
Biological and Pharmacological Properties of Carbon Monoxide: A General Overview


Chair of Medical Biochemistry, Medical College, Jagiellonian University, 31-034 Cracow, Poland
*
Author to whom correspondence should be addressed.
Oxygen 2022, 2(2), 130-151; https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen2020012
Submission received: 28 February 2022
/
Revised: 12 May 2022
/
Accepted: 19 May 2022
/
Published: 24 May 2022
(This article belongs to the Special Issue Review Papers in Oxygen)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Carbon monoxide (CO) is one of the most common causes of inhalation poisoning worldwide. However, it is also well known that CO is produced endogenously in the heme degradation reaction catalyzed by heme oxygenase (HO) enzymes. HO catalyzes the degradation of heme to equimolar quantities of CO, iron ions (Fe2+), and biliverdin. Three oxygen molecules (O2) and the electrons provided by NADPH-dependent cytochrome P450 reductase are used in the reaction. HO enzymes comprise three distinct isozymes: the inducible form, heme oxygenase-1 (HO-1); the constitutively expressed isozyme, heme oxygenase-2 (HO-2); and heme oxygenase-3 (HO-3), which is ubiquitously expressed but possesses low catalytic activity. According to some authors, HO-3 is rather a pseudogene originating from the HO-2 transcript, and it has only been identified in rats. Therefore, cellular HO activity is provided by two major isoforms—the inducible HO-1 and the constitutively expressed HO-2. For many years, endogenously generated CO was treated as a by-product of metabolism without any serious physiological or biochemical significance, while exogenous CO was considered only as an extremely toxic gas with lethal effects. Research in recent years has proven that endogenous and exogenous CO (which may be surprising, given public perceptions) acts not only as an agent that affects many intracellular pathways, but also as a therapeutic molecule. Hence, the modulation of the HO/CO system may be one option for a potential therapeutic strategy. Another option is the administration of CO by exogenous inhalation. As alternatives to gas administration, compounds known as CO-releasing molecules (CORMs) can be administered, since they can safely release CO in the body. The aim of this article is to provide a brief overview of the physiological and biochemical properties of CO and its therapeutic potential.
1. Introduction
Carbon monoxide (chemical formula CO) is the simplest molecule of the oxocarbon family (oxocarbon is a chemical compound consisting only of carbon and oxygen atoms). The most common source of CO is the incomplete combustion of products containing hydrocarbons when the supply of oxygen is insufficient. Formally, CO is the anhydride of formic acid, but its solubility in water is only 27.6 mg/L at 25 °C. CO has a molar mass of 28.0, which makes it slightly less dense than air, which has an average molar mass of 28.8. It is an important compound in the production of many other compounds, ranging from drugs through fragrances to fuels. As a curiosity, it may be recalled that—next to molecular hydrogen—CO is the second most abundant molecule in space. For humans, CO is a toxic gas; it is often called “the silent killer” because it is an odorless, tasteless, and colorless gas and has no irritating properties and is, therefore, organoleptically undetectable by humans. Its undesirable action is related to its direct and irreversible binding to hemoglobin (which is significantly stronger than oxygen binding), the formation of carboxyhemoglobin (HbCO), and the induction of tissue hypoxia. Moreover, the toxicity of CO can be caused by binding to other heme proteins, i.e., cytochrome c oxidase or myoglobin. Each of the Hb heme groups can bind one CO molecule, but oxygen (O2) and CO molecules cannot bind to the same heme simultaneously. The affinity of CO for Hb is 200–250 times greater than the affinity of O2 for Hb. The binding of CO to Hb is strong but reversible, while HbCO dissociation is 10 times slower than the dissociation of oxyhemoglobin (HbO2). Staying in an atmosphere containing 0.1% CO for 1 h leads to the binding of CO by almost half of the Hb heme groups in erythrocytes, which usually results in death.
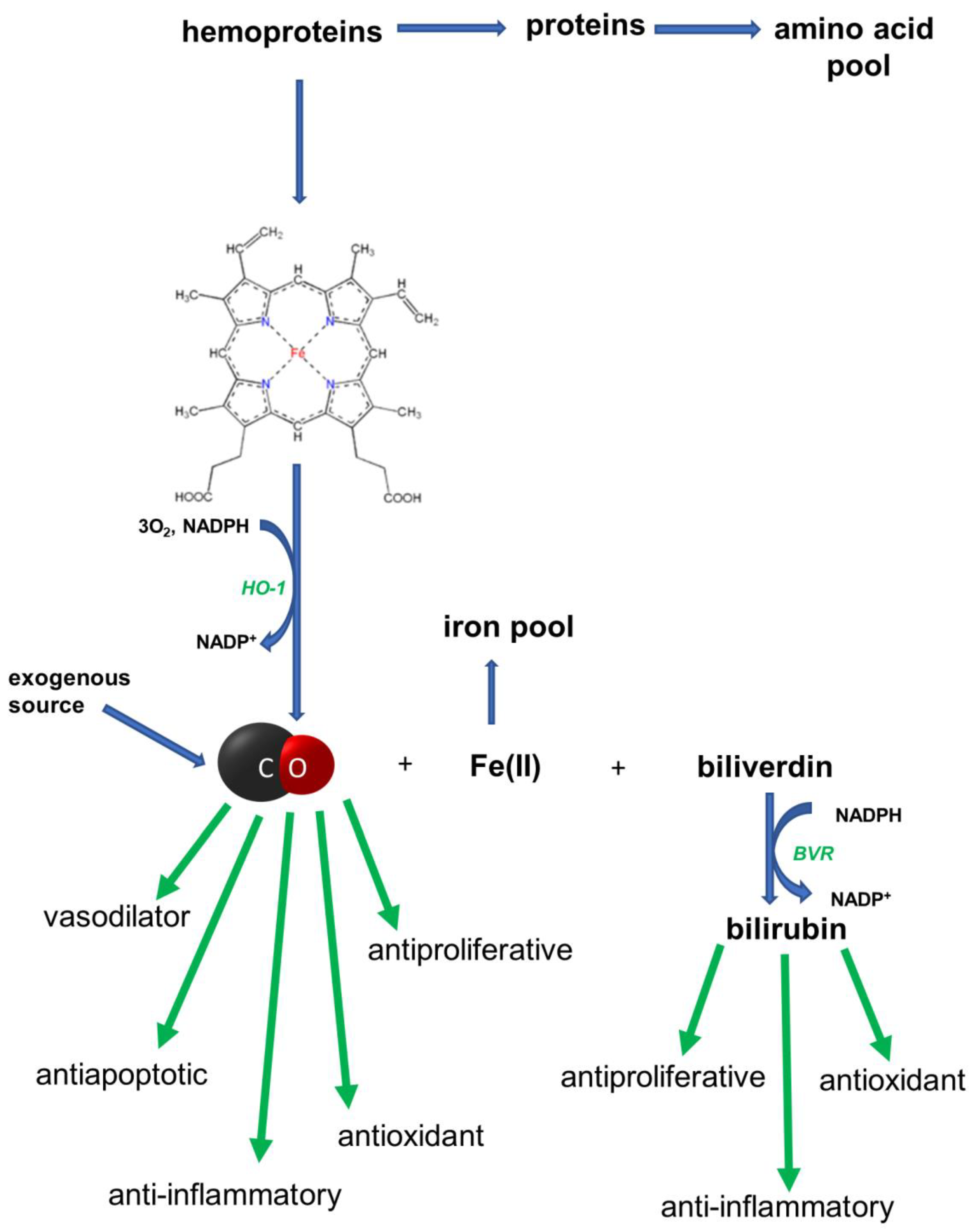
In 1952, it was shown that the degradation of Hb in vivo led to CO production [1]. It is now known that the synthesis of CO in the human body is catalyzed by the microsomal heme oxygenase (HO) system of reticuloendothelial cells and is associated with the catabolism of heme to biliverdin. This process requires O2 and NADPH. HO catalyzes the cleavage of the α-methene bridge, which links the two pyrrole residues that contain vinyl substituents. The α-methene carbon is converted quantitatively to CO. The oxygen present in the CO and in the newly derivatized lactam rings of biliverdin comes directly from O2. The stoichiometry of this reaction requires three moles of O2 for each ring cleavage. In short, the heme oxygenase in the presence of O2 and NADPH catalyzes three successive oxygenations, which results in the opening of the porphyrin ring (converting the cyclic heme to linear biliverdin), the production of CO, and the release of ferrous ion (Fe2+). Biliverdin is converted to bilirubin by biliverdin reductase (BVR). After conjugation with glucuronic acid, bilirubin is excreted into bile. The bile pigments obtained from heme, biliverdin, and bilirubin have a strong antioxidant effect. Iron is used up to synthesize other hemoproteins, or it is stored in complexes with a protein, e.g., ferritin. The formation of CO from heme and its biological role is presented in Figure 1.
According to the literature and textbooks, reactions catalyzed by HO are the only endogenous sources of CO in humans. However, research indicates that, in pathological conditions, lipid peroxidation, the photo-oxidation of organic compounds, and the metabolic activity of intestinal bacteria become additional sources of CO [2].
It is known that HO is present in mammals in three active isoforms, HO-1, HO-2, and HO-3, and the HO-4 isoform has been identified in plants. The isoforms of HO are encoded by various genes. The isoform HO-1, with a molecular weight of 32 kDa, also known as heat-shock protein 32 (hsp32), is an inducible isoform whose expression can be upregulated by different stress conditions and can be activated by multiple stimuli, including high heme concentration, hypoxia, nitric oxide, cytokines, heavy metals, radiation ultraviolet (UV), lipopolysaccharide (LPS), and oxidized low-density lipoprotein (LDL) [3,4,5]. In addition, transcription factors (AP-1, NF-κB) and the kinases cooperating with them (ERK, JUN, p38MAPK) are involved in the expression of the HO-1 isoform [6,7]. The induction of HO-1 can lead to a hundredfold increase in the enzyme’s activity [6,7,8,9].
HO-2 is the constitutive form of the enzyme, with a molecular weight of 36 kDa. This enzyme shows its greatest activity in the brain. HO-3 is most abundant in the liver, prostate, and kidneys. However, there is evidence that HO-3 is rather a pseudogene originating in the HO-2 transcript, and it has only been identified in rats [10,11].
Thus, cellular HO activity is provided by two major isoforms: inducible HO-1 and constitutively expressed HO-2. These isoforms are products of separate genes and differ in their biochemical properties [12,13]. Among the different HO isoforms, the HO-1 isoform kindles the greatest interest among researchers [14,15].
The rate of CO formation in the body is about 20 μmol/h, which is approximately 500 μmol per day, and corresponds to 12 mL per volume. Under physiological conditions, CO occurs in tissues in nanomolar concentrations [16,17]. Research data indicate that only 1% of formed CO is oxidized to CO2 [18]. Thus, it can be assumed that CO is practically not metabolized. It binds to Hb, and then it is excreted through the respiratory system. Thus, the measurement of CO in exhaled air is a good measure of heme destruction.
For many years, however, endogenously generated CO was treated as a by-product of metabolism without any serious physiological significance. The physiological properties of CO were not considered until the regulatory properties of nitric oxide (NO) became apparent in the 1980s.
Contemporary research shows that endogenous CO and HO, the enzyme that produces it, are important actors in the body’s metabolism. In light of the accumulated research, CO also seems to be a promising drug in the treatment of many diseases. Therefore, there is a rapid increase in the research on a means to increase the activity of HO on the one hand and, on the other, on the possibility of using exogenous CO donors as drugs.
In this review, we briefly summarize the current knowledge about the biological and pharmacological properties of CO.
