
Asymmetric Dinuclear Lanthanide(III) Complexes from the Use of a Ligand Derived from 2-Acetylpyridine and Picolinoylhydrazide: Synthetic, Structural and Magnetic Studies †

 and
and
Abstract
:1. Introduction
2. Results and Discussion
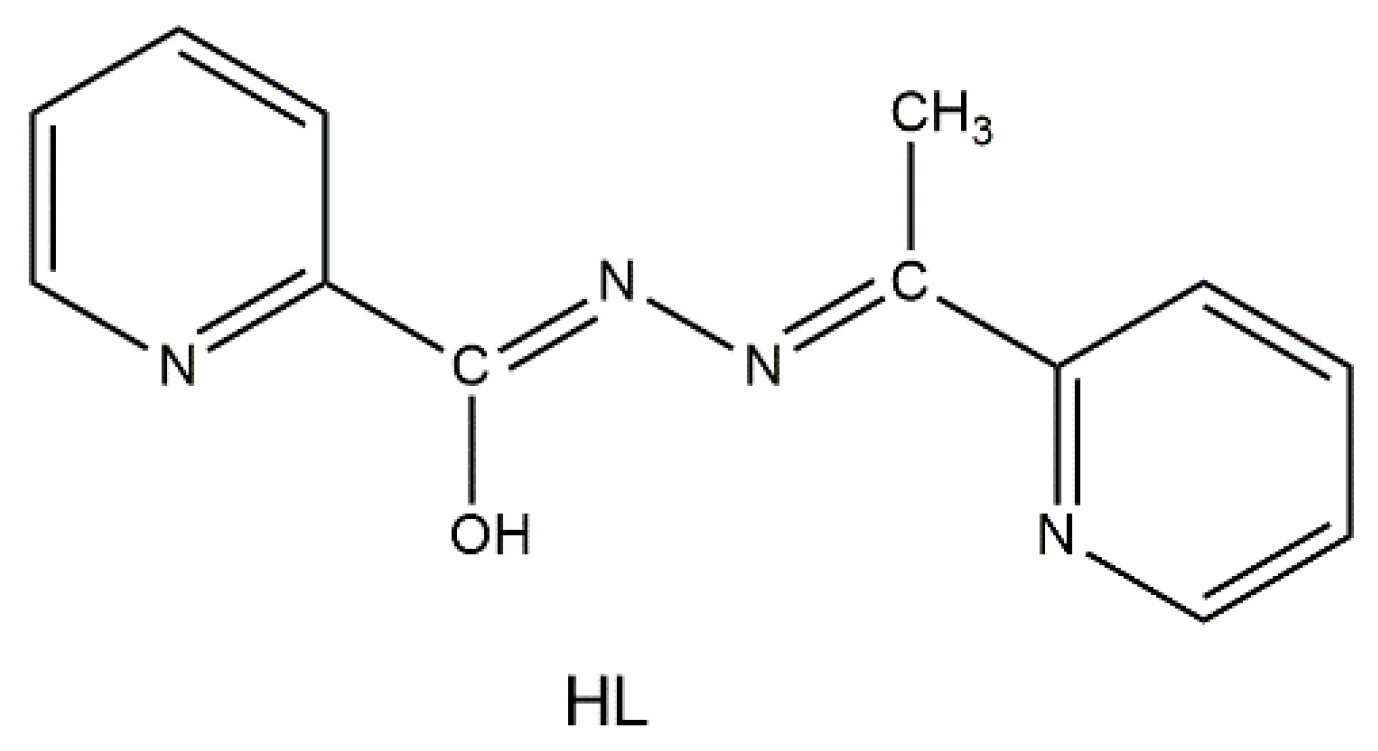
2.1. Synthetic Comments and Conventional