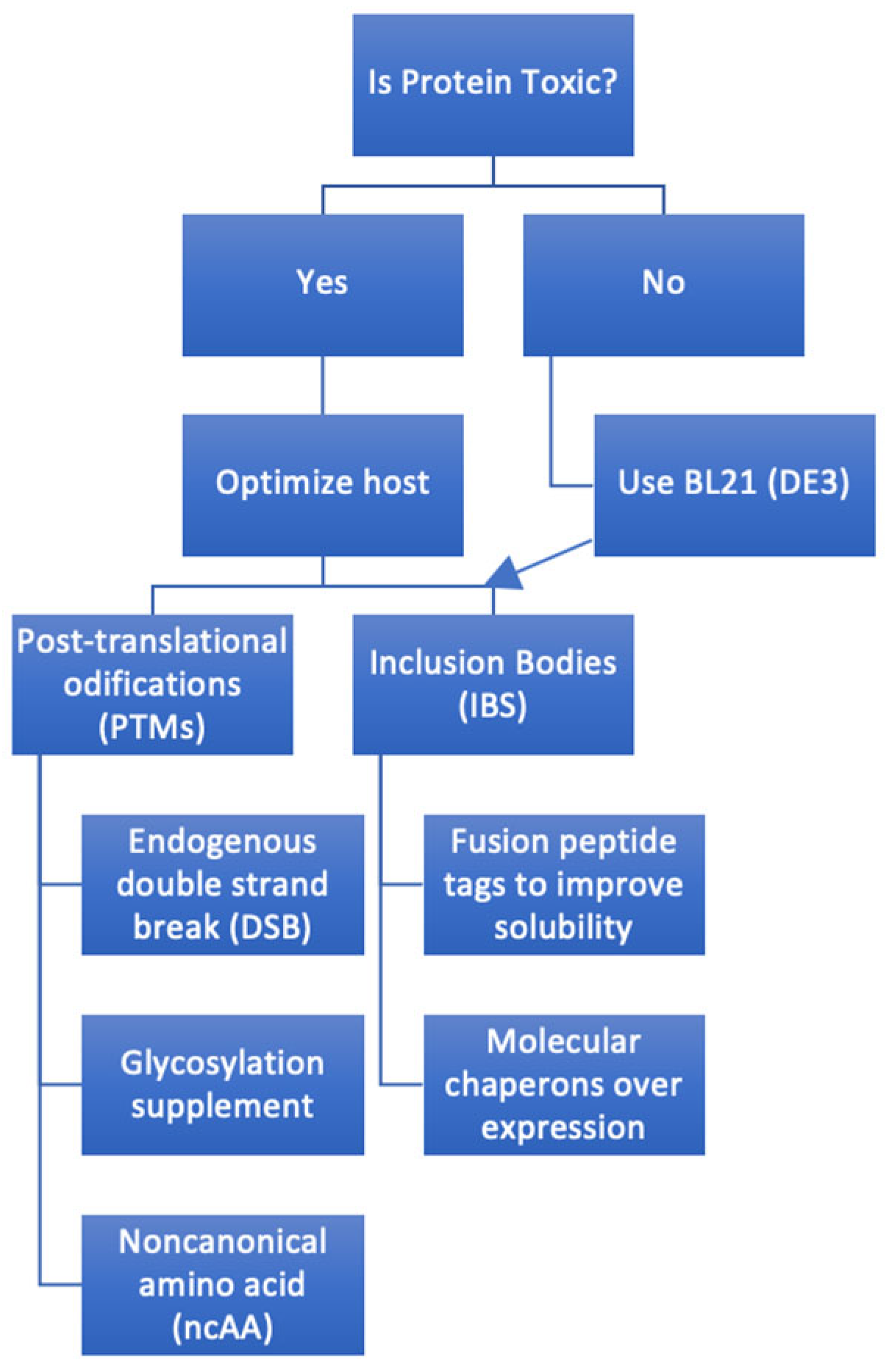
4. Challenges
- Proteins overexpressed in E. coli may form insoluble aggregates known as inclusion bodies, requiring specific solubilization and refolding steps, adding complexity to the purification process compared to eukaryotic cells [19]; additional purification steps if inclusion bodies are formed. A frequently encountered challenge is the formation of inclusion bodies—insoluble aggregates of misfolded proteins. Several tactics have been developed to address this. Incorporating solubility-enhancing fusion tags, such as SUMO or maltose-binding protein (MBP), has proven to enhance the solubility of certain target proteins [20]. Additionally, co-expressing the protein of interest with molecular chaperones can help in its proper folding, making inclusion body formation less likely [21]. Like adjusting the temperature or the IPTG concentration, fine-tuning expression conditions can also modulate protein synthesis rates and improve solubility [22]. Even if inclusion bodies form, there is a workaround: the proteins can be solubilized with denaturants and then refolded, salvaging the protein for further use [23]. These adaptive strategies emphasize the versatility and adaptability of the E. coli expression system, showcasing the myriad tools researchers have at their disposal to optimize protein production.
- E. coli lacks the machinery for many eukaryotic PTMs, such as glycosylation, which may affect protein stability, folding, and activity [24].
- Unlike eukaryotic systems, E. coli produces endotoxin contamination from its lipopolysaccharide, which must be removed during purification [25].
- The toxicity of overexpressed proteins to E. coli often forces the expression of toxic protein fragments or domains retaining essential functions [26]. One strategy involves using signal sequences attached to the protein’s N-terminus, directing the protein’s export to the periplasm, and decreasing cytoplasmic accumulation, thereby reducing potential toxicity [27].
- Regulating the expression through weak promoters or controlled induction can temper any adverse impacts on the host cells. This requires codon optimization to enhance translation efficiency [28].
- Expressing monoclonal antibodies (mAbs) in Escherichia coli (E. coli) presents multiple challenges, stemming primarily from the intricacy of these proteins. One of the main hurdles is ensuring the proper folding of mAbs, especially since they possess multiple domains. E. coli often struggles to correctly fold such large eukaryotic proteins, especially when they have multiple disulfide bonds. Furthermore, bacteria lack the machinery for certain post-translational modifications like glycosylation, which are vital for the function of mAbs. This absence can compromise the mAb’s efficacy [29]. The reducing environment of the E. coli cytoplasm also makes disulfide bond formation problematic, while protein degradation can occur if the expressed proteins are unstable or perceived as foreign. Several strategies can be employed to counter these challenges. One approach is the expression of single-chain variable fragments (scFvs), which comprise the variable regions of the mAb’s heavy and light chains connected by a short peptide linker. Researchers can also leverage specialized E. coli strains designed for disulfide bond formation in the cytoplasm, such as SHuffle strains [30]. Directing mAbs or scFv expression to the periplasmic space of E. coli, which is more oxidizing than the cytoplasm, can also encourage proper disulfide bond formation. Adjustments in expression conditions, co-expression with molecular chaperones, and codon optimization for E. coli are additional strategies to improve yields [31]. The ability of bispecific antibodies (BsAbs) [32] to effectively target two entities concurrently enhances the practicality of antibody-based treatments. Genentech has successfully devised a periplasmic expression system in Escherichia coli, known as the BsAb expression system. This system utilizes either the Knobs-into-Holes (KiH) [33] technology or Fc domain HC heterodimerization [34]. Genentech has made significant advancements in the production process of bispecific antibodies (BsAbs), including two distinct heavy chains (HCs) and two distinct light chains (LCs). These improvements have been achieved by utilizing either a two-culture or a coculture strategy in Escherichia coli (E. coli) systems [35].