2. CO in the Circulatory System
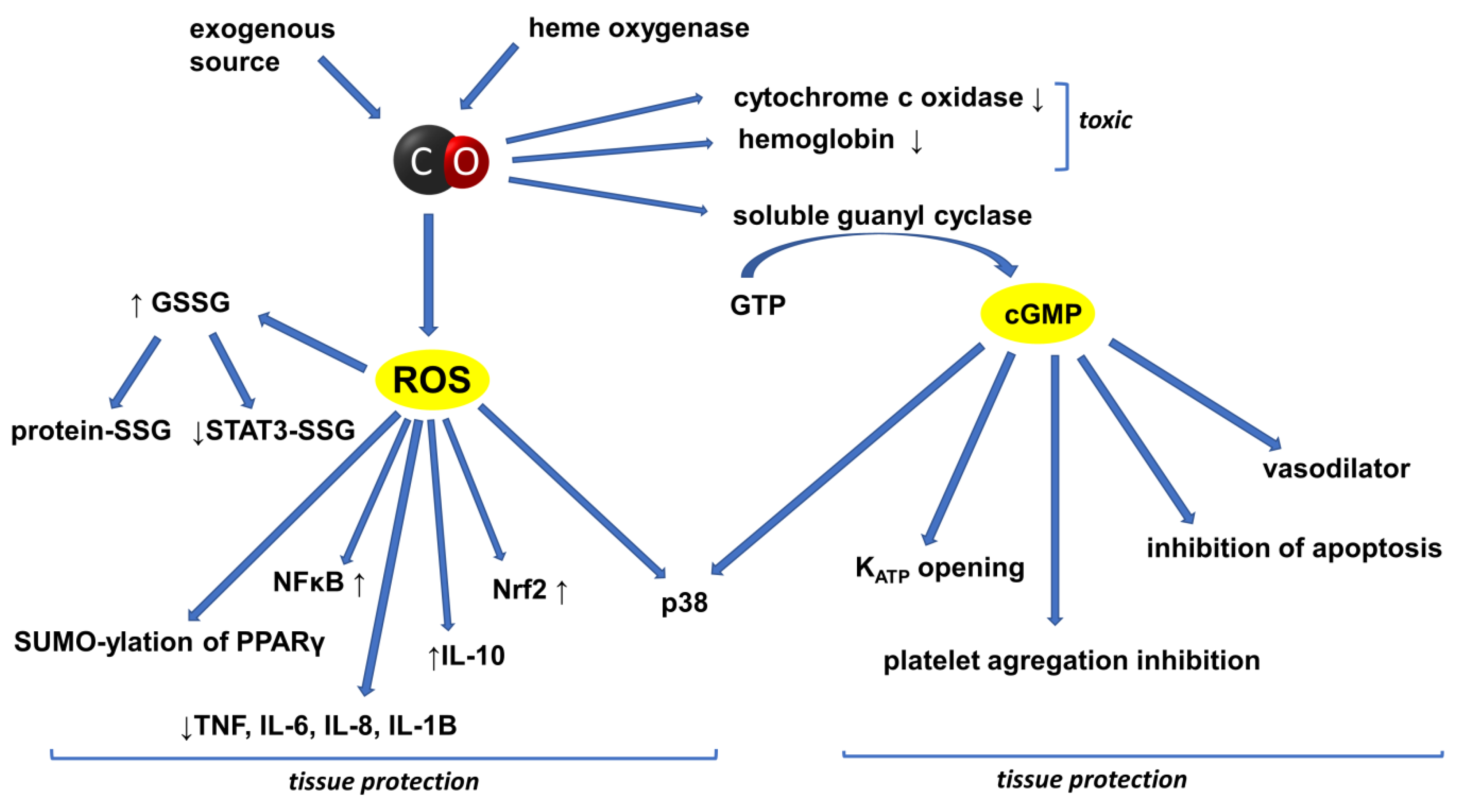
According to research data, the main activity of CO in the circulatory system is related to its vasodilating effects. The results reported by Wang et al. indicate that CO may activate the soluble guanylate cyclase (sGC; EC 4.6.1.2), as demonstrated for nitric oxide (NO) [19]. This enzyme is a heme protein that catalyzes the conversion of guanosine 5′-triphosphate (GTP) to guanosine 3′,5′-cyclic monophosphate (cGMP). On the other hand, many scientists believe that although there is a parallelism between the biological actions and functions of the CO- and NO-generating systems and their regulation, the mechanism of the biological activity of NO and CO may differ [12]. A simple comparison of the physicochemical properties of both gases is sufficient to reveal the similarities and differences between them. Both CO and NO are diatomic molecules. CO has ten valence electrons, so it is a stable molecule. NO has eleven valence electrons, so it has one unpaired electron; therefore, it is a highly reactive free radical. It is known that the presence of the reduced Fe2+ in heme moieties is one major prerequisite for the NO-induced activation of sGC. Under normal conditions, the Fe2+ ion in heme molecules is bound by four pyrrole rings and the imidazole group of one of the histidine residues. After forming a coordination bond with the Fe2+ ion, NO breaks the bond with the imidazole group and the Fe2+ ion changes its position—it is “projected” above the plane of the heme molecule. In the case of CO, the heme–histidine bond does not break, so the Fe2+ ion does not change its position, but stays in the plane of the heme molecule. It is known that NO is a potent activator of sCG and increases cGMP production in vitro ≈ 130 fold, whereas the increase in cGMP induced by CO is ≈4.4 fold. Therefore, the cleavage of the heme–histidine bond is the molecular switch that leads to an increase of over a hundredfold in the activity of sGC [20,21,22,23,24].
The activation of sGC causes an increase in the intracellular concentration of cGMP, which activates the cGMP-dependent protein kinase. Intracellular calcium levels decrease and smooth muscles relax. Thus, the vasodilatory effect of CO is weaker than that of NO. Nonetheless, CO may be of particular importance under oxidative stress conditions, because, as already mentioned, it is a stable molecule and, unlike NO, it does not react with other free radicals. However, in the case of NO, its reaction with the superoxide radical anion (O2•−) leads to formation of toxic peroxynitrite (ONO−). Under oxidative stress, this reaction leads to the rapid inactivation of NO, which limits its vasodilating effect, while the activity of CO is not attenuated under these conditions [25].
The cGMP-dependent processes in which CO is involved are also implicated—in addition to vasodilation—in the inhibition of platelet aggregation and neurotransmission [26,27,28,29]. Studies show that the vasodilating effect of CO is also associated with the activation of the potassium channels (KCa) of the smooth vascular muscles. This is because CO directly activates KCa, leading to vascular relaxation through Ca desensitization [24,30].
Pharmacological research focuses on the search for compounds that either increase the activity of HO or are safe donors of CO. In this respect, tin (II) chloride, also known as stannous chloride (SnCl2), which has been reported to increase the expression of HO-1, shows positive results. Escalante et al. reported that the short-term treatment of spontaneously hypertensive rats (SHRs) and normotensive Wistar–Kyoto rats (WKY) with SnCl2 increased the expression of HO-1 and restored blood pressure (BP) to normal in young, but not in adult, SHRs. The authors also indicated that SnCl2 did not affect the BP of either young or adult WKY rats. Their study also demonstrated that SnCl2 administration—which started at 5 weeks of age and continued for the next 10 weeks—prevented the development of hypertension in the SHRs. It should be noted that the suspension of SnCl2 treatment after 8 weeks did not result in the return of BP to hypertensive levels in the SHRs [31]. It is known that hemin (ferric chloride heme) is a natural inducer of the HO-1 gene. Hemin is an iron-containing porphyrin with chlorine that can be formed from a heme group, which is a part of the structure of hemoglobin and other hemoproteins. Research data indicated that the administration of hemin caused a drop in blood pressure in young SHRs [32]. It has also been shown that HO-1 improves glucose metabolism in essential hypertension in SHRs. Ndisang and Mishra demonstrated that the administration of hemin improved insulin signaling and decreased blood levels of total cholesterol and triglycerides in SHRs. These effects were accompanied by an increased activity of HO-1 and increased levels of cGMP. The authors also pointed out that the administration of chromium-mesoporphyrin (CrMP) to rats reduced the effect of the hemin [33]. Studies in the murine venous thrombosis model induced by the ligation of the inferior vena cava (IVCL) indicated that the administration of hemin significantly decreased clot sizes [34]. It is worth mentioning at this point that heme is now recognized as a procoagulant. Sparkenbaugh et al. indicated that an excess of heme promoted systemic thrombin generation in mice that was dependent on the extrinsic coagulation pathway, but not the intrinsic pathway. The authors also demonstrated that hemopexin partially attenuated thrombin generation in sickle-cell mice, suggesting that excess heme influences coagulation and might contribute in part to the thrombotic complications of sickle-cell disease [35]. Hemopexin is the plasma glycoprotein with the highest binding affinity for heme. Therefore, it is a scavenger of labile heme. Research by other authors also provided evidence for the antithrombotic effect of hemopexin [34,36].
An important role in the history of the research on the biological role of CO is thought to have been played by studies carried out by Motterlini et al., published in 2002. These authors examined the characterization of transition-metal carbonyls as potential CO-releasing molecules (CO-RMs or CORMS). These compounds contain a transition metal, such as cobalt (Co), iron (Fe), manganese (Mn), nickel (Ni), or ruthenium (Ru), surrounded by carbonyl (CO) groups as coordinated ligands. The results obtained by the authors showed that dimanganese decacarbonyl—Mn2(CO)10 and tricarbonyldichlororuthenium (II) dimer—[Ru(CO)3Cl2]2 released CO under appropriate conditions and could elicit specific vascular activities that were reminiscent of those mediated by the HO-1/CO pathway [37]. It was the beginning of a period of highly intensive research on CORMs.
In the aforementioned murine venous thrombosis model, IVCL, it was demonstrated that the administration of tricarbonylchloro(glycinato)ruthenium (II) (C5H4ClNO5Ru, CORM-3) reduced the clot weight, clot length, and clot weight-to-length ratio at day 2 after IVCL compared with inactivated CORM-3, which does not release CO (iCORM-3) [34]. Evidence for the antithrombotic activity of CORM-3 was also provided by the studies of Kramkowski et al., who suggested that the antithrombotic activity of CORM-3 was associated with the inhibition of platelet activation and the stimulation of the fibrinolysis process by inhibiting the activity of the plasminogen activator inhibitor-1 (PAI-1) [38]. It has also been shown that CORM-3 functions as a vasodilator through mechanisms that involve sGC and KCa activation [39]. It is necessary to add that both CORM-1 and CORM-2 are antihypertensive and vasodilatory, and reduce coronary vasoconstriction [37]. CORM-3 is completely soluble in water, unlike CORM 1 and CORM-2, which are soluble in organic solvents. It rapidly liberates CO in vitro, ex vivo, and in vivo in various animal models [40,41,42]. Many authors reported that CORM-3 protected myocardial tissues against ischemia-reperfusion injury and prolonged the survival of cardiac allografts in mice [41,42]. Hence, according to the aforementioned reports, numerous studies have confirmed that transition-metal carbonyls meet the criteria of biologically and pharmacologically active CO carriers.
In 2001, Alberto et al. prepared and described a new water-soluble CO releaser—CORM-A1. It is sodium boranocarbonate (Na2[H3BCO2] that does not contain a transition metal but a boron atom, to which a carboxyl group (COO−) is covalently bound. CORM-A1 releases CO at a slower rate compared with CORM-3 [43]. In 2005, Motterlini et al. examined the biochemical reactivity and pharmacological effects of CORM-A1 and discussed how its “slow” CO-releaser properties could be exploited in the therapeutic context. Comparing the properties of CORM-3 and CORM-A1, the authors noticed that CORM-3 is a compound that releases CO very rapidly (“fast releaser”) in biological systems and, therefore, it can be used in therapeutic applications in which CO is needed as a high-speed signaling mediator. On the other hand, CORM-A1, which releases CO with slow kinetics (“slow releaser”) may be advantageous in the treatment of chronic conditions that require CO to be delivered in a carefully controlled manner [44]. It has recently been shown that CORM-A1 inhibited platelet aggregation through the inhibition of platelet energy metabolism [45]. A study in which the authors evaluated efficacy and safety of CORM-A1 in reducing the infarct size in a clinically relevant porcine model of acute myocardial infarction (AMI) is also worth noting. The obtained results demonstrated that CORM-A1-treated animals showed a significant reduction in absolute infarct area and infarct area corrected for the area at risk; furthermore, in the CORM-A1 treated group, the biochemical markers of myocardial injury also tended to be lower, and left ventricular function tended to recover better compared to the sodium-borate control group. Moreover, CORM-A1 reduced infarct size and improved left ventricular remodeling and the function of reperfused myocardial infarction by a reduction in inflammation [46].
In conclusion, there is a large body of evidence that CO plays an important role in the circulatory system, suggesting that this gaseous transmitter may become a promising target for new treatment strategies for cardiovascular diseases.