Spectroscopic Characterization
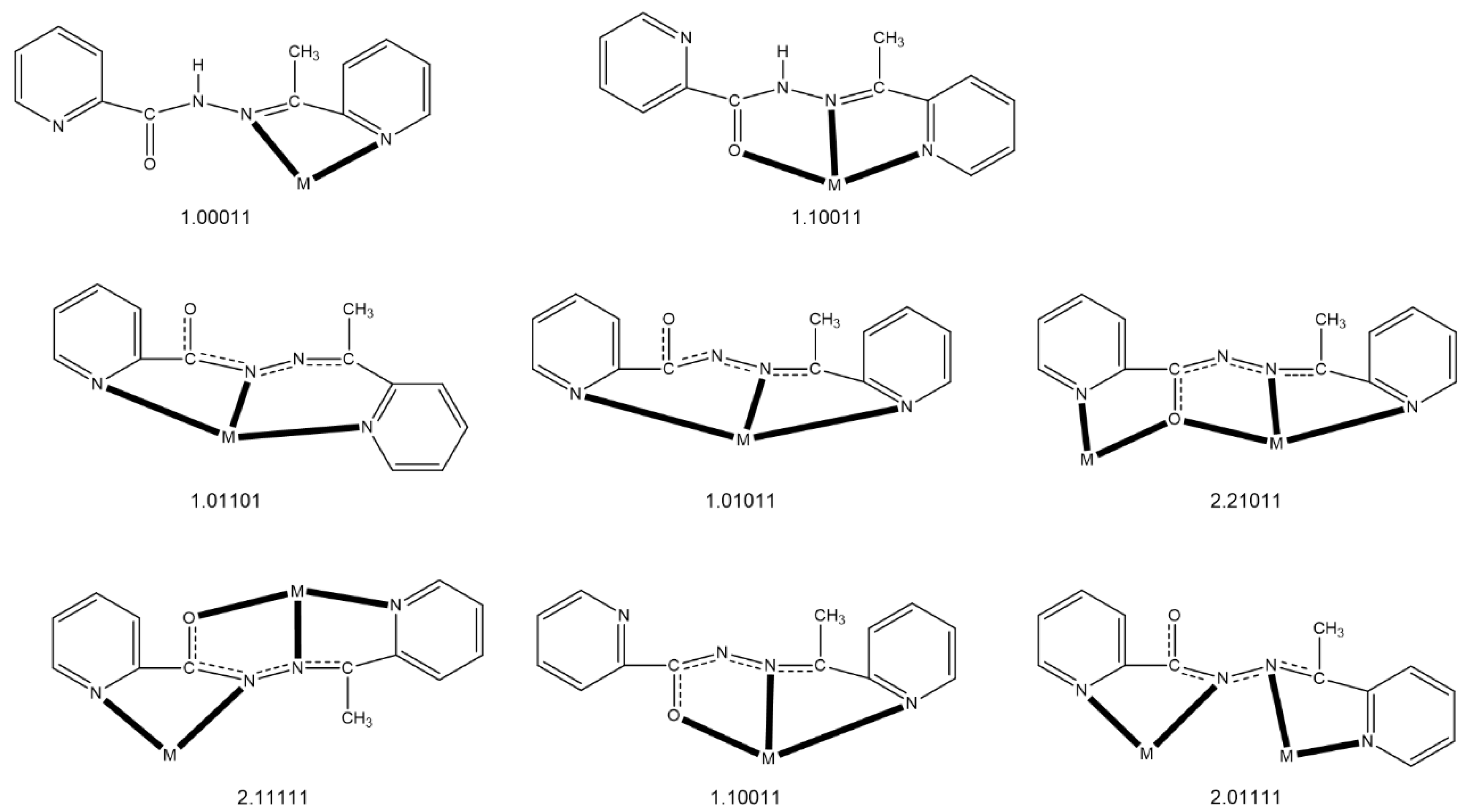
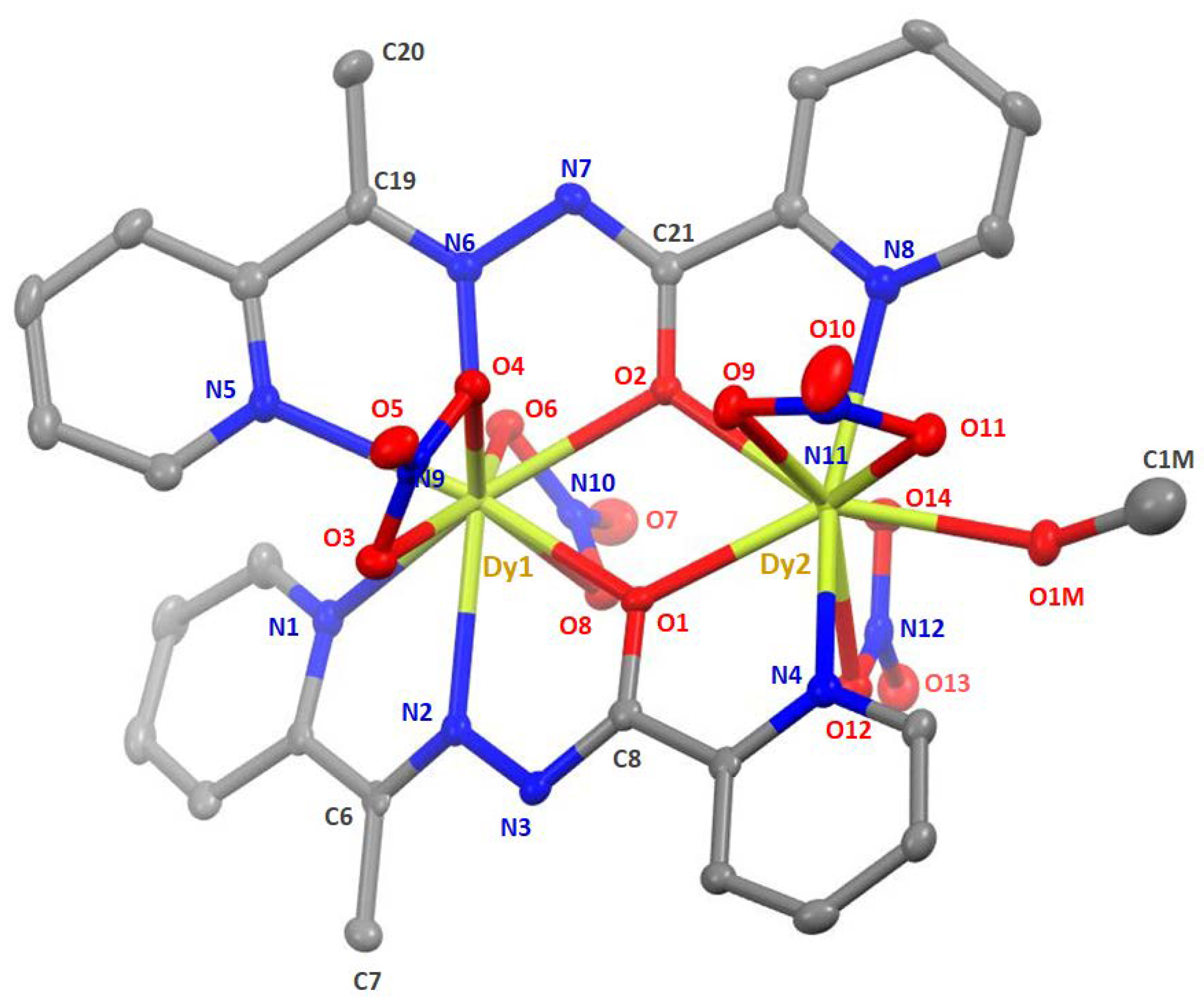
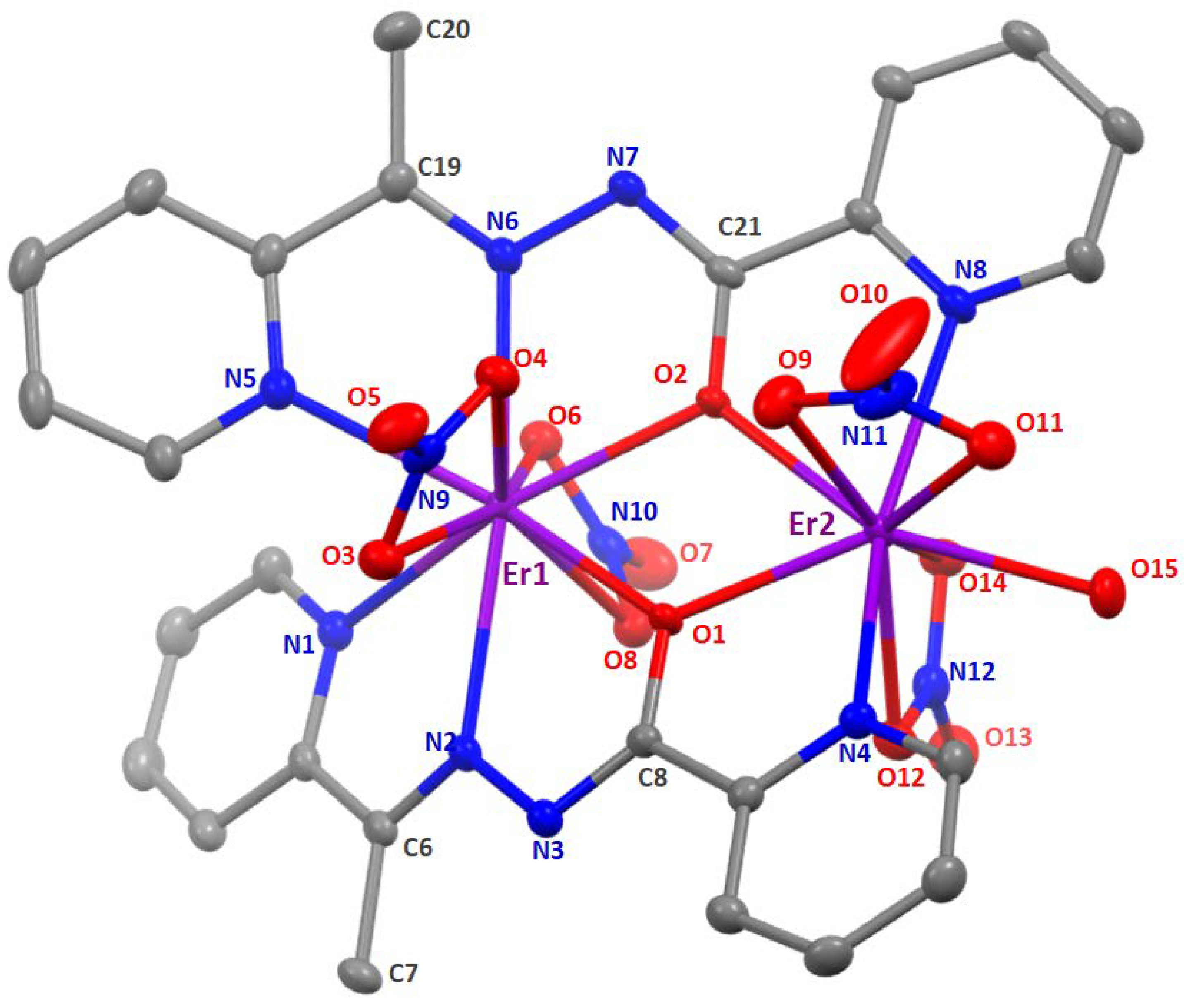