5. Bioinformatics Applications
- Exploiting the use of bioinformatics tools to determine the biophysical characteristics of the protein [41]. It is a complex process that involves various computational methods. These methods utilize algorithms and statistical models to analyze the protein’s primary sequence, infer its three-dimensional structure, and predict its interactions and functions.
- ○
- Sequence analysis involves comparing the amino acid sequence of a protein with known sequences in databases to identify conserved domains, motifs, or families [42];
- ○
- Structure prediction includes methods like homology modeling, ab initio modeling, and threading to predict a protein’s three-dimensional (3D) structure based on its sequence [43];
- ○
- Functional prediction identifies the biological role of a protein by assessing its structural and sequential features, often in conjunction with known protein–protein interactions and pathway analyses [44];
- ○
- Molecular dynamics simulations and related techniques are used to study the movement and interactions of proteins, providing insight into their behavior in the cellular environment [45];
- ○
- Specific bioinformatics tools are designed to predict sites in proteins likely to undergo post-translational modifications (PTMs) such as phosphorylation or glycosylation [46];
- ○
- Predicting how proteins interact with other proteins or ligands can be achieved through docking simulations and other modeling techniques [47].
- Accurate delineation [48]:
- ○
- Identifying the boundaries of protein domains is essential for understanding the function and evolution of proteins [49];
- ○
- Signal sequences are crucial for the targeting of proteins to specific cellular locations. Identifying these sequences helps in understanding the transportation and localization of proteins [50];
- ○
- Transmembrane regions anchor proteins in membranes, playing essential roles in cellular communication, signaling, and transport. Accurate prediction of these regions aids in understanding membrane protein structure and function [51];
- ○
- Identifying obligate oligomeric complexes is essential for understanding protein–protein interactions and the assembly of multi-protein complexes [52];
- ○
- Identification of PTMs is vital for understanding protein regulation and signaling [53].
6. Gene Cloning and Design
6.1. Ribosomes
- The characteristics and location of the ribosome binding site (RBS) and the disparities in translation rates observed in prokaryotic and eukaryotic organisms [70]. The ribosome binding site (RBS) plays a crucial role in the translation initiation. The sequence and position of a gene relative to the initiation codon can influence the translation efficiency. Customizing the RBS to the host organism might enhance the efficiency of translating the desired protein [71];
- Correct use of the strain and media to optimize production, though with many limitations [72]. The optimization of production in E. coli strains through proper selection of the strain and media is a common strategy in biotechnology but comes with certain limitations;
- Optimization in E. coli can vary widely depending on the protein or other manufactured product. Selecting the right strain of E. coli, determining the optimal temperature, and choosing the appropriate culture media are crucial considerations for recombinant protein expression.
6.2. Promoter
- To create the promoter variation lac1G, the promoter lacUV5 and lac were joined again. (G was substituted for A at position +1) [82];
- The expression of T7 RNA polymerase (RNAP) is effectively regulated to prevent leakage by the presence of a mutant form of the Lac repressor protein (LacI), specifically the V192F variant. This mutant variant cannot bind to isopropyl β-D-1-thiogalactopyranoside (IPTG), hence preventing its activation. Consequently, the mutant LacI dynamically governs the levels of transcripts produced by T7 RNAP [83];