3. The Effect of CO on the Central Nervous System
The main group of brain diseases comprises mental diseases. Another group comprises neurodegenerative diseases. Other brain diseases include stroke, traumatic brain injury, and brain tumors. It is estimated that around 0.5 billion people worldwide are affected by brain diseases; therefore, the scale of the problem is extensive. Moreover, contemporary medicine does not have effective drugs, unlike, for example, for cardiovascular diseases, which in many cases can be controlled and effectively treated.
It is now known that the neurotoxic activity of microglia is seen as one of the contributing factors in the development of brain diseases. It must be remembered that the microglial cells are derived from monocytes and are macrophages of the central nervous system (CNS). Microglia cells, which make up about 5–10% of the population of non-neuronal cells in the brain, are normally found in a resting form. The main physiological functions of microglia involve monitoring the tissue microenvironment, the removal of dying neurons, and reactions as a result of the presence of foreign antigens (for reviews, see [47,48]). In the pathological conditions of various etiologies, microglia cells are activated and, in an uncontrolled way, produce pro-inflammatory and toxic cytokines, as well as numerous inflammatory mediators that induce the death of other cells, including neuronal cells, aggravating the damage to the CNS.
Dias-Pedroso et al. showed that ALF-826A, a novel molybdenum-based CORM, inhibited the LPS-induced inflammation in BV2 microglial cells. The neuroprotective effect of CO released from the ALF-826A was related to the fact that CO inhibited: (1) secretion of TNF-α; (2) the activity of inducible nitric oxide synthase (iNOS); and (3) the production of NO. The study also demonstrated that the CO stimulated the release of interleukin-10 (IL-10), is an anti-inflammatory cytokine that inhibits the production of pro-inflammatory cytokines, such as interferon-gamma, IL-2, IL-3, TNF-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF). It is known that GM-CSF, by acting on natural killer cells (NK), enhances their cytotoxicity. It is also interesting that the authors demonstrated for the first time that the anti-inflammatory activity of CO in microglia depended on neuroglobin (Ngb), which is a hemoprotein, like Hb [49]. Other studies conducted using the microglial culture have shown that the CO released from ALF-826 molecules decreased LPS-induced inflammation through the inhibition of (1) the expression of integrin CD11b, which is known as a pro-inflammatory molecule, (2) the production of ROS, and (3) the secretion of TNF-α, as well as (4) reducing the level of nitrites, which are pro-inflammatory. The authors also demonstrated for the first time that CO improved microglial neurotrophism under non-inflammatory conditions and had an important role in the communication between microglia cells and neurons. In our opinion, this is an important observation, pointing to the previously unknown physiological role of CO in the CNS [50].
The group of the most serious human diseases of the 21st century, which still cannot be effectively prevented or cured, also includes cerebral ischemia/reperfusion injury (IRI). Zeynalov and Doré, by examining mice with transient occlusion of the cerebral middle artery (MCAo), showed that the inhalation of CO at concentrations of 125 or 250 ppm by animals significantly improved the neurological deficit scores. The authors also showed that in animals inhaling CO at a concentration of 125 ppm, the total infarct volume was reduced by ca. 32%, and that at a CO concentration of 250 ppm, this reduction was over 60% [51]. Panahian et al. demonstrated that the stroke volume in transgenic mice overexpressing HO-1 with permanent occlusion of the MCAo was nearly 45% lower compared to the stroke volume in control mice. Therefore, further studies confirming the effectiveness of HO-1 activators in clinical therapy for ischemic stroke patients are needed [52]. What is the mechanism of protective action of the CO/HO system on neurons during cerebral IRI? Many authors emphasize that the CO produced by HOs protects neurons against apoptosis. In the study cited above, the authors showed an increase in the expression of bcl-2 protein (anti-apoptotic protein) in neurons in transgenic HO-1-overexpressing mice. It is worth noting that anti-apoptotic proteins from the Bcl-2 family inhibit the release of cytochrome c and the formation of apoptosomes. Moreover, the same authors demonstrated the inhibition of the nuclear localization of the p53 protein in the neurons of transgenic HO-1-overexpressing mice [52]. Vieira et al., in studies using primary cultures of cerebellar granule cells, demonstrated that the endogenous CO was crucial for the anti-apoptotic activity of HO. In particular, the authors showed that adding glutamate and zinc protoporphyrin IX (which is an inhibitor of HO) to the cell culture led to an increase in cell-death levels. However, the pretreatment of cell cultures with CO reversed the harmful effects triggered by HO inhibition (especially at glutamate concentrations of 20 and 30 μM). This means that the HO activity was restored by exogenous CO [53]. In our opinion, this is important information because the anti-apoptotic activity of HO is commonly associated with the antioxidant activity of bilirubin. In turn, studies by Biermann et al. carried out on rats showed that CO inhalation (250 ppm) before IRI reduced inflammation and apoptosis in retinal ganglion cells (RGC). The results obtained by these authors showed that seven days after IRI, RGC death decreased by 52% in the CO-inhaling group compared to the control group breathing the ambient air. Additionally, the analysis of the proteins in retinal tissues showed that CO inhalation resulted, among other effects, in attenuated TNF-alpha protein expression and caspase-3 activity [54]. The other CO-releasing molecule, ALF-186, also exerts neuroprotective effects by reducing the expression of inflammatory proteins, such as NF-κB, TNF-α, and IL-6, via the sCG in RGCs after IRI [55].
Based on the literature data, it is known that molecules with the ability to activate ATP-sensitive potassium (KATP) channels have a protective effect on neurons [56] The studies of Vieira et al. indicated that CO was able to activate KATP channels in neurons. The authors even emphasized that the anti-apoptotic activity of CO would not be possible without the activation of these channels [53].
The question is how CO influences the production of ROS, because it seems that, to date, the literature is inconsistent with regard to this problem. As is often the case, the truth lies somewhere in the middle: on the one hand, CO has antioxidant properties; on the other, a low level of ROS is essential for CO-induced cytoprotection [53,57,58,59].
Lin et al., in research conducted on rat-brain astrocytes (RBA-1), indicated that CORM3 activated the Keap1-Nrf2 (Kelch-like ECH-associated protein 1—nuclear factor-erythroid-2-related factor 2) signaling pathway, which, consequently, led to an increase in the expression of the HO-1 gene in these cells (Figure 2) [60].
Yabluchanskiy et al. proved that CORM3 reduced inflammation and brain damage in an animal model of hemorrhagic stroke (HS). The authors also observed that the animals receiving CORM3 had a significantly lower level of plasma tumor necrosis factor-α (TNF-α). It also seems important that the reduction in brain damage observed in the rats receiving CORM3 was correlated with the inhibition of microglial activity. On this basis, the authors concluded that pharmacological treatment with microglial inhibitors should be considered in HS patients [61]. Similar results were obtained by Chen et al. in studies conducted using an intracerebral hemorrhage (ICH) rat model. The authors showed that the microglia participated in post-ICH inflammation. The results obtained by the authors showed that CORM-2 attenuated ICH-mediated inflammation by inhibiting microglial activation, which—according to the authors—might be related to the NF-κB signaling pathway [62]. Yang et al. observed, in turn, that CO reduces the activity of the signal transducer and activator of transcription 3 (STAT3) via S-glutathionylation [63].
Currently, an increasing body of data also indicates the role of microglia in the pathogenesis of Alzheimer’s disease (AD). It is suggested that in the initial stage of AD development, microglia have a protective function, cleansing the brain of excess amyloid-beta (βA), whereas prolonged contact of microglia with βA leads to the excessive production of pro-inflammatory cytokines, which, in the later stage of AD development, have a neurotoxic effect [64]. Recent studies have demonstrated the ability of CORM-2 and CORM-3 to protect neurons and astrocytes from self-assembled Aβ, particularly Aβ1–42. Moreover, CORM-3 showed the ability to stabilize Aβ in its soluble, unordered conformation, thereby preventing its misfolding and aggregation [65]. Moreover, Hettiarachchi et al. demonstrated that Aβ1–42-mediated neuronal cell toxicity could be diminished by transient HO-1 overexpression or by CORM-2 [66]. In light of the cited data, it is also possible that the beneficial effect of CO in the course of AD is related to its inhibitory effect on the activity of microglia.
To conclude this section, it is worth adding that the results of studies by INO Therapeutics LLC Company, discussed in Mahan’s review, indicated that inhaled CO was safe and tolerable in humans [67]. Based on the database of privately and publicly funded clinical studies conducted around the world (www.clinicaltrials.gov, accessed on 29 February 2000), it is known that clinical trials using inhaled CO were performed on many different diseases, while its clinical application in CNS disorders remains unexplored; however, anecdotal data suggest that smokers have a very low incidence of Alzheimer’s disease [68].
Multiple sclerosis (MS) is a chronic, incurable disease of the CNS. The disease begins mainly in young adults with the appearance of clinically isolated syndrome (CIS), which is the result of inflammation and demyelination in one or more parts of the CNS. Symptoms may include weakness in the extremities, pain, and inflammation of the optic nerve. In this context, the results of studies showing that CO is capable of suppressing autoimmune neuroinflammation are also interesting. Some authors conducted research on the animal model of MS, which was obtained by the subcutaneous administration of myelin oligodendrocyte glycoprotein (MOG35-55) and the amino acid residue 139 to 151 of myelin proteolipid protein (PLP139-151) to mice. Hmox1+/+, Hmox1+/−, and Hmox1−/− mice were used. The authors hypothesized that the expression of “protective genes” might promote MS remission. According to the authors, the HO-1 gene (HMOX1/HO-1) is the protective gene. The results seemed to confirm this hypothesis. It was shown that the increased activity of HO-1 was associated with the inhibition of the major histocompatibility complex (MHC) class II expression in the activated antigen-presenting cells (APCs). It was also observed that the increased activity of HO-1 was correlated with the reversal of paralysis during the progression of EAE, which was associated with the inhibition of CNS demyelination. The fact that exogenous CO mimicked these effects means that CO contributes to the protective action of HO-1 [69].
4. Inflammation and Cancer: The Role of the HO/CO System
Inflammation is the physiological response of the body to tissue damage caused by injury (wounds, breakages, and bruising) and harmful chemical (toxic compounds) or biological (viruses, bacteria, pathogenic mushrooms, etc.) factors. Many studies point to a strong association between chronic inflammation and cancer (for a review, see [70]). The first stage of tumor development is the complex of interactions between the tumor that arises and the surrounding tissue, which becomes the tumor’s microenvironment. The tumor microenvironment, largely formed by cell inflammation, is an indispensable participant in the neoplastic process, promoting the proliferation, survival, and migration of cancer cells. It is currently estimated that the etiology of over 15% of all malignant neoplasms may be related to the presence of chronic inflammation caused by viral infections (for a review, see [71]). Molecules, such as selectins, interleukins, chemokines, interferons, growth factors, and their receptors are assigned important roles in the cascades of processes aimed at limiting tissue destruction, the activation of repair processes, and the restoration of systemic homeostasis. However, cancer cells “skillfully use” these signaling molecules for invasion, migration, and metastasis. This knowledge supports new anti-inflammatory therapeutic approaches to cancer development (for a review, see [72]). According to several authors, a molecule that plays a crucial role both in regulating the inflammatory response and in the process of oncogenesis and tumor progression is the nuclear factor NF-κB. The mammalian NF-κB family of proteins consists of five transcription factors, which can be divided into two groups. One group includes the proteins RelA (p65), RelB, and cRel, while the second group includes the proteins NF-κB1 (p50/p105) and NF-κB2 (p52/p100). The proteins belonging to the second group are synthesized as precursor and inactive proteins: p105 is the precursor of the p50 protein, and p100 is the precursor of the p52 protein. Both precursor proteins have a protein-binding domain that inhibits their activity. These proteins belong to the family of inhibitors called IκBs (Inhibitor of κB), which includes IκBα, IκBβ, IκBε, and Bcl-3. The best known of these is the 37 kDa protein, IκBα. There are currently over 150 known stimuli capable of activating NF-kappa B. The activation of NF-κB occurs through the canonical (classical) pathway, initiated by NF-κB1, and the non-canonical (alternative) initiation pathway, driven by NF- κB2. Different stimuli activate NF-κB via the classical and alternative signaling pathways. Active NF-κB is translocated into the nucleus where it can “carry out its program” ([73], for reviews, see [74,75,76]). What is this program like? It is commonly known that NF-κB is a transcription factor enhancing the expression of genes encoding the proteins IL-6, IL-8, and TNF-α. Current evidence also shows that the uncontrolled activation of the NF-κB signaling pathway is related to the pathogenesis of diseases such as atherosclerosis, asthma, AIDS, diabetes, inflammatory bowel disease, septic shock, rheumatoid arthritis, and many autoimmune diseases ([77,78,79]; for a review, see [80]).