- Building a T7 RNAP RBS library quickly involves using the base editor and CRISPR/Cas9 to screen potential expression hosts [84];
- The ability of T7 RNA polymerase to bind to the PT7 promoter was impaired due to a specific amino acid substitution (A102D), resulting in an alteration in the rate of RNA production. The T7 RNA polymerase (T7 RNAP) was fragmented into two segments and co-expressed with a light-responsive dimerization domain, exhibiting functional behavior upon exposure to blue light [85].
6.3. Codons
6.4. Protein Folding
7. Enhanced Efficiency
7.1. Solubilization
7.2. Disulfide Bond
7.3. Post-Translational Modifications
7.4. Strain and Media
7.5. Fermentation Conditions
7.6. Purification
8. Cell-Free Protein Synthesis System (CFPS)
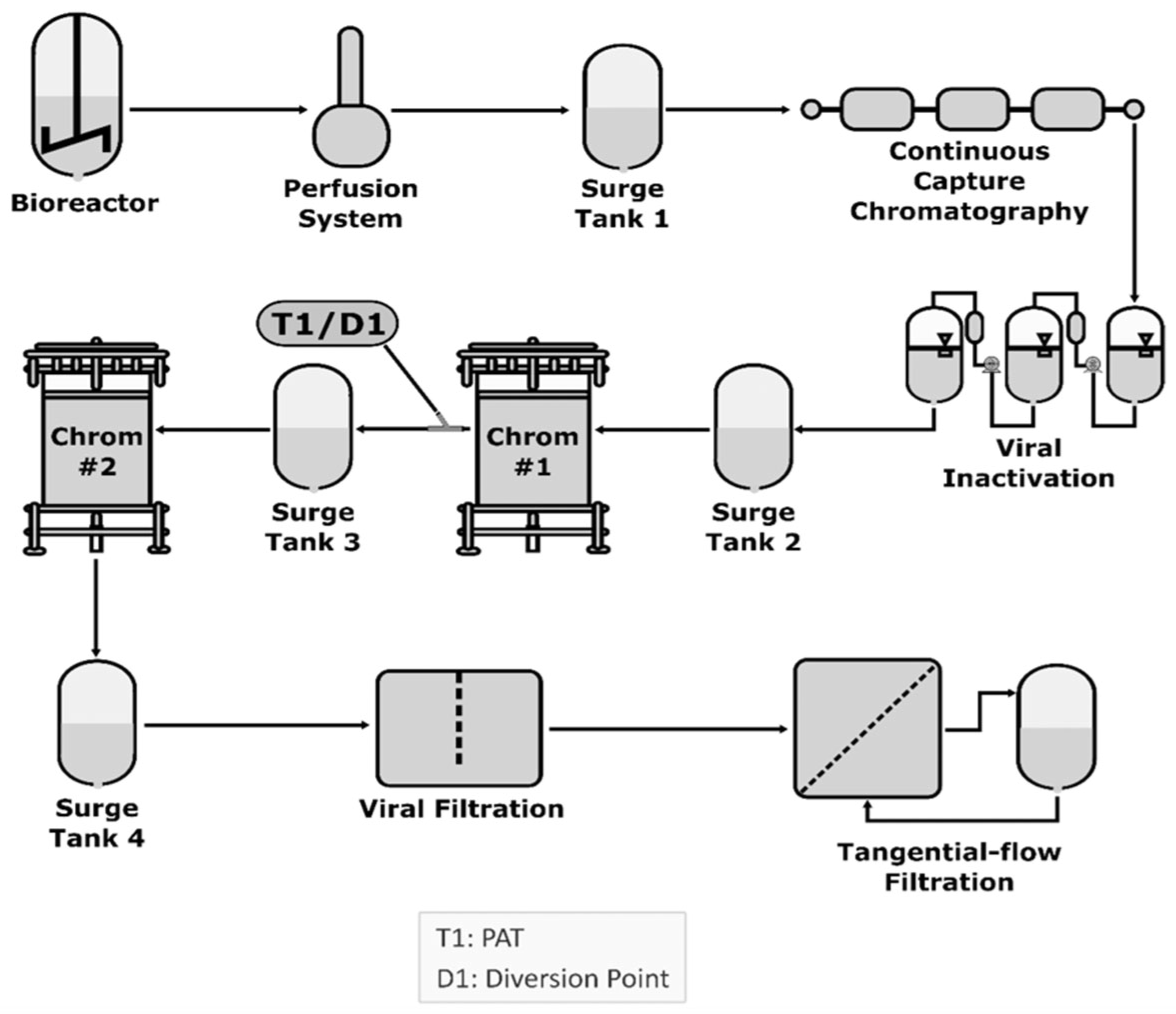
9. Continuous Manufacturing (CM)
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Landgraf, W.; Sandow, J. Recombinant Human Insulins–Clinical Efficacy and Safety in Diabetes Therapy. Eur. Endocrinol. 2016, 12, 12–17. [Google Scholar] [CrossRef]
- Approved Protein Drugs in the US and EU. Available online: https://drugs.ncats.io/ (accessed on 10 July 2023).
- Dimitrov, D.S. Therapeutic proteins. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2012; Volume 899, pp. 1–26. [Google Scholar]
- Zhang, Z.X.; Nong, F.T.; Wang, Y.Z.; Yan, C.-X.; Gu, Y.; Song, P.; Sun, X.-M. Strategies for efficient production of recombinant proteins in Escherichia coli: Alleviating the host burden and enhancing protein activity. Microb. Cell Fact. 2022, 21, 191. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Cao, Y.; Yu, Z.; Wang, G.; Li, Y.; Ye, X.; Li, C.; Lin, X.; Song, H. Synthetic sRNA-based engineering of Escherichia coli for enhanced production of full-length immunoglobulin G. Biotechnol. J. 2020, 15, e1900363. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.A.; Symons, R.H.; Berg, P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909. [Google Scholar] [CrossRef]
- Cohen, S.N.; Chang, A.C.; Boyer, H.W.; Helling, R.B. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.P.; Colaianni, A.; Liu, K. Lessons from the Commercialization of the Cohen-Boyer Patents: The Stanford University Licensing Program. Handbook of Best Practices. In Intellectual Property Management in Health and Agricultural Innovation: A Handbook of Best Practices; MIHR: Oxford, UK; PIPRA: Davis, CA, USA, 2007; Available online: https://maryannfeldman.web (accessed on 10 July 2023).
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983, 47, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Wei, Q.; Wu, W.; He, Y.; Gao, J.; Zhou, R.; Jiang, L.; Qu, J.; Xia, J. Phase-Separated Multienzyme Compartmentalization for Terpene Biosynthesis in a Prokaryote. Angew. Chem. Int. Ed. 2022, 8, 61–69. [Google Scholar]
- Wei, S.-P.; Qian, Z.-G.; Hu, C.-F.; Pan, F.; Chen, M.-T.; Lee, S.Y.; Xia, X.-X. Formation and functionalization of membraneless compartments in Escherichia coli. Nat. Chem. Biol. 2020, 16, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- McElwain, L.; Phair, K.; Kealey, C.; Brady, D. Current trends in biopharmaceuticals production in Escherichia coli. Biotechnol. Lett. 2022, 44, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Grauschopf, U.; Maier, T.; Glockshuber, R.; Ban, N. The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature 2009, 459, 726–730. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/drugs/regulatory-science-action/impact-continuous-manufacturing-processes-viral-safety-therapeutic-proteins (accessed on 10 July 2023).