Additionally, the constitutive expression and activation of NF-κB has been observed in several types of human cancer, leading to increased cell proliferation, blocking apoptosis and promoting angiogenesis, invasiveness, and metastatic capacity ([81]; for a review, see [82]).
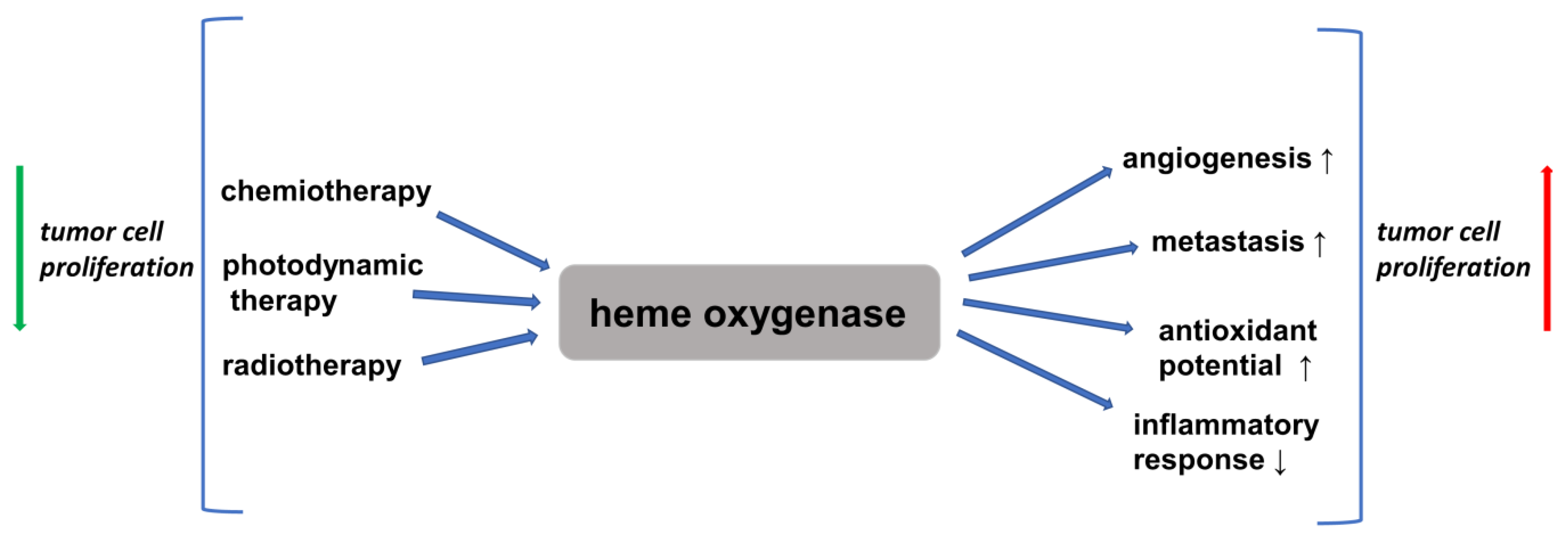
Therefore, the question arises as to whether and how the HO/CO system affects the NF-κB signaling pathway. The literature to date shows that CO activates NF-κB via ROS generation and the protein kinase B (PKB, also known as Akt kinase) pathways [59]. Moreover, HO-1 overexpression is observed in many human cancers, such as brain tumors, chronic myleoid leukemia, melanoma, pancreatic cancer, prostate cancer, and renal adenocarcinoma (for a review, see [83]). In this context, it is worth focusing on the research of Waś et al., carried out on the B16 (F10) murine melanoma cell line. Test cells were transfected with HO-1 cDNA (B16-HO-1), which resulted in HO-1 overexpression. The B16-HO-1 cells proliferated three times faster compared to the wild type (B16-WT) and, moreover, showed increased resistance to the oxidative stress induced by H2O and increased angiogenic activity compared to B16-WT cells. Mice subcutaneously injected with B16-HO-1 cancer cells survived 22 days, while animals injected with wild-type B16-WT cells survived for 38 days. This means that the cancer cells overexpressing HO-1 shortened the life of the animals by more than 40%. The authors also observed that the B16-HO-1 cells were more tightly packed in the tumors, which confirmed that the HO-1 accelerated the division of these cells. In addition, the wild-type tumors were infiltrated by a greater number of inflammatory cells, and high levels of the proinflammatory factor TNF-α were observed in the sera of the mice with these tumors, while the sera of the mice injected with the B16-HO-1 cells contained practically trace amounts of TNF, and the number of inflammatory cells in their tumor tissues was low. This means that the overexpression of HO-1 inhibits the inflammatory process, which additionally “supports” melanoma cells. A further observation made by the authors is that after the injection of the tumor cells of both lines into the tail veins of the mice, a significantly greater number of secondary tumors was found in the lungs of the animals that received the B16-HO-1 cells compared to the mice that received the wild-type tumor cells. This indicates that the overexpression of HO-1 increases the ability of cancer cells to metastasize [84]. The studies of other authors also confirm that an increase in the activity of HO-1 in cancer cells results in faster tumor growth, decreased apoptosis, and increased proliferative, angiogenic, and metastatic potential [85].
Therefore, significant literature data indicate that HO-1 activation may play a role in carcinogenesis and can potently influence the growth and metastasis of tumors. Moreover, HO-1 expression is further increased in response to therapies (for reviews, see [86,87]). However, it is believed that the cancer-cell proliferation induced by the upregulation of HO-1 is not connected with NF-κB activation [86].
Therefore, an obvious conclusion is that the inhibitors of HO-1 may have anti-cancer properties. This has been confirmed by the research of many authors. The zinc protoporphyrin (ZnPP) IX, an HO-1 inhibitor, inhibited the growth of the LL/2 murine lung-cancer cell line. The authors also conducted studies on C57BL mice inoculated with LL/2 cells. Tumors developed from inoculated LL/2 cells in nearly 90% of the control mice and in 25% of the mice that received 20 µg of (ZnPP) IX. By contrast, LL/2 tumors formed in all the mice (100%) that were administered cobalt zinc protoporphyrin (CoPP) IX, an activator of HO-1. The authors also found a significant reduction in the level of vascular endothelial growth factor (VEGF) in homogenized cancer tissues taken from the mice that received the HO-1 inhibitor (ZnPP) IX [88]. McAllister et al. conducted research using dermal microvascular endothelial cells (DMVECs) infected with Kaposi-sarcoma-associated herpes virus (KSHV). The authors demonstrated that HO-1 mRNA and protein were up-regulated in the KSHV-infected cultures, which resulted in a proliferative advantage of infected cells compared with uninfected cells. The treatment of the infected DMVECs with chromium mesoporphyrin IX (CrMP) IX), an HO-1 inhibitor, abolished this increase in cell division [89]. Recently, a novel imidazole-based inhibitor (SLV-11199) of HO-1 was developed, which was demonstrated to produce the pharmacological inhibition of HO activity in human pancreatic (PANC-1) and prostate (DU-145) cancer-cell lines. Moreover, it was shown that in PANC-1 cells, diminished HO-1 activity resulted in the down-regulation of pro-angiogenic factors, such as interleukin 8 (IL-8), which is the strongest chemotactic factor in the human body. These investigations also revealed that the treatment with SLV-11199 decreased cell migration and inhibited the expression of metalloproteinases (MMPs) [90]. It is worth recalling that in the process of cancer progression, cancer cells have to overcome many barriers, the most important of which are the basement membrane and extracellular matrix (ECM). In this process, neoplastic cells are “assisted” by the MMPs, a group of Zn2+-dependent endopeptidases involved in the degradation of basement membrane proteins and the ECM. MMP-9 is a special member of the MMP family, and it is known that the increased expression of MMP-9 demonstrated in colorectal cancer tissue is correlated to greater invasiveness and shorter patient survival times ([91]; for reviews, see [92,93,94,95]).
On the other hand, some researchers suggest that HO-1 may be seen as an anticancer agent. Yokoyama et al. indicated resistance to radiotherapy in more than 80% of patients with esophageal cancer who had low HO-1 activity in the tumor tissue [96]. Gandini et al. demonstrated that the pharmacological and/or genetic increase in the activity of HO-1 in some breast-cancer cell lines induced the apoptosis of cancer cells and reduced the rate of their migration and metastatic potential through pathways involved in the epithelial–mesenchymal transition. It has also been shown that increased levels of HO-1 are closely related with reduced tumor size and longer overall survival times for patients [97].
Phorbol esters are thought to be tumor-promoting compounds. Among them 12-O-tetradecanoylphorbol-13-acetate (TPA) is often used in many studies to activate the protein kinase C (PKC) signaling pathway. PKC is a serine/threonine kinase involved in the transduction of cellular signals in response to stimulation with growth factors. Its activity depends on the second messenger, i.e., diacylglycerol (DAG). The use of phorbol esters to activate PKC in tumor cells leads to the activation of pro-angiogenic and pro-proliferative signal transduction cascades “favorable” to tumor cells [98]. It was observed that the ability of TPA to increase the production of ROS and the activity of MMP-9 was inhibited by the overexpression of HO-1 protein in the human breast-cancer cell line, MCF-7. The use of ferric protoporphyrin (FePP) IX, an inducer of HO-1, suppressed the TPA-induced invasion and migration of MCF-7 cells, while in the presence of tin protoporphyrin (SnPP) IX, an inhibitor of HO-1, the activity of tumor cells was restored [99]. It has also been observed that the focal HO-1 expression 1 in colorectal cancer samples correlated with a reduced number of cancer cells in the blood and lymph vessels. The presence of cancer cells in vascular or lymphatic spaces is defined as lymphovascular invasion (LVI). LVI is a key factor in cancer’s progression and its ability to spread throughout the body. Therefore, the authors showed that the colonic expression of HO-1 was associated with better long-term survival rates in patients with colorectal cancer [100]. The aim of the research by Tsuji et al. was to investigate neoplastic cells from patients with oral squamous cell carcinoma (SCC) and to examine the correlation between the expression of HO-21 in these cells and the clinical condition of the patients. The obtained results showed that high expressions of HO-1 were found in the cells taken from the patients with no nodal metastases. Furthermore, cancer cells with high expressions of HO-1 were well differentiated [101].
These conflicting reports on the effect of HO-1 on tumor cell metabolism indicate that our knowledge on this subject remains limited [102]. In answer to this question, a study entitled, ‘Heme oxygenase-1 in tumors: is it a false friend?’ Józkowicz et al. state: “…HO-1 can be considered a ‘friend’ protecting healthy tissues from induction of some types of cancers. However, if, despite this protection, the disease starts to develop, HO-1 turns into a ‘false friend’ as it will protect the tumor cells, improving their survival and resistance to treatments” [83].
In light of the information provided above, questions arise about CO, which is one of products of the reaction catalyzed by HO enzymes. Is CO a pro-inflammatory or anti-inflammatory agent? Is it a pro-cancerous or antitumor factor?
Many studies have shown that CO is anti-inflammatory. Otterbein et al. have reported that CO inhibits the expression of the lipopolysaccharide-induced pro-inflammatory cytokines TNF-α and IL-1β and macrophage inflammatory protein-1β (MIP-1β, CCL4), and that it stimulates the production of IL-10, the anti-inflammatory cytokine. According to the authors, the anti-inflammatory activity of CO is related to the p38-mitogen-activated protein kinase signaling pathway (p38 MAP, p38 MAPK), independently of cGMP and NO activity [103]. In studies conducted with the human bronchial epithelial cell line, 16HBE14o, it was shown that the cationic peptide poly-l-arginine (PLA) induced bronchial epithelial damage, while HO-1 induction or CORMs suppressed PLA-induced IL-6 and IL-8 release [104]. Tsui et al. demonstrated that HO-1 induced the expression of the anti-apoptotic protein, Bcl-xL, in human hepatocytes by activating the p38 MAPK signaling pathway. Moreover, CO has been shown to significantly increase ATP levels in hepatocytes, which, in turn, increases the activation of the p38 MAPK signaling pathway [105]. Haschemi et al. showed that the anti-inflammatory effect of carbon monoxide was related to the process of the peroxisome proliferator-activated receptor-γ (PPARγ) sumoylation and the blockage of the expression of the uncoupling protein 2(UCP2) gene in macrophages [106]. Sumoylation is one of the post-translational modifications of proteins, consisting in the covalent, reversible attachment of the small ubiquitin-like modifier protein (SUMO) to the ε-amino group of lysine in the target protein. The participation of PPARγ in the mechanism of the anti-inflammatory activity of CO was also noted by Bilban et al. [58]. The mechanisms of the anti-inflammatory action of CO—indicated by the authors—also include blocking the aforementioned signal pathway involving the STAT 3 protein, which is a key transcription factor in the human body. Shin et al. demonstrated that CO inhibited the expression of hepcidin during inflammation through a mechanism related to the inhibition of STAT3. Hepcidin is a hepatic hormone that inhibits iron absorption and release from cells. Hepcidin is also an acute-phase protein because its concentration is enhanced during inflammation. Its expression increases under the influence of, inter alia, the pro-inflammatory cytokine IL-6 though the activation of STAT-3 [107]. In turn, studies conducted on bovine aortic endothelial cells (BAECs) have shown that CORMs increase the level of ROS and oxidized glutathione (GSGG). The GSSG reacts with STAT3 protein, yielding a mixed disulfide STAT3-S-S-glutathione. This process, called S-glutathionylation, is seen as one of the forms of reversible, covalent protein modification of regulatory importance. Therefore, under the influence of the CO released from CORMS, the activity of the STAT3 protein via S-glutathionylation is inhibited [63].
Our next question is whether CO is a pro-cancerous or antitumor factor. As in the case of HO-1, there is no clear answer. Currently, there are studies showing both the carcinogenic and anticancer effects of CO (for a review, see [108]). Research by Fang et al. showed that CORM-2 could increase tumor blood flow, thereby exerting a proangiogenic effect on solid tumors [109]. Other papers report similar results, according to which CO could induce vascular endothelial growth factor (VEGF) expression, which stimulates the formation of blood vessels ([110] for a review, see [111]). This means that CO has a positive effect on the angiogenesis process. However, there are also reports, which, by contrast, describe an antiangiogenic effect of HO-1 and CO on cancer cells [112,113,114]. An in vitro study performed on human pancreatic cancer cells (CAPAN-2, BxPc3, and PaTu-8902) treated with CORM-2 or exposed to CO gas was the first to show that CO inhibited tumor proliferation and reduced microvascular density in human pancreatic cancer cells. Moreover, the authors observed a substantial decrease in Akt (protein kinase B) phosphorylation (a significant contributor to cancer neovascularization) by 30–50% in CO-treated cancer cells. The authors also demonstrated, in vivo, using an animal pancreatic cancer model, that CORM-2 treatment led to an increase in the survival rate of animals [113]. Jackson et al. described the synthesis of a new stable iron carbonyl complex [FeII(CO)(N4Py)](ClO4)2 that, upon irradiation with UV light with a wavelength of 365 nm, released CO. It was shown that this compound was active against PC-3 prostate cancer cells [114]. It is also worth noting that there are studies in which a series of CORMs based on carbohydrates was synthesized and evaluated. The test results showed that all the complexes displayed anticancer activity [115,116]. Furthermore, it is noteworthy that there are studies comparing the risk of developing breast cancer between a group of female patients diagnosed with carbon monoxide poisoning (COP) over the period between 2002 and 2009, from the from the Nationwide Poisoning Database of Taiwan, with a group of female patients without COP from the National Health Insurance Research Database. The authors followed up the two cohorts until the end of 2014 and compared their risk of developing breast cancer. The obtained results showed that the participants with COP were at a lower risk of developing breast cancer than those without COP (0.7% vs. 1.0%, p < 0.001) [117].
Since different studies report conflicting results regarding the impact of CO on cancer cells, which conclusion should be drawn? In our opinion, CO has either a carcinogenic or an antitumoral effect, depending on the type of cell involved. This means that the clinical application of CO as a drug in cancer therapy requires further, highly intensive research over time. It is also possible that CO will never be introduced into practical clinical medicine as a drug for the treatment of neoplastic diseases.
5. The Effect of CO on Metabolic Processes
In recent years, we have seen growing evidence that CO is not only a signaling molecule, but also has a direct impact on the metabolic processes of cells. In order to acquaint readers with this issue, we would like to present some examples of research on this subject.
It was shown that the precise delivery of CO to isolated heart mitochondria using CORM-3 uncouples respiration. The uncoupling effect mediated by CORM-3 resulted in increased basal respiration and decreased mitochondrial membrane potential (ψ) in the absence of ADP. This CO-induced uncoupling effect was reversed by malonate, a well-known inhibitor of complex II, and by the inhibitors of uncoupling proteins or adenine nucleotide transporters. This suggests a partial role for complex II of the respiratory chain in the uncoupling effect mediated by CO. The authors also showed that the addition of CORM-3 at concentrations of 1–20 μM significantly increased the mitochondrial oxygen consumption rate (state 2 respiration) in a concentration-dependent manner. By contrast, CORM-3 at a concentration of 100 μM suppressed ADP-dependent respiration through the inhibition of cytochrome c oxidase. The uncoupling effect mediated by CORM-3 was inhibited in the presence of the CO scavenger myoglobin [118].
In 2014, Long et al. suggested a molecular mechanism through which CO could induce mitochondrial uncoupling. The authors showed that the blockade of the dicarboxylate carrier (DIC) (which functions to export malate from the mitochondria in exchange for inorganic phosphate) decreased the effects induced by CORM-3. However, the direct inhibition of the phosphate carrier (PiC, a mitochondrial solute carrier protein, which delivers phosphate, a key substrate of oxidative phosphorylation, across the inner mitochondrial membrane) with N-ethylmaleimide (NEM, an organic compound that is derived from maleic acid) completely abolished the effects of the CORM-3. Finally, the results obtained by the authors indicate that CORM-3 activates the PiC, leading to an increase in phosphate and proton transport inside the mitochondria, both of which could contribute to the non-classical uncoupling effect mediated by CORM-3 [119].
It is known that plasma-membrane large-conductance calcium-regulated potassium channels (BKCa) contain a heme-binding domain [120]. Other experimental evidence suggests that CO induces the uncoupling of mitochondrial respiration via the activation of mitoBKCa channels [121].
Glycolysis is a metabolic pathway that converts glucose (C6H12O6, Glu) into pyruvic acid (CH3COCOOH; PA). This pathway is employed by all tissues for the oxidation of Glu to provide energy in the form of ATP and its intermediates for other metabolic pathways. Phosphofructokinase-1 (PFK-1) catalyzes an important, “committed”, step in glycolysis, namely, the conversion of fructose 6-phosphate and ATP to fructose 1,6-bisphosphate and ADP. This irreversible phosphorylation reaction catalyzed by PFK-1 is the key control point and the rate-limiting step in glycolysis. Glycolytic flux is correlated with the levels of one of the most potent activators of PFK-1, i.e., fructose-2,6-bisphosphate (F-2,6-BP). F-2,6-BP is synthesized and degraded by a family of bifunctional enzymes, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB) [122]. The conducted research allowed the identification of phosphofructokinase/fructose bisphosphatase type-3 (PFKFB3), which is a protein with methylated arginine (Arg) residues in the polypeptide chain. The methyl groups in this reaction are derived from S-adenosyl methionine (SAM), which is produced from adenosine triphosphate (ATP) and methionine (Met) by methionine adenosyltransferase. It has been shown that Arg-methylated PFKFB3 activates glycolytic flux by increasing F-2,6-BP, while its unmethylated form accelerates the pentose phosphate pathway (PPP), which benefits the NADPH/GSH system, which “fights” oxidative stress. The results of some studies provide the first evidence for the crucial role of the site-specific asymmetric dimethylarginine modification of the carbohydrate-metabolizing enzyme in modulating the directional biotransformation of glucose towards PPP in cells [123]. In the same study, the authors showed that CO was the factor that reduced the methylation of PFKFB3. The loss of PFKFB3 methylation depends on the inhibitory effects of CO on heme-containing cystathionine β-synthase (CBS), which is the rate-limiting enzyme determining the ratio between remethylation and the transsulfuration metabolism of sulfur amino acid (SAA). The remethylation cycle includes homocysteine (Hcy), Met, SAM, and S-adenosylhomocysteine (SAH). CBS can up- and/or down-regulate Arg methylation in proteins by regulating the cellular levels of SAM, a methyl donor, and SAH, an inhibitor of protein methyltransferases. The authors’ results showed that CBS knockdown by siRNA significantly suppressed the basal F-2,6-BP content in the cells. Moreover, the cells were rendered insensitive to the CO. Next, the authors showed that the CO treatment significantly inhibited the intracellular conversion of 15N-labelled Met to 15N-cystathionine by CBS. Therefore, with a whole series of very well designed and elegant experiments, the authors finally showed that CO modulated the glucose metabolic pathways through the post-translational mechanisms involving the methylation of PFKFB3 Arg in the cells [92]. It is worth recalling that CBS can also generate hydrogen sulfide (H2S) through reactions utilizing cysteine (Cys) and Hcy as substrates. Thus, the CO/CBS-assisted signaling system does not only serve as a switch for decreasing methylated PFKFB3, but also accounts for the mechanism of H2S/HS-mediated bioregulation [124,125].
It is worth noting that in the context of the influence of CO on metabolic pathways, currently, many researchers focus on a chemical compound called CORM-401. Its chemical formula is [Mn(CO)4(S2CNMeCH2CO2H)]. CORM-401 is a CO-releasing molecule that provides at least three moles of CO per mole of compound. The synthesis and characterization of CORM-401 are based on the research conducted by Crook et al. [126]. Using CORM-401, Kaczara et al. also showed that CO inhibited glycolysis (extracellular acidification rate, ECAR) and decreased ATP turnover [121]. Furthermore, it was shown that CORM-401 inhibited cytochrome P450-dependent monooxygenase (CYP) activity and modulated xenobiotic metabolism [127].
Currently, obesity is the most common metabolic disease, acquiring the dimensions of a global epidemic. Many studies show that obesity leads to the inflammation of adipose tissue, which maintained over time by dysfunctional adipocytes. Adipose tissue is an endocrine organ, and its secretion products, adipocytokines (adipokines), play an important role in the metabolism of the entire organism. Leptin, an adipokine first discovered in 1994, plays a fundamental role in the regulation of appetite and body weight. In general, adipocytokines are peptides, or low-molecular-weight proteins, that are involved in the regulation of appetite and satiety, energy homeostasis, and carbohydrate and fat metabolism. They also participate in the process of coagulation, angiogenesis and vascular remodeling, including the formation of atherosclerotic plaques, and regulate blood pressure. Moreover, they influence the activity of the immune and reproductive systems, as well as osteogenesis. Adipokines are active at the central, tissue, and cellular levels, showing endo-, para-, and autocrine activity. The secretion profiles of adipocytokines show specific changes in obese patients, and, therefore, it is believed that they may be the link between obesity and disorders such as insulin resistance, hypertension, metabolic syndrome, and atherosclerosis. Adipose tissue is also a source of pro-inflammatory cytokines, such as TNF-α, Il-6, IL-8, VEGF, plasminogen activator inhibitor 1 (PAI-1), resistin, and transforming growth factor ß (TGF-ß). In addition, adipose tissue generates pro-inflammatory cytokines, such as TNF-α, Il-6, IL-8, VEGF, plasminogen activator inhibitor 1 (PAI-1), resistin, and transforming growth factor ß (TGF-ß). In obesity, adipocytes are dysfunctional, and the production of pro-inflammatory cytokines increases, while the formation of protective adipokines, such as adiponectin, decreases. The reduced production of adiponectin entails the intensification of the pro-inflammatory effect of TNF-α ([128]; for reviews, see [129,130]). It has also been shown that IL-6 and TNF-α activate the inflammatory kinases, such as c-jun N-terminal kinase (JNK) and IκB kinase (IKK), which leads to both insulin resistance and ß-cell dysfunction (for reviews, see [131,132]).