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef]
- Fahnert, B.; Lilie, H.; Neubauer, P. Inclusion bodies: Formation and utilisation. Adv. Biochem. Eng./Biotechnol. 2004, 89, 93–142. [Google Scholar] [PubMed]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.G.; Baneyx, F. Protein misfolding and inclusion body formation in recombinant Escherichia coli cells overexpressing Heat-shock proteins. J. Biol. Chem. 1996, 271, 11141–11147. [Google Scholar] [CrossRef]
- Vasina, J.A.; Baneyx, F. Recombinant protein expression at low temperatures under the transcriptional control of the major Escherichia coli cold shock promoter cspA. Appl. Environ. Microbiol. 1996, 62, 1444–1447. [Google Scholar] [CrossRef]
- Singh, S.M.; Panda, A.K. Solubilization and refolding of bacterial inclusion body proteins. J. Biosci. Bioeng. 2005, 99, 303–310. [Google Scholar] [CrossRef]
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241–1252. [Google Scholar] [CrossRef]
- Magalhães, P.O.; Lopes, A.M.; Mazzola, P.G.; Rangel-Yagui, C.; Penna, T.C.V.; Pessoa, A., Jr. Methods of endotoxin removal from biological preparations: A review. J. Pharm. Pharm. Sci. 2007, 10, 388–404. [Google Scholar]
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef]
- Mergulhão, F.J.; Summers, D.K.; Monteiro, G.A. Recombinant protein secretion in Escherichia coli. Biotechnol. Adv. 2005, 23, 177–202. [Google Scholar] [CrossRef]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 2012, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Skretas, G.; Georgiou, G. Simple genetic selection protocol for isolation of overexpressed genes that enhance accumulation of membrane-integrated human G protein-coupled receptors in Escherichia coli. Appl. Environ. Microbiol. 2009, 75, 3853–3863. [Google Scholar] [CrossRef]
- Wang, S.; Chen, K.; Lei, Q.; Ma, P.; Yuan, A.Q.; Zhao, Y.; Jiang, Y.; Fang, H.; Xing, S.; Fang, Y.; et al. The state of the art of bispecific antibodies for treating human malignancies. EMBO Mol. Med. 2021, 13, e14291. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Ma, X.; Maun, H.R.; Zheng, Z.; Peng, J.; Romero, M.; Huang, A.; Yang, N.Y.; Nishimura, M.; Greve, J.; et al. Monovalent antibody design and mechanism of action of Onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. USA 2013, 110, E2987–E2996. [Google Scholar] [CrossRef]
- Spiess, C.; Bevers, J.; Jackman, J.; Chiang, N.; Nakamura, G.; Dillon, M.; Liu, H.; Molina, P.; Elliott, J.M.; Shatz, W.; et al. Development of a human IgG4 bispecific antibody for dual targeting of interleukin-4 (IL-4) and interleukin-13 (IL-13) cytokines. J. Biol. Chem. 2013, 288, 26583–26593. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Darlington, P.J.; Kirchhof, M.G.; Criado, G.; Sondhi, J.; Madrenas, J. Hierarchical Regulation of CTLA-4 Dimer-Based Lattice Formation and Its Biological Relevance for T Cell Inactivation. J. Immunol. 2005, 175, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Boyd, D.; Berkmen, M. Disulfide Bond Formation in the Periplasm of Escherichia coli. EcoSal Plus 2019, 8, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual synergistic folding in recruitment of cbp/p300 by p160 nuclear receptor coactivators. Nature 2002, 415, 549–553. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Radivojac, P.; Clark, W.T.; Oron, T.R.; Schnoes, A.M.; Wittkop, T.; Sokolov, A.; Graim, K.; Funk, C.; Verspoor, K.; Ben-Hur, A.; et al. A large-scale evaluation of computational protein function prediction. Nat. Methods 2013, 10, 221–227. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Vakser, I.A. Protein docking for low-resolution structures. Protein Eng. 1995, 8, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 5th ed.; Garland Science: New York, NY, USA, 2008; ISBN 978-0-8153-4105-5. [Google Scholar]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- von Heijne, G. The signal peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.D.; Teichmann, S.A. Structural, evolutionary, and assembly principles of protein oligomerization. Prog. Mol. Biol. Transl. Sci. 2013, 117, 25–51. [Google Scholar]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef]
- Subramanian, S.; Ross, P.D. Dye-ligand affinity chromatography: The interaction of cibacron blue f3GA® with proteins and enzyme. Crit. Rev. Biochem. Mol. Biol. 1984, 16, 169–205. [Google Scholar] [CrossRef]
- Kish, W.S.; Roach, M.K.; Sachi, H.; Naik, A.D.; Menegatti, S.; Carbonell, R.G. Purification of human erythropoietin by affinity chromatography using cyclic peptide ligands. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1085, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Young, C.L.; Britton, Z.T.; Robinson, A.S. Recombinant protein expression and purification: A comprehensive review of affinity tags and microbial applications. Biotechnol. J. 2012, 7, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, K.F.; Dixon, H.B.F.; Rosner, P.J.; Hoth, L.R.; Lanzetti, A.J.; Borzilleri, K.A.; Marr, E.S.; Pezzullo, L.H.; Martin, L.B.; LeMotte, P.K.; et al. Spontaneous α-N-6-phosphogluconoylation of a “His tag” in Escherichia coli: The cause of extra mass of 258 or 178 Da in fusion proteins. Anal. Biochem. 1999, 267, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.N.; Smith, K.D.; Ream, J.A.; Lewis, L.K. Enhancing yields of low and single copy number plasmid DNAs from Escherichia coli cells. J. Microbiol. Methods 2017, 133, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Lee, S.Y. High-level production of human leptin by fed-batch cultivation of recombinant Escherichia coli and its purification. Appl. Environ. Microbiol. 1999, 65, 3027–3032. [Google Scholar] [CrossRef]
- Sharma, A.K.; Shukla, E.; Janoti, D.S.; Mukherjee, K.J.; Shiloach, J. A novel knock out strategy to enhance recombinant protein expression in Escherichia coli. Microb. Cell Fact. 2020, 19, 148. [Google Scholar] [CrossRef]
- Ganoza, M.C.; Kiel, M.C.; Aoki, H. Evolutionary conservation of reactions in translation. Microbiol. Mol. Biol. Rev. 2002, 66, 460–485. [Google Scholar] [CrossRef]
- Irastortza-Olaziregi, M.; Amster-Choder, O. Coupled Transcription-Translation in Prokaryotes: An Old Couple with New Surprises. Front. Microbiol. 2021, 11, 624830. [Google Scholar] [CrossRef]
- Hui, A.; De Boer, H.A. Specialized ribosome system: Preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 4762–4766. [Google Scholar] [CrossRef]
- Dixon, N.; Robinson, C.J.; Geerlings, T.; Duncan, J.N.; Drummond, S.P.; Micklefield, J. Orthogonal Riboswitches for Tuneable Coexpression in Bacteria. Angew. Chem. Int. Ed. 2012, 51, 3620–3624. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Carlson, E.D.; Szal, T.; Florin, T.; Jewett, M.C.; Mankin, A.S. Protein synthesis by ribosomes with tethered subunits. Nature 2015, 524, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Morra, R.; Shankar, J.; Robinson, C.J.; Halliwell, S.; Butler, L.; Upton, M.; Hay, S.; Micklefield, J.; Dixon, N. Dual transcriptional-Translational cascade permits cellular level tuneable expression control. Nucleic Acids Res. 2016, 44, 21. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.D.; d’Aquino, A.E.; Kim, D.S.; Fulk, E.M.; Hoang, K.; Szal, T.; Mankin, A.S.; Jewett, M.C. Engineered ribosomes with tethered subunits for expanding biological function. Nat. Commun. 2019, 10, 3920. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Zhang, G.; Darst, S.A. Structure of the Escherichia coli RNA polymerase α subunit amino-terminal domain. Science 1998, 281, 262–266. [Google Scholar] [CrossRef]
- Chen, H.; Bjerknes, M.; Kumar, R.; Jay, E. Determination of the optimal aligned spacing between the shine-dalgarno sequence and the translation initiation codon of Escherichia coli m RNAs. Nucleic Acids Res. 1994, 22, 4953–4957. [Google Scholar] [CrossRef]
- Shepard, H.M.; Yelverton, E.; Goeddel, D.V. Increased Synthesis in E. coli of Fibroblast and Leukocyte Interferons Through Alterations in Ribosome Binding Sites. DNA 1982, 1, 125–131. [Google Scholar]
- Shine, J.; Dalgarno, L. The 3′ terminal sequence of Escherichia coli 16S ribosomal RNA: Complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. USA 1974, 71, 1342–1346. [Google Scholar] [CrossRef]
- Jeong, H.; Barbe, V.; Lee, C.H.; Vallenet, D.; Yu, D.S.; Choi, S.-H.; Couloux, A.; Lee, S.-W.; Yoon, S.H.; Cattolico, L. Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J. Mol. Biol. 2009, 394, 644–652. [Google Scholar] [CrossRef]
- Du, F.; Liu, Y.-Q.; Xu, Y.S.; Li, Z.J.; Wang, Y.Z.; Zhang, Z.X.; Sun, X.M. Regulating the T7 RNA polymerase expression in E. coli BL21(DE3) to provide more host options for recombinant protein production. Microb. Cell Fact. 2021, 20, 189. [Google Scholar] [CrossRef]
- Khlebnikov, A.; Risa, Ø.; Skaug, T.; Carrier, T.A.; Keasling, J.D. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J. Bacteriol. 2000, 182, 7029–7034. [Google Scholar] [CrossRef]
- Lutz, R.; Bujard, H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997, 25, 1203–1210. [Google Scholar] [CrossRef]
- Mueller, K.L.; Simon, J.D.; Elf, J. Design, construction, and implementation of a fully repressible bistable genetic switch in E. coli. Nucleic Acids Res. 2019, 47, 6307–6317. [Google Scholar]
- Sun, X.M.; Zhang, Z.X.; Wang, L.R.; Wang, J.G.; Liang, Y.; Yang, H.F.; Tao, R.S.; Jiang, Y.; Yang, J.J.; Yang, S. Downregulation of T7 RNA polymerase transcription enhances pET-based recombinant protein production in Escherichia coli BL21(DE3) by suppressing autolysis. Biotechnol. Bioeng. 2021, 118, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Lee, D.-H.; Kim, O.C.; Kim, J.F.; Yoon, S.H. Tunable control of an Escherichia coli expression system for the overproduction of membrane proteins by titrated expression of a mutant lac repressor. ACS Synth. Biol. 2017, 6, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Zhang, Z.X.; Xu, Y.; Shi, T.Q.; Ye, C.; Sun, X.M.; Huang, H. CRISPR-Based Construction of a BL21 (DE3)-derived variant strain library to rapidly improve recombinant protein production. ACS Synth. Biol. 2022, 11, 343–352. [Google Scholar] [CrossRef]
- Baumschlager, A.; Aoki, S.K.; Khammash, M. Dynamic blue light-inducible T7 RNA polymerases (Opto-T7RNAPs) for precise spatiotemporal gene expression control. ACS Synth. Biol. 2017, 6, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Rouches, M.V.; Xu, Y.; Cortes, L.B.G.; Lambert, G. A plasmid system with tunable copy number. Nat. Commun. 2022, 13, 3908. [Google Scholar] [CrossRef]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981, 146, 1–21. [Google Scholar] [CrossRef]
- Boël, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.H.; Su, M.; Luff, J.D.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, M.; Hausherr, A.; Ferbitz, L.; Schödl, T.; Heitzer, M.; Hegemann, P. Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Mol. Biol. 2004, 55, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Kleber-Janke, T.; Becker, W.M. Use of modified BL21(DE3) Escherichia coli cells for high-level expression of recombinant peanut allergens affected by poor codon usage. Protein Expr. Purif. 2000, 19, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Novy, R.; Drott, D.; Yaeger, K.; Mierendorf, R. Overcoming the codon bias of E. coli for enhanced protein expression. Innovations 2001, 12, 1–3. [Google Scholar]
- Komar, A.A. The Yin and Yang of codon usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef]
- Chemla, Y.; Peeri, M.; Heltberg, M.L.; Eichler, J.; Jensen, M.H.; Tuller, T.; Alfonta, L. A possible universal role for mRNA secondary structure in bacterial translation revealed using a synthetic operon. Nat. Commun. 2020, 11, 4827. [Google Scholar] [CrossRef]
- Lenz, G.; Doron-Faigenboim, A.; Ron, E.Z.; Tuller, T.; Gophna, U. Sequence Features of E. coli mRNAs Affect Their Degradation. PLoS ONE 2011, 6, e28544. [Google Scholar]
- Siller, E.; DeZwaan, D.C.; Anderson, J.F.; Freeman, B.C.; Barral, J.M. Slowing Bacterial Translation Speed Enhances Eukaryotic Protein Folding Efficiency. J. Mol. Biol. 2010, 396, 1310–1318. [Google Scholar] [CrossRef]
- Angov, E.; Hillier, C.J.; Kincaid, R.L.; Lyon, J.A. Heterologous Protein Expression Is Enhanced by Harmonizing the Codon Usage Frequencies of the Target Gene with those of the Expression Host. PLoS ONE 2008, 3, e2189. [Google Scholar] [CrossRef] [PubMed]