Many authors suggest that CO has a beneficial effect on glucose metabolism in adipose tissue. Studies have shown that the oral administration of CORM-401 reduced weight gain and increased insulin sensitivity in high-fat-diet (HFD)-fed obese C57BL6 mice. The mechanism of action of CO released from CORM-401 proposed by the authors involves CO acting as an uncoupling factor in the respiratory chain, leading to an increase in glycolysis to maintain ATP levels. In the authors’ opinion, this can be considered as a metabolic switch, associated with an increase in the phosphorylation of the insulin signaling effector Akt, which, in turn, enhances the sensitivity of adipose tissue to insulin [133]. The beneficial effect of CO on metabolism in animal models of obesity is also confirmed by the studies by Hosick et al., which show that chronic treatment with CORM-A1 reverses diet-induced obesity, hyperglycemia, and insulin resistance in mice, which, in the authors’ opinion, results from the ability of CO to increase the metabolic rate [134]. It was also observed that the chronic administration of CORM-A1 significantly increased the level of nuclear respiratory factor 1 (Nrf1) in the epididymal fat of the tested mice. This protein is a transcription factor that activates, inter alia, some of the nuclear genes required for mitochondrial respiration. It was also found that in the epididymal fat of mice receiving CORM-A1, the level of peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1α), which plays a key role in the regulation of cellular energy metabolism, was also increased. The uncoupling protein 1 (UCP1), a protein in the form of a hydrogen ion-permeable ion channel, is another protein whose increased level in the epididymal fat of mice receiving of CORM-A1 was observed [135]. Zheng et al. demonstrated, using in vitro studies, that CO-releasing molecules reversed the leptin resistance induced by endoplasmic reticulum stress (ER). The authors also showed that CO inhalation normalized body weight and inhibited food intake in HFD-fed obese mice [136]. There are also reports indicating the influence of CO on the activity of the signal transducer and activator of transcription 3 (STAT3). Shin et al. demonstrated that CORM-2 treatment reduced IL-6-induced STAT3 phosphorylation in the HepG2 hepatoma cell line [107]. However, there are also studies in which CORM-2 has been shown to increase STAT3 phosphorylation [136]. Hosick et al. showed that inhalation of CO not only caused weight loss in HFD-fed obese mice but also prevented obesity in mice despite this diet [137]. Studies on mice also showed that CO activated the R-like endoplasmic reticulum kinase (PERK)—transcription factor 4 (ATF4) pathway through increased ROS production. The activation of the PERK-ATF4 pathway, in turn, increased the expression of the fibroblast growth factor 21 (FGF21). Thus, the authors showed that CO improved metabolic dysfunction in animals by increasing the activity of FGF21 protein [138].
It is well known that overweight and obesity are risk factors for the development of type 2 diabetes. Type 2 diabetes is the most common dysfunction of carbohydrate metabolism. The disease is a common and still unsolved health problem worldwide. According to the World Health Organization (WHO), the prevalence of diabetes is increasing dramatically: in 1980, there were about 180 million diabetes victims; in 2014, there were 422 million; and estimates for 2045 predict that the disease will affect as many as 628 million people. Due to the mass occurrence and the dramatic increase in the number of cases, it is the only non-communicable disease recognized by the WHO as an epidemic in the 21st century [139,140]. In an animal, streptozotocin-induced diabetes model, CORM-A1 was shown to reduce the activity of T helper cells (Th). Th cells contain and release cytokines. Due to the profiles of secreted cytokines, two main subclasses of Th lymphocytes are distinguished: Th1 cells, which mainly produce interferon-γ and IL-2; and Th2 cells, which produce many cytokines, including IL-4, IL-5 IL-10, and IL-13. There are also Th17 lymphocytes, which are strong inducers of autoimmune and autoinflammatory diseases. Th17s produce a number of cytokines and chemokines, including the most characteristic of type, IL-17. The authors observed a reduction in Th1 and Th17 cell responses and an increase in Th2 cell differentiation. In in vitro studies, the authors showed that CORM-A1 interfered with the cytokine-induced apoptosis of beta cells by reducing the levels of cytochrome c and caspase 3 [141].
6. Conclusions
Carbon monoxide (CO) is an odorless, tasteless, and colorless gas that is generated by heme oxygenase enzymes (HOs). As with nitric oxide, CO activates SCG and elevates cGMP in target tissues, which dilates blood vessels. For many years, endogenously generated CO was treated as a by-product of metabolism without any serious physiological or biochemical significance. Exogenous CO was for a long time the subject of publications and textbooks in the field of toxicology. Currently, CO is still seen as a toxic gas; it is the most common cause of fatal poisoning in many countries and accounts for approximately 75% of all suicide poisonings worldwide.
However, increasing attention is also now paid to the beneficial aspects of CO activity. The physiological and biochemical properties of CO only began to be considered in the 1980s. Over the last two decades, significant progress has been made in determining the therapeutic potential of the HOs/CO system in a number of preclinical models for many diseases and lesions. The history of CO research has indicated several ways to increase the concentration of this gas in the bodies of experimental animals. The simplest method involves the inhalation of CO alone or gas mixtures containing CO. Another method is based on inducing HO-1 by using appropriate activators of this CO-producing enzyme. In addition, interest in CO as a pharmacological factor has grown significantly since the development of methods for synthesizing a new group of compounds capable of releasing this gas, termed carbon-oxide-releasing molecules (CORMs). The protective effect of endogenous or exogenous CO on tissues has been observed in many preclinical and a few clinical studies so far. The different, intricately related and interpenetrating pro-and anti-inflammatory signaling cascades underlie the biological activity of both endogenously generated and exogenously administered CO. The schematic summary of the basic signaling pathways through which CO affects cell activity is shown in Figure 3.
Intensive research is aimed at better understanding the mechanisms of interaction between key signaling pathways and the effects of CO on these signaling pathways. Preclinical and clinical studies aiming to establish the biochemical and physiological properties of CO and its therapeutic potential are underway.
In summary, there is ample evidence that CO plays an important role in the body, suggesting that this gaseous transmitter may become a promising target for new treatment strategies for many diseases. Consequently, the authors are in agreement with the opinion of Dulak and Józkowicz that CO, the “notoriously infamous” by-product of HO activity entered on stage as a key player in biochemistry, physiology and medicine [142].
Author Contributions
Conceptualization, A.B.-W.; writing—original draft preparation, A.B.-W.; literature search, M.I.; production of graphic material, A.B.-W. and M.G.; review and editing, A.B.-W., M.G. and M.I.; supervision, A.B.-W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sjöstrand, T. The formation of carbon monoxide by the decomposition of haemoglobin in vivo. Acta Physiol. Scand. 1952, 26, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Role of carbon monoxide in cardiovascular function. J. Cell. Mol. Med. 2006, 10, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme oxygenases in cardiovascular health and disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Q.; Li, L.J.; Wang, X.Y.; Sun, Y.Y.; Wu, J. Research Progress in Understanding the Relationship Between Heme Oxygenase-1 and Intracerebral Hemorrhage. Front. Neurol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Ning, W.; Choi, A.M.; Li, C. Carbon monoxide inhibits IL-17-induced IL-6 production through the MAPK pathway in human pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L268–L273. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.P.; Choi, A.M. A new road to induce heme oxygenase-1 expression by carbon monoxide. Circ. Res. 2007, 101, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T.; Samoylenko, A.; Immenschuh, S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J. Biol. Chem. 2003, 278, 17927–17936. [Google Scholar] [CrossRef] [Green Version]
- Zapora, E.; Jarocka, I. Hemoglobin—Source of reactive oxygen. Postepy Hig. Med. Dosw. 2013, 67, 214–220. (In Polish) [Google Scholar] [CrossRef]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Nowak, S.; Gilun, P.; Kozioł, K.; Romerowicz-Misielak, M.; Koziorowska-Gilun, M.; Wąsowska, B. Carbon Monoxide (CO) as a Retinal Regulator of Heme Oxygenases-1, and-2 (HO’s) Expression. Biomedicines 2022, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Cruse, I.; Maines, M.D. Evidence suggesting that the two forms of heme oxygenase are products of different genes. J. Biol. Chem. 1988, 263, 3348–3353. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J.; Jamroz, A.; Borkowska, E. Heme oxygenase and carbon monoxide in the physiology and pathology of the cardiovascular system. Postepy Hig. Med. Dosw. 2004, 58, 83–99. (In Polish) [Google Scholar]
- Jasnos, K.; Magierowski, M.; Kwiecień, S.; Brzozowski, T. Carbon monoxide in human physiology—Its role in the gastrointestinal tract. Postepy Hig. Med. Dosw. 2014, 68, 101–109. (In Polish) [Google Scholar] [CrossRef]
- Thom, S.R. Carbon Monoxide pathophysiology and treatment. In Physiology and Medicine of Hyperbaric Oxygen Therapy; Neuman, T.S., Thom, S.R., Eds.; Elsevier Inc.: Philadelphia, PA, USA, 2008; pp. 321–347. [Google Scholar]
- Wang, R.; Wang, Z.; Wu, L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br. J. Pharmacol. 1997, 121, 927–934. [Google Scholar] [CrossRef]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Haskó, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Furchgott, R.F.; Jothianandan, D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: Relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessel 1991, 28, 52–61. [Google Scholar] [CrossRef]
- Kozma, F.; Johnson, R.A.; Zhang, F.; Yu, C.; Tong, X.; Nasjletti, A. Contribution of endogenous carbon monoxide to regulation of diameter in resistance vessels. Am. J. Physiol. 1999, 276, R1087–R1094. [Google Scholar] [CrossRef] [PubMed]
- Katada, K.; Bihari, A.; Mizuguchi, S.; Yoshida, N.; Yoshikawa, T.; Fraser, D.D.; Potter, R.F.; Cepinskas, G. Carbon monoxide liberated from CO-releasing molecule (CORM-2) attenuates ischemia/reperfusion (I/R)-induced inflammation in the small intestine. Inflammation 2010, 33, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Morita, T. Heme oxygenase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1786–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubos, E.; Handy, D.E.; Loscalzo, J. Role of oxidative stress and nitric oxide in atherothrombosis. Front. Biosci. 2008, 13, 5323–5344. [Google Scholar] [CrossRef] [Green Version]
- Chlopicki, S.; Olszanecki, R.; Marcinkiewicz, E.; Lomnicka, M.; Motterlini, R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc. Res. 2006, 71, 393–401. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Thorup, C.; Jones, C.L.; Gross, S.S.; Moore, L.C.; Goligorsky, M.S. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am. J. Physiol. 1999, 277, F882–F889. [Google Scholar] [CrossRef]