- Gasser, B.; Saloheimo, M.; Rinas, U.; Dragosits, M.; Rodríguez-Carmona, E.; Baumann, K.; Giuliani, M.; Parrilli, E.; Branduardi, P.; Lang, C.; et al. Protein folding and conformational stress in microbial cells producing recombinant proteins: A host comparative overview. Microb. Cell Fact. 2008, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Goemans, C.; Denoncin, K.; Collet, J.F. Folding mechanisms of periplasmic proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A.; Pluckthun, A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 1988, 240, 1038–1041. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Snedecor, B.; Nishihara, J.C.; Joly, J.C.; McFarland, N.; Andersen, D.C.; Battersby, J.E.; Champion, K.M. High-level accumulation of a recombinant antibody fragment in the periplasm of Escherichia coli requires a triple-mutant (degP prc spr) host strain. Biotechnol. Bioeng. 2004, 85, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Kelley, R.F.; Rodrigues, M.L.; Snedecor, B.; Covarrubias, M.; Velligan, M.D.; Wong, W.L.T.; Rowland, A.M.; Kotts, C.E.; Carver, M.E.; et al. High level Escherichia coli expression and production of a bivalent humanized antibody fragment. Biotechnology 1992, 10, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.N.; Lu, X.Y.; Zong, H.; Bin, Z.G.; Shen, W. Bioproduction of trans-10, cis-12-Conjugated Linoleic Acid by a Highly Soluble and Conveniently Extracted Linoleic Acid Isomerase and an Extracellularly Expressed Lipase from Recombinant Escherichia coli Strains. J. Microbiol. Biotechnol. 2018, 28, 739–747. [Google Scholar] [CrossRef]
- Guzmán, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Jo, B.H. An intrinsically disordered peptide tag that confers an unusual solubility to aggregation-prone proteins. Appl. Environ. Microbiol. 2022, 88, e00097-22. [Google Scholar] [CrossRef]
- Choi, S.W.; Pangeni, R.; Jung, D.H.; Kim, S.J.; Park, J.W. Construction and characterization of cell-penetrating peptide-fused fibroblast growth factor and vascular endothelial growth factor for an enhanced percutaneous delivery system. J. Nanosci. Nanotechnol. 2018, 18, 842–847. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.-J.; Han, M.-H.; Yoon, N.-K.; Kim, Y.-C.; Ahn, J. Effective production of human growth factors in Escherichia coli by fusing with small protein 6HFh8. Microb. Cell Fact. 2021, 20, 9. [Google Scholar] [CrossRef]
- Chowdhury, T.; Chien, P.; Ebrahim, S.; Sauer, R.T.; Baker, T.A. Versatile modes of peptide recognition by the ClpX N domain mediate alternative adaptor-binding specificities in different bacterial species. Protein Sci. 2010, 19, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Cabilly, S.; Riggs, A.D.; Pande, H.; Shively, J.E.; Holmes, W.E.; Rey, M.; Perry, L.J.; Wetzel, R.; Heyneker, H.L. Generation of antibody activity from immunoglobulin polypeptide chains produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 3273–3277. [Google Scholar] [CrossRef] [PubMed]
- Saaranen, M.J.; Ruddock, L.W. Applications of catalyzed cytoplasmic disulfide bond formation. Biochem. Soc. Trans. 2019, 47, 1223–1231. [Google Scholar] [CrossRef]
- Guerrero Montero, I.; Richards, K.L.; Jawara, C.; Browning, D.F.; Peswani, A.R.; Labrit, M.; Allen, M.; Aubry, C.; Davé, E.; Humphreys, D.P. Escherichia coli “TatExpress” strains export several g/L human growth hormone to the periplasm by the Tat pathway. Biotechnol. Bioeng. 2019, 116, 3282–3291. [Google Scholar]
- de Marco, A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb. Cell Fact. 2009, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Karyolaimos, A.; de Gier, J.W. Strategies to Enhance Periplasmic Recombinant Protein Production Yields in Escherichia coli. Front Bioeng Biotechnol. 2021, 9, 797334. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov, S.M.; Little, M. Affinity purification of tagged recombinant proteins using immobilized single chain Fv fragments. Anal Biochem. 1997, 244, 189–191. [Google Scholar] [CrossRef]
- Menon, V.; Thomas, R.; Ghale, A.R.; Reinhard, C.; Pruszak, J. Flow cytometry protocols for surface and intracellular antigen analyses of neural cell types. J. Vis. Exp. 2014, 18, 52241. [Google Scholar]
- Brinkmann, U.; Mattes, R.E. High-level expression of a human immunoglobulin fragment Fab in Escherichia coli. Gene 1989, 85, 517–521. [Google Scholar]
- Ma, Y.; Lee, C.J.; Park, J.S. Strategies for Optimizing the Production of Proteins and Peptides with Multiple Disulfide Bonds. Antibiotics 2020, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Derman, A.I.; Prinz, W.A.; Belin, D.; Beckwith, J. Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 1993, 262, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.emdmillipore.com/US/en/product/Origami-BDE3-Competent-Cells-Novagen,EMD_BIO-70837 (accessed on 10 July 2023).
- Zhang, W.; Zheng, W.; Mao, M.; Yang, Y. Highly efficient folding of multidisulfide proteins in superoxidizing Escherichia coli cytoplasm. Biotechnol. Bioeng. 2014, 111, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Ruddock, L.W. Topological plasticity of enzymes involved in disulfide bond formation allows catalysis in either the periplasm or the cytoplasm. J. Mol. Biol. 2013, 425, 3268–3276. [Google Scholar] [CrossRef] [PubMed]
- Sohail, A.A.; Gaikwad, M.; Khadka, P.; Saaranen, M.J.; Ruddock, L.W. Production of extracellular matrix proteins in the cytoplasm of E. coli: Making giants in tiny factories. Int. J. Mol. Sci. 2020, 21, 688. [Google Scholar] [CrossRef]
- Lapteva, Y.S.; Vologzhannikova, A.A.; Sokolov, A.S.; Ismailov, R.G.; Uversky, V.N.; Permyakov, S.E. In Vitro N-Terminal Acetylation of Bacterially Expressed Parvalbumins by N-Terminal Acetyltransferases from Escherichia coli. Appl. Biochem. Biotechnol. 2021, 193, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J.; Koomey, M. Sweet new roles for protein glycosylation in prokaryotes. Trends Microbiol. 2017, 25, 662–672. [Google Scholar] [CrossRef]
- Harding, C.M.; Feldman, M.F. Glycoengineering bioconjugate vaccines, therapeutics, and diagnostics in E. coli. Glycobiology 2019, 29, 519–529. [Google Scholar] [CrossRef]
- Wacker, M.; Linton, D.; Hitchen, P.G.; Nita-Lazar, M.; Haslam, S.M.; North, S.J.; Panico, M.; Morris, H.R.; Dell, A.; Wren, B.W. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 2002, 298, 1790–1793. [Google Scholar] [CrossRef]
- Silverman, J.M.; Imperiali, B. Bacterial N-glycosylation efficiency is dependent on the structural context of target sequence. J. Biol. Chem. 2016, 291, 22001–22010. [Google Scholar] [CrossRef]
- Ollis, A.A.; Zhang, S.; Fisher, A.C.; DeLisa, M.P. Engineered oligosaccharyltransferases with greatly relaxed acceptor-site specificity. Nat. Chem. Biol. 2014, 10, 816–822. [Google Scholar] [CrossRef]
- Kightlinger, W.; Warfel, K.F.; DeLisa, M.P.; Jewett, M.C. Synthetic glycobiology: Parts, systems, and applications. ACS Synth. Biol. 2020, 9, 1534–1562. [Google Scholar] [CrossRef] [PubMed]
- Pratama, F.; Linton, D.; Dixon, N. Genetic and process engineering strategies for enhanced recombinant N-glycoprotein production in bacteria. Microb. Cell Fact. 2021, 20, 198. [Google Scholar] [CrossRef]
- Valderrama-Rincon, J.D.; Fisher, A.C.; Merritt, J.H.; Fan, Y.-Y.; Reading, C.A.; Chhiba, K.; Heiss, C.; Azadi, P.; Aebi, M.; DeLisa, M.P. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat. Chem. Biol. 2012, 8, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Buenbrazo, N.; Kell, L.; Rahmani, S.; Sim, L.; Withers, S.G.; DeFrees, S.; Wakarchuk, W. A bacterial expression platform for production of therapeutic proteins containing human-like O-linked glycans. Cell Chem. Biol. 2019, 26, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kasari, M.; Kasari, V.; Kärmas, M.; Jõers, A. Decoupling growth and production by removing the origin of replication from a bacterial chromosome. ACS Synth. Biol. 2022, 11, 2610–2622. [Google Scholar] [CrossRef] [PubMed]
- Leabman, M.K.; Meng, Y.G.; Kelley, R.F.; DeForge, L.E.; Cowan, K.J.; Iyer, S. Effects of altered FcγR binding on antibody pharmacokinetics in cynomolgus monkeys. mAbs 2013, 5, 896–903. [Google Scholar] [CrossRef]
- Tytgat, H.L.; Lin, C.-W.; Levasseur, M.D.; Tomek, M.B.; Rutschmann, C.; Mock, J.; Liebscher, N.; Terasaka, N.; Azuma, Y.; Wetter, M. Cytoplasmic glycoengineering enables biosynthesis of nanoscale glycoprotein assemblies. Nat. Commun. 2019, 10, 5403. [Google Scholar] [CrossRef]
- Gallwitz, M.; Enoksson, M.; Thorpe, M.; Hellman, L. The extended cleavage specificity of human thrombin. PLoS ONE 2012, 7, e31756. [Google Scholar] [CrossRef]
- Gasser, B.; Mattanovich, D. Antibiotic resistance marker genes in Saccharomyces cerevisiae: An overview on different mechanisms and new approaches for the elimination. FEMS Yeast Res. 2004, 4, 235–245. [Google Scholar]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Miroux, B.; Walker, J.E. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Inada, T.; Kimata, K.; Aiba, H. Mechanism responsible for glucose-lactose diauxie in Escherichia coli: Challenge to the cAMP model. Genes Cells 1996, 1, 293–301. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, T.; Pérez-Bermúdez, P.; Gavidia, I. Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. Springerplus 2013, 2, 89. [Google Scholar] [PubMed]
- Bertani, G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951, 62, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, F.C.; Bloch, P.L.; Smith, D.F. Culture medium for enterobacteria. J. Bacteriol. 1974, 119, 736–747. [Google Scholar] [CrossRef]
- Lipinszki, Z.; Vernyik, V.; Farago, N.; Sari, T.; Puskas, L.G.; Blattner, F.R.; Posfai, G.; Gyorfy, Z. Enhancing the translational capacity of E. coli by resolving the codon bias. ACS Synth. Biol. 2018, 7, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Serra, L.R.; Hardy, E.; Ubieta, R.; Vispo, N.S.; Fernandez, C.; Besada, V.; Falcon, V.; Gonzalez, M.; Santos, A.; Perez, G.; et al. Expression and folding of an interleukin-2-proinsulin fusion protein and its conversion into insulin by a single step enzymatic removal of the C-peptide and the N-terminal fused sequence. FEBS Lett. 1996, 378, 171–176. [Google Scholar] [CrossRef]
- Ludeman, J.P.; Pike, R.N.; Bromfield, K.M.; Duggan, P.J.; Cianci, J.; Le Bonniec, B.; Whisstock, J.C.; Bottomley, S.P. Determination of the P′1, P′2 and P′3 subsite-specificity of factor Xa. Int. J. Biochem. Cell Biol. 2003, 35, 221–225. [Google Scholar] [CrossRef]
- Bianchini, E.P.; Louvain, V.B.; Marque, P.E.; Juliano, M.A.; Juliano, L.; Le Bonniec, B.F. Mapping of the catalytic groove preferences of factor Xa reveals an inadequate selectivity for its macromolecule substrates. J. Biol. Chem. 2002, 277, 20527–20534. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Smolskaya, S.; Logashina, Y.A.; Andreev, Y.A. Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis. Int. J. Mol. Sci. 2020, 21, 928. [Google Scholar] [CrossRef]
- Chong, S. Overview of Cell-Free Protein Synthesis: Historic Landmarks, Commercial Systems, and Expanding Applications. Curr. Protoc. Mol. Biol. 2013, 108, 16.30.1–16.30.11. [Google Scholar] [CrossRef] [PubMed]
- Spirin, A.S.; Baranov, V.I.; Ryabova, L.A.; Ovodov, S.; Alakhov, Y.B. A continuous cell-free translation system capable of producing polypeptides in high yield. Science 1988, 242, 1162–1164. [Google Scholar] [CrossRef]
- Wang, S.; Majumder, S.; Emery, N.J.; Liu, A.P. Simultaneous monitoring of transcription and translation in mammalian cell-free expression in bulk and in cell-sized droplets. Synth. Biol. 2018, 3, ysy005. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Noireaux, V. An E. coli cell-free expression toolbox: Application to synthetic gene circuits and artificial cells. ACS Synth. Biol. 2012, 1, 29–41. [Google Scholar]
- Kwon, Y.C.; Jewett, M.C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 2015, 5, 8663. [Google Scholar] [CrossRef]
- Ge, X.; Luo, D.; Xu, J. Cell-free protein expression under macromolecular crowding conditions. PLoS ONE. 2011, 6, e28707. [Google Scholar] [CrossRef]
- Ho, D.; Quake, S.R.; McCabe, E.R.B.; Chng, W.J.; Chow, E.K.; Ding, X.; Gelb, B.D.; Ginsburg, G.S.; Hassenstab, J.; Ho, C.M.; et al. Enabling Technologies for Personalized and Precision Medicine. Trends Biotechnol. 2020, 38, 497–518. [Google Scholar] [CrossRef]
- Caschera, F.; Noireaux, V. Integration of biological parts toward the synthesis of a minimal cell. Curr. Opin. Chem. Biol. 2014, 22, 85–91. [Google Scholar] [CrossRef]
- Klammt, C.; Lohr, F.; Schafer, B.; Haase, W.; Dotsch, V.; Ruterjans, H.; Glaubitz, C.; Bernhard, F. High-level cell-free expression and specific labeling of integral membrane proteins. Eur. J. Biochem. 2006, 273, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Forster, A.C. Synthesis and applications of DNA constructs for cell-free expression systems. Nucleic Acids Res. 2006, 34, e9. [Google Scholar]
- Jewett, M.C.; Swartz, J.R. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol Bioeng. 2004, 86, 19–26. [Google Scholar] [CrossRef]
- Pardee, K.; Slomovic, S.; Nguyen, P.Q.; Lee, J.W.; Donghia, N.; Burrill, D.; Ferrante, T.; McSorley, F.R.; Furuta, Y.; Vernet, A.; et al. Portable, on-demand biomolecular manufacturing. Cell 2016, 167, 248–259. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine; Division on Earth and Life Studies; Board on Chemical Sciences and Technology. Continuous Manufacturing for the Modernization of Pharmaceutical Production: Proceedings of a Workshop; National Academies Press: Washington, DC, USA, 2019. [Google Scholar]
- FDA. Q13 Continuous Manufacturing of Drug Substances and Drug Products. Available online: https://www.fda.gov/media/165775/download (accessed on 19 July 2023).