- Xue, L.; Farrugia, G.; Miller, S.M.; Ferris, C.D.; Snyder, S.H.; Szurszewski, J.H. Carbon monoxide and nitric oxide as coneurotransmitters in the enteric nervous system: Evidence from genomic deletion of biosynthetic enzymes. Proc. Natl. Acad. Sci. USA 2000, 97, 1851–1855. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wu, L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 8222–8226. [Google Scholar] [CrossRef] [Green Version]
- Escalante, B.; Sacerdoti, D.; Davidian, M.M.; Laniado-Schwartzman, M.; McGiff, J.C. Chronic treatment with tin normalizes blood pressure in spontaneously hypertensive rats. Hypertension 1991, 17, 776–779. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Wu, L.; Zhao, W.; Wang, R. Induction of heme oxygenase-1 and stimulation of cGMP production by hemin in aortic tissues from hypertensive rats. Blood 2003, 101, 3893–3900. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F.; Mishra, M. The heme oxygenase system selectively suppresses the proinflammatory macrophage m1 phenotype and potentiates insulin signaling in spontaneously hypertensive rats. Am. J. Hypertens. 2013, 26, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Grande, J.P.; Belcher, J.D.; Garovic, V.D.; Croatt, A.J.; Hillestad, M.L.; Barry, M.A.; Nath, M.C.; Regan, R.F.; Vercellotti, G.M. Antithrombotic effects of heme-degrading and heme-binding proteins. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H671–H681. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Wang, S.; Jonas, W.; Kirchhofer, D.; Gailani, D.; Gruber, A.; Kasthuri, R.; Key, N.S.; Mackman, N.; et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 2015, 100, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Detzel, M.S.; Schmalohr, B.F.; Steinbock, F.; Hopp, M.T.; Ramoji, A.; Paul George, A.A.; Neugebauer, U.; Imhof, D. Revisiting the interaction of heme with hemopexin. Biol. Chem. 2021, 402, 675–691. [Google Scholar] [CrossRef]
- Motterlini, R.; Clark, J.E.; Foresti, R.; Sarathchandra, P.; Mann, B.E.; Green, C.J. Carbon monoxide-releasing molecules: Characterization of biochemical and vascular activities. Circ. Res. 2002, 90, E17–E24. [Google Scholar] [CrossRef] [Green Version]
- Kramkowski, K.; Mogielnicki, A.; Motterilini, R.; Chłopicki, S.; Buczko, W. Carbon monoxide releasing molecule 3 (CORM-3) inhibits arterial thrombosis in rats. J. Thromb. Haemost. 2009, 7 (Suppl. 2), 2149–2157. [Google Scholar]
- Foresti, R.; Hammad, J.; Clark, J.E.; Johnson, R.A.; Mann, B.E.; Friebe, A.; Green, C.J.; Motterlini, R. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br. J. Pharmacol. 2004, 142, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Mann, B.E.; Johnson, T.R.; Clark, J.E.; Foresti, R.; Green, C.J. Bioactivity and pharmacological actions of carbon monoxide-releasing molecules. Curr. Pharm. Des. 2003, 9, 2525–2539. [Google Scholar] [CrossRef]
- Clark, J.E.; Naughton, P.; Shurey, S.; Green, C.J.; Johnson, T.R.; Mann, B.E.; Foresti, R.; Motterlini, R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ. Res. 2003, 93, e2–e8. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Stein, A.B.; Wu, W.J.; Tan, W.; Zhu, X.; Li, Q.H.; Dawn, B.; Motterlini, R.; Bolli, R. Administration of a CO-Releasing Molecule at the Time of Reperfusion Reduces Infarct Size In Vivo. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1649–H1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberto, R.; Ortner, K.; Wheatley, N.; Schibli, R.; Schubiger, A.P. Synthesis and properties of boranocarbonate: A convenient in situ CO source for the aqueous preparation of [(99m)Tc(OH(2))3(CO)3]+. J. Am. Chem. Soc. 2001, 123, 3135–3136. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Sawle, P.; Hammad, J.; Bains, S.; Alberto, R.; Foresti, R.; Green, C.J. CORM-A1: A new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005, 19, 284–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczara, P.; Sitek, B.; Przyborowski, K.; Kurpinska, A.; Kus, K.; Stojak, M.; Chlopicki, S. Antiplatelet effect of carbon monoxide is mediated by NAD+ and ATP depletion. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2376–2390. [Google Scholar] [CrossRef]
- Iqbal, J.; Chamberlain, J.; Alfaidi, M.; Hughes, M.; Alizadeh, T.; Casbolt, H.; Evans, P.; Mann, B.; Motterlini, R.; Francis, S.; et al. Carbon Monoxide Releasing Molecule A1 Reduces Myocardial Damage After Acute Myocardial Infarction in a Porcine Model. J. Cardiovasc. Pharmacol. 2021, 78, e656–e661. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Nakajima, K.; Kohsaka, S. Functional roles of microglia in the brain. Neurosci. Res. 1993, 17, 187–203. [Google Scholar] [CrossRef]
- Dias-Pedroso, D.; Ramalho, J.S.; Sardão, V.A.; Jones, J.G.; Romão, C.C.; Oliveira, P.J.; Vieira, H.L.A. Carbon Monoxide-Neuroglobin Axis Targeting Metabolism against Inflammation in BV-2 Microglial Cells. Mol. Neurobiol. 2022, 59, 916–931. [Google Scholar] [CrossRef]
- Soares, N.L.; Paiva, I.; Bravo, J.; Queiroga, C.S.F.; Melo, B.F.; Conde, S.V.; Romão, C.C.; Summavielle, T.; Vieira, H.L.A. Carbon Monoxide Modulation of Microglia-Neuron Communication: Anti-Neuroinflammatory and Neurotrophic Role. Mol. Neurobiol. 2022, 59, 872–889. [Google Scholar] [CrossRef]
- Zeynalov, E.; Doré, S. Low doses of carbon monoxide protect against experimental focal brain ischemia. Neurotox. Res. 2009, 15, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem. 1999, 72, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- Vieira, H.L.; Queiroga, C.S.; Alves, P.M. Pre-conditioning induced by carbon monoxide provides neuronal protection against apoptosis. J. Neurochem. 2008, 107, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Biermann, J.; Lagrèze, W.A.; Dimitriu, C.; Stoykow, C.; Goebel, U. Preconditioning with inhalative carbon monoxide protects rat retinal ganglion cells from ischemia/reperfusion injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3784–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrich, F.; Hagmann, C.; Buerkle, H.; Romao, C.C.; Schallner, N.; Goebel, U.; Biermann, J. The Carbon monoxide releasing molecule ALF-186 mediates anti-inflammatory and neuroprotective effects via the soluble guanylate cyclase ß1 in rats’ retinal ganglion cells after ischemia and reperfusion injury. J. Neuroinflamm. 2017, 14, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhu, Q.L.; Wang, G.Z.; Deng, T.Z.; Chen, R.; Liu, M.H.; Wang, S.W. The protective roles of mitochondrial ATP-sensitive potassium channels during hypoxia-ischemia-reperfusion in brain. Neurosci. Lett. 2011, 49, 63–67. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Bilban, M.; Bach, F.H.; Otterbein, S.L.; Ifedigbo, E.; d’Avila, J.C.; Esterbauer, H.; Chin, B.Y.; Usheva, A.; Robson, S.C.; Wagner, O.; et al. Carbon monoxide orchestrates a protective response through PPARγ. Immunity 2006, 24, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Loughran, P.A.; Rao, J.; Billiar, T.R.; Zuckerbraun, B.S. Carbon monoxide activates NF-κB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Yang, C.C.; Hsiao, L.D.; Yang, C.M. Carbon Monoxide Releasing Molecule-3 Enhances Heme Oxygenase-1 Induction via ROS-Dependent FoxO1 and Nrf2 in Brain Astrocytes. Oxidative Med. Cell. Longev. 2021, 2021, 5521196. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Sawle, P.; Homer-Vanniasinkam, S.; Green, C.J.; Foresti, R.; Motterlini, R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit. Care Med. 2012, 40, 544–552. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Zhou, J.; Stone, C.; Ding, Y.; Zhang, Y.; Ren, C.; Yin, X.; Meng, R. CORM-2 inhibits intracerebral hemorrhage-mediated inflammation. Neurol. Res. 2021, 43, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Huang, Y.T.; Hsieh, C.W.; Yang, P.M.; Wung, B.S. Carbon monoxide induces heme oxygenase-1 to modulate STAT3 activation in endothelial cells via S-glutathionylation. PLoS ONE 2014, 9, e100677. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, A.; Naldi, M.; Tedesco, D.; Milelli, A.; Bartolini, M.; Davani, L.; Widera, D.; Dallas, M.L.; Andrisano, V. Investigating in Vitro Amyloid Peptide 1–42 Aggregation: Impact of Higher Molecular Weight Stable Adducts. ACS Omega 2019, 4, 12308–12318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-beta(1–42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [Green Version]
- Mahan, V.L. Neuroprotective, neurotherapeutic, and neurometabolic effects of carbon monoxide. Med. Gas Res. 2012, 2, 32. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Zhao, Y.; Lee, C.W.; Ganguli, M. Smoking, death, and Alzheimer disease: A case of competing risks. Alzheimer Dis. Assoc. Disord. 2012, 26, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Chora, A.A.; Fontoura, P.; Cunha, A.; Pais, T.F.; Cardoso, S.; Ho, P.P.; Lee, L.Y.; Sobel, R.A.; Steinman, L.; Soares, M.P. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Investig. 2007, 117, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Stasik, Z.; Skotnicki, P.; Nowak-Sadzikowska, J.; Kulpa, J.K. C-reactive protein in cancer patients. Nowotw. J. Oncol. 2008, 58, 441–446. (In Polish) [Google Scholar]
- Persing, D.H.; Prendergast, F.G. Infection, immunity, and cancer. Arch. Pathol. Lab. Med. 1999, 123, 1015–1022. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-κB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, W.C. Regulation of immune responses by NF-kappa B/Rel transcription factor. J. Exp. Med. 1998, 187, 143–146, Erratum in J. Exp. Med. 1998, 187, 661. [Google Scholar] [CrossRef] [PubMed]
- Skórka, K.; Giannopoulos, K. The structure and the role of NF-κB proteins and their significance in chronic lymphocytic leukemia. Acta Haematol. Pol. 2012, 43, 54–62. (In Polish) [Google Scholar] [CrossRef]
- Li, X.; Stark, G.R. NFkappaB-dependent signaling pathways. Exp. Hematol. 2002, 30, 285–2896. [Google Scholar] [CrossRef]
- Marok, R.; Winyard, P.G.; Coumbe, A.; Kus, M.L.; Gaffney, K.; Blades, S.; Mapp, P.I.; Morris, C.J.; Blake, D.R.; Kaltschmidt, C.; et al. Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue. Arthritis Rheum. 1996, 39, 583–591. [Google Scholar] [CrossRef]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Mammana, S.; Cavalli, E.; Di Marco, R.; Barcellona, M.L.; Salvatorelli, L.; Magro, G.; Nicoletti, F. Carbon monoxide-releasing molecule-A1 (CORM-A1) improves clinical signs of experimental autoimmune uveoretinitis (EAU) in rats. Clin. Immunol. 2015, 157, 198–204. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Guttridge, D.C.; Mayo, M.W.; Baldwin, A.S., Jr. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell Biol. 1999, 19, 5923–5929. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Józkowicz, A.; Waś, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waś, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Wang, B.; Yin, Y.; Chen, G.; Wang, W.; Gao, X.; Wang, P.; Zhou, H. Overexpressions of HO-1/HO-1G143H in C57/B6J mice affect melanoma B16F10 lung metastases rather than change the survival rate of mice-bearing tumours. Exp. Biol. Med. 2013, 238, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Castruccio Castracani, C.; Longhitano, L.; Distefano, A.; Di Rosa, M.; Pittalà, V.; Lupo, G.; Caruso, M.; Corona, D.; Tibullo, D.; Li Volti, G. Heme Oxygenase-1 and Carbon Monoxide Regulate Growth and Progression in Glioblastoma Cells. Mol. Neurobiol. 2020, 57, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, M.M. Heme-Oxygenase-1. Antioxid. Redox Signal. 2020, 32, 1239–1242. [Google Scholar] [CrossRef]
- Hirai, K.; Sasahira, T.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int. J. Cancer 2007, 120, 500–505. [Google Scholar] [CrossRef]
- McAllister, S.C.; Hansen, S.G.; Ruhl, R.A.; Raggo, C.M.; DeFilippis, V.R.; Greenspan, D.; Früh, K.; Moses, A.V. Kaposi sarcoma-associated herpesvirus (KSHV) induces heme oxygenase-1 expression and activity in KSHV-infected endothelial cells. Blood 2004, 103, 3465–3473. [Google Scholar] [CrossRef] [Green Version]