- Bielser, J.M.; Wolf, M.; Souquet, J.; Broly, H.; Morbidelli, M. Perfusion mammalian cell culture for recombinant protein manufacturing-A critical review. Biotechnol. Adv. 2018, 36, 1328–1340. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, H.; Kim, E.-Y.; Kim, J.F.; Lee, S.Y.; Yoon, S.H. Genomic and transcriptomic landscape of Escherichia coli BL21(DE3). Nucleic Acids Res. 2017, 45, 5285–5293. [Google Scholar] [CrossRef] [PubMed]
- Packiam, K.A.R.; Ramanan, R.N.; Ooi, C.W.; Krishnaswamy, L.; Tey, B.T. Stepwise optimization of recombinant protein production in Escherichia coli utilizing computational and experimental approaches. Appl. Microbiol. Biotechnol. 2020, 104, 3253–3266. [Google Scholar] [CrossRef] [PubMed]




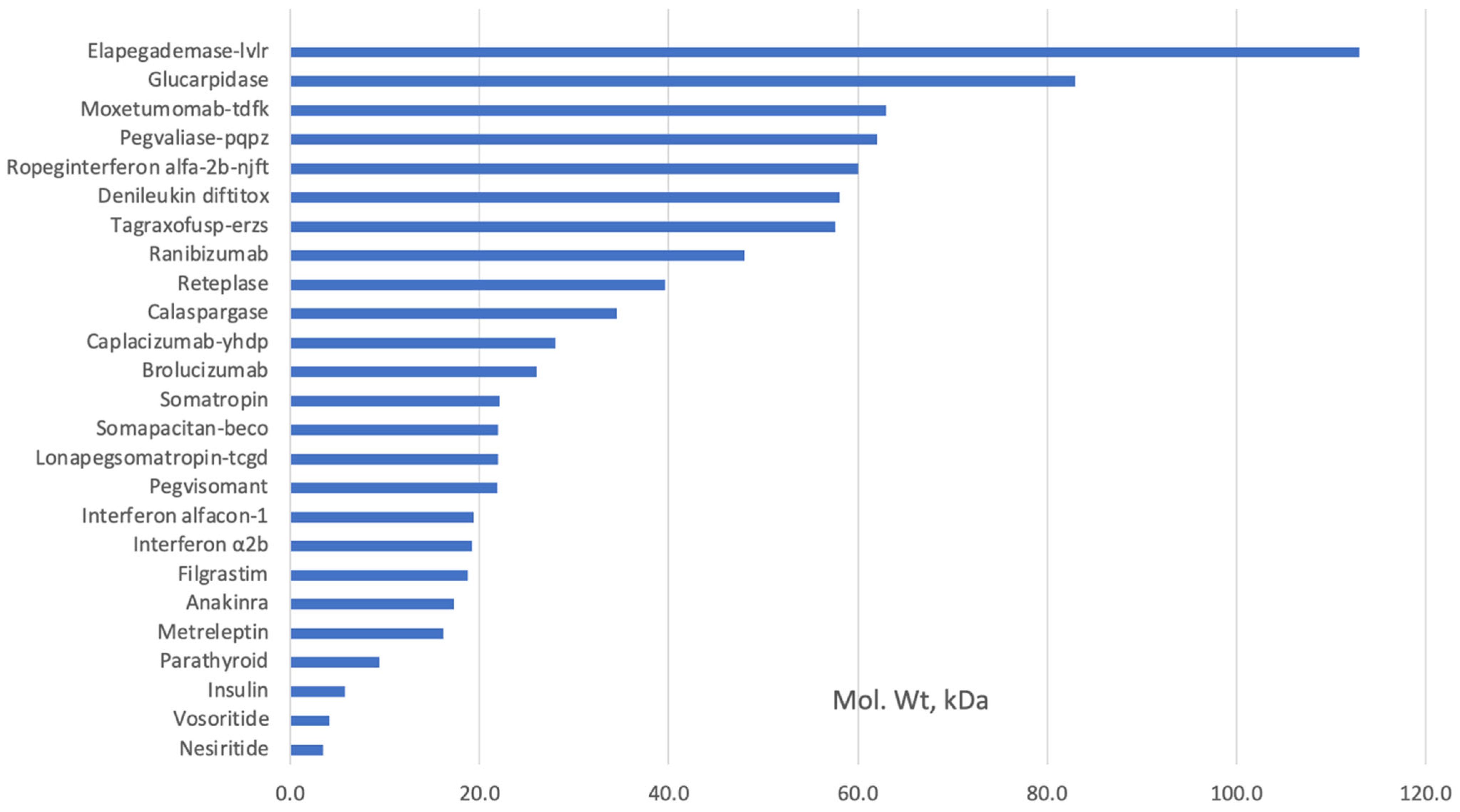
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Listed Protein Class | Number |
|---|---|
| Monoclonal Antibody | 94 |
| Hormone | 10 |
| Enzyme | 8 |
| Monoclonal Antibody Conjugate | 8 |
| Cytokine | 4 |
| Bispecific Antibody | 3 |
| Coagulation Factor | 3 |
| Growth Factor | 3 |
| Peptide | 3 |
| Carrier Protein | 1 |
| Enzyme Inhibitor | 1 |
| Fab | 1 |
| Fusion Proteins | 1 |
| Single-Domain Antibody | 1 |
| Toxin | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K.; Magoola, M. Advances in Escherichia coli-Based Therapeutic Protein Expression: Mammalian Conversion, Continuous Manufacturing, and Cell-Free Production. Biologics 2023, 3, 380-401. https://0-doi-org.brum.beds.ac.uk/10.3390/biologics3040021
Niazi SK, Magoola M. Advances in Escherichia coli-Based Therapeutic Protein Expression: Mammalian Conversion, Continuous Manufacturing, and Cell-Free Production. Biologics. 2023; 3(4):380-401. https://0-doi-org.brum.beds.ac.uk/10.3390/biologics3040021
Chicago/Turabian StyleNiazi, Sarfaraz K., and Matthias Magoola. 2023. "Advances in Escherichia coli-Based Therapeutic Protein Expression: Mammalian Conversion, Continuous Manufacturing, and Cell-Free Production" Biologics 3, no. 4: 380-401. https://0-doi-org.brum.beds.ac.uk/10.3390/biologics3040021