- Mucha, O.; Podkalicka, P.; Mikulski, M.; Barwacz, S.; Andrysiak, K.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Ryszawy, D.; Białas, A.; et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch. Biochem. Biophys. 2019, 671, 130–142. [Google Scholar] [CrossRef]
- Haq, M.; Shaeii, A.E.; Zervos, E.E.; Rosemurgy, A.S. In Vitro and In Vivo matrix metalloproteinase production by pancreatic cancer cells and by distant organs. Int. J. Surg. Investig. 2000, 1, 459–465. [Google Scholar]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Kołomecki, K.; Stępień, H.; Narębski, J.M. Matrix metalloproteinase serum levels in surgically treated adrenal tumours. J. Endocrinol. Investig. 1999, 22, 63. [Google Scholar]
- Wysocka, A.; Gizinski, S.; Lechowski, R. Matrix metalloproteinases—Their structure and function. Życie Weter. 2014, 89, 223–227. (In Polish) [Google Scholar]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef]
- Yokoyama, S.; Mita, S.; Okabe, A.; Abe, M.; Ogawa, M. Prediction of radiosensitivity in human esophageal squamous cell carcinomas with heme oxygenase-1: A clinicopathological and immunohistochemical study. Oncol. Rep. 2001, 8, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Gandini, N.A.; Alonso, E.N.; Fermento, M.E.; Mascaró, M.; Abba, M.C.; Coló, G.P.; Arévalo, J.; Ferronato, M.J.; Guevara, J.A.; Núñez, M.; et al. Heme Oxygenase-1 Has an Antitumor Role in Breast Cancer. Antioxid. Redox Signal. 2019, 30, 2030–2049. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Yoon, S.H.; Choe, E.Y.; Cho, S.H.; Woo, C.H.; Rho, J.Y.; Kim, J.H. PMA-induced up-regulation of MMP-9 is regulated by a PKCa-NF-nB cascade in human lungepithelial cells. Exp. Mol. Med. 2007, 39, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.W.; Shen, S.C.; Hou, W.C.; Yang, L.Y.; Chen, Y.C. Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9. Mol. Cancer Ther. 2008, 7, 1195–1206. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.C.; Fukui, H.; Imai, Y.; Sekikawa, A.; Kimura, T.; Yamagishi, H.; Yoshitake, N.; Pohle, T.; Domschke, W.; Fujimori, T. Colonic expression of heme oxygenase-1 is associated with a better long-term survival in patients with colorectal cancer. Scand. J. Gastroenterol. 2007, 42, 852–858. [Google Scholar] [CrossRef]
- Tsuji, M.H.; Yanagawa, T.; Iwasa, S.; Tabuchi, K.; Onizawa, K.; Bannai, S.; Toyooka, H.; Yoshida, H. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999, 138, 53–59. [Google Scholar] [CrossRef]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.G.; Pan, K.; Hao, Y.; Yip, C.Y.; Ko, W.H. Anti-inflammatory action of HO-1/CO in human bronchial epithelium in response to cationic polypeptide challenge. Mol. Immunol. 2019, 105, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Tsui, T.Y.; Siu, Y.T.; Schlitt, H.J.; Fan, S.T. Heme oxygenase-1-derived carbon monoxide stimulates adenosine triphosphate generation in human hepatocyte. Biochem. Biophys. Res. Commun. 2005, 336, 898–902. [Google Scholar] [CrossRef]
- Haschemi, A.; Chin, B.Y.; Jeitler, M.; Esterbauer, H.; Wagner, O.; Bilban, M.; Otterbein, L.E. Carbon monoxide induced PPARγ SUMOylation and UCP2 block inflammatory gene expression in macrophages. PLoS ONE 2011, 6, e26376. [Google Scholar] [CrossRef]
- Shin, D.Y.; Chung, J.; Joe, Y.; Pae, H.O.; Chang, K.C.; Cho, G.J.; Ryter, S.W.; Chung, H.T. Pretreatment with CO-releasing molecules suppresses hepcidin expression during inflammation and endoplasmic reticulum stress through inhibition of the STAT3 and CREBH pathways. Blood 2012, 119, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Kourti, M.; Jiang, W.G.; Cai, J. Aspects of Carbon Monoxide in Form of CO-Releasing Molecules Used in Cancer Treatment: More Light on the Way. Oxidative Med. Cell. Longev. 2017, 2017, 9326454. [Google Scholar] [CrossRef]
- Fang, J.; Qin, H.; Nakamura, H.; Tsukigawa, K.; Shin, T.; Maeda, H. Carbon monoxide, generated by heme oxygenase-1, mediates the enhanced permeability and retention effect in solid tumors. Cancer Sci. 2012, 103, 535–541. [Google Scholar] [CrossRef]
- Józkowicz, A.; Huk, I.; Nigisch, A.; Weigel, G.; Dietrich, W.; Motterlini, R.; Dulak, J. Heme oxygenase and angiogenic activity of endothelial cells: Stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid. Redox Signal. 2003, 5, 155–162. [Google Scholar] [CrossRef]
- Dulak, J.; Deshane, J.; Józkowicz, A.; Agarwal, A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: Focus on angiogenesis. Circulation 2008, 117, 231–241. [Google Scholar] [CrossRef]
- Ahmad, S.; Hewett, P.W.; Fujisawa, T.; Sissaoui, S.; Cai, M.; Gueron, G.; Al-Ani, B.; Cudmore, M.; Ahmed, S.F.; Wong, M.K.; et al. Carbon monoxide inhibits sprouting angiogenesis and vascular endothelial growth factor receptor-2 phosphorylation. Thromb. Haemost. 2015, 113, 329–337. [Google Scholar] [PubMed] [Green Version]
- Vítek, L.; Gbelcová, H.; Muchová, L.; Váňová, K.; Zelenka, J.; Koníčková, R.; Suk, J.; Zadinova, M.; Knejzlík, Z.; Ahmad, S.; et al. Antiproliferative effects of carbon monoxide on pancreatic cancer. Dig. Liver Dis. 2014, 46, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, M.; Gueron, G.; Elguero, B.; Giudice, J.; Salles, A.; Leskow, F.C.; Jares-Erijman, E.A.; Colombo, L.; Meiss, R.; Navone, N.; et al. Heme oxygenase 1 (HO-1) challenges the angiogenic switch in prostate cancer. Angiogenesis 2011, 14, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.S.; Schmitt, S.; Dou, Q.P.; Kodanko, J.J. Synthesis, characterization, and reactivity of the stable iron carbonyl complex [Fe(CO)(N4Py)](ClO4)2: Photoactivated carbon monoxide release, growth inhibitory activity, and peptide ligation. Inorg. Chem. 2011, 50, 5336–5338. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, J.; Zhang, Q.; Bai, Z.; Zhao, Q.; He, D.; Wang, Z.; Chen, Y.; Liu, B. Syntheses and anti-cancer activity of CO-releasing molecules with targeting galactose receptors. Org. Biomol. Chem. 2018, 16, 8115–8129. [Google Scholar] [CrossRef]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Hsu, C.C.; Lin, H.J.; Tian, Y.F.; Wang, J.J.; Guo, H.R. Impact of carbon monoxide poisoning on the risk of breast cancer. Sci. Rep. 2020, 10, 20450. [Google Scholar] [CrossRef]
- Lo Iacono, L.; Boczkowski, J.; Zini, R.; Salouage, I.; Berdeaux, A.; Motterlini, R.; Morin, D.A. Carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 1556–1564. [Google Scholar] [CrossRef] [Green Version]
- Long, R.; Salouage, I.; Berdeaux, A.; Motterlini, R.; Morin, D. CORM-3, a water soluble CO-releasing molecule, uncouples mitochondrial respiration via interaction with the phosphate carrier. Biochim. Biophys. Acta 2014, 1837, 201–209. [Google Scholar] [CrossRef]
- Tang, X.D.; Xu, R.; Reynolds, M.F.; Garcia, M.L.; Heinemann, S.H.; Hoshi, T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 2003, 425, 531–535. [Google Scholar] [CrossRef]
- Kaczara, P.; Motterlini, R.; Rosen, G.M.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takano, N.; Ishiwata, K.; Ohmura, M.; Nagahata, Y.; Matsuura, T.; Kamata, A.; Sakamoto, K.; Nakanishi, T.; Kubo, A.; et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat. Commun. 2014, 17, 3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishiki, T.; Yamamoto, T.; Morikawa, T.; Kubo, A.; Kajimura, M.; Suematsu, M. Carbon monoxide: Impact on remethylation/transsulfuration metabolism and its pathophysiologic implications. J. Mol. Med. 2012, 90, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Kabil, O.; Weeks, C.L.; Carballal, S.; Gherasim, C.; Alvarez, B.; Spiro, T.G.; Banerjee, R. Reversible heme-dependent regulation of human cystathionine β-synthase by a flavoprotein oxidoreductase. Biochemistry 2011, 50, 8261–8263. [Google Scholar] [CrossRef] [Green Version]
- Crook, S.H.; Mann, B.E.; Meijer, A.J.; Adams, H.; Sawle, P.; Scapens, D.; Motterlini, R. [MnCO)4{S2CNMe(CH2CO2H)}], a new water-soluble CO-releasing molecule. Dalton Trans. 2011, 40, 4230–4235. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Stahl, W.; Brenneisen, P.; Reichert, A.S.; Stucki, D. Carbon monoxide releasing molecule 401 (CORM-401) modulates phase I metabolism of xenobiotics. Toxicol. In Vitro 2019, 59, 215–220. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S53–S55. [Google Scholar] [CrossRef] [Green Version]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [Green Version]
- Zick, Y. Role of Ser/Thr kinases in the uncoupling of insulin signaling. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S56–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braud, L.; Pini, M.; Muchova, L.; Manin, S.; Kitagishi, H.; Sawaki, D.; Czibik, G.; Ternacle, J.; Derumeaux, G.; Foresti, R.; et al. Carbon monoxide-induced metabolic switch in adipocytes improves insulin resistance in obese mice. JCI Insight 2018, 3, e123485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosick, P.A.; AlAmodi, A.A.; Hankins, M.W.; Stec, D.E. Chronic treatment with a carbon monoxide releasing molecule reverses dietary induced obesity in mice. Adipocyte 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hosick, P.A.; AlAmodi, A.A.; Storm, M.V.; Gousset, M.U.; Pruett, B.E.; Gray, W., 3rd; Stout, J.; Stec, D.E. Chronic carbon monoxide treatment attenuates development of obesity and remodels adipocytes in mice fed a high-fat diet. Int. J. Obes. 2014, 38, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Zhang, Q.; Joe, Y.; Kim, S.K.; Uddin, M.J.; Rhew, H.; Kim, T.; Ryter, S.W.; Chung, H.T. Carbon monoxide-releasing molecules reverse leptin resistance induced by endoplasmic reticulum stress. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E780–E788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosick, P.A.; Ahmed, E.K.; Gousset, M.U.; Granger, J.P.; Stec, D.E. Inhalation of carbon monoxide is ineffective as a long-term therapy to reduce obesity in mice fed a high fat diet. BMC Obes. 2014, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Joe, Y.; Kim, S.; Kim, H.J.; Park, J.; Chen, Y.; Park, H.J.; Jekal, S.J.; Ryter, S.W.; Kim, U.H.; Chung, H.T. FGF21 induced by carbon monoxide mediates metabolic homeostasis via the PERK/ATF4 pathway. FASEB J. 2018, 32, 2630–2643. [Google Scholar] [CrossRef] [Green Version]
- Diabetes, Fact Sheet No. 312, Media Centre, World Health Organization. 2011. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 31 August 2011).
- WHO. The Top 10 Causes of Death; Fact Sheet No. 310; World Health Organization: Geneva, Switzerland, 2015; Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 9 December 2020).
- Nikolic, I.; Saksida, T.; Mangano, K.; Vujicic, M.; Stojanovic, I.; Nicoletti, F.; Stosic-Grujicic, S. Pharmacological application of carbon monoxide ameliorates islet-directed autoimmunity in mice via anti-inflammatory and anti-apoptotic effects. Diabetologia 2014, 57, 980–990. [Google Scholar] [CrossRef]
- Dulak, J.; Józkowicz, A. Carbon monoxide—A “new” gaseous modulator of gene expression. Acta Biochim. Pol. 2003, 50, 31–47. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Heme oxygenase-1 (HO-1)-catalyzed reaction.

Figure 2.
Schematic illustration of the effect of HO-1 on tumor progression.

Figure 3.
The schematic summary of the basic signaling pathways through which CO affects cell activity.
Figure 3.
The schematic summary of the basic signaling pathways through which CO affects cell activity.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bilska-Wilkosz, A.; Górny, M.; Iciek, M. Biological and Pharmacological Properties of Carbon Monoxide: A General Overview. Oxygen 2022, 2, 130-151. https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen2020012
AMA Style
Bilska-Wilkosz A, Górny M, Iciek M. Biological and Pharmacological Properties of Carbon Monoxide: A General Overview. Oxygen. 2022; 2(2):130-151. https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen2020012
Chicago/Turabian StyleBilska-Wilkosz, Anna, Magdalena Górny, and Małgorzata Iciek. 2022. "Biological and Pharmacological Properties of Carbon Monoxide: A General Overview" Oxygen 2, no. 2: 130-151. https://0-doi-org.brum.beds.ac.uk/10.3390/oxygen2020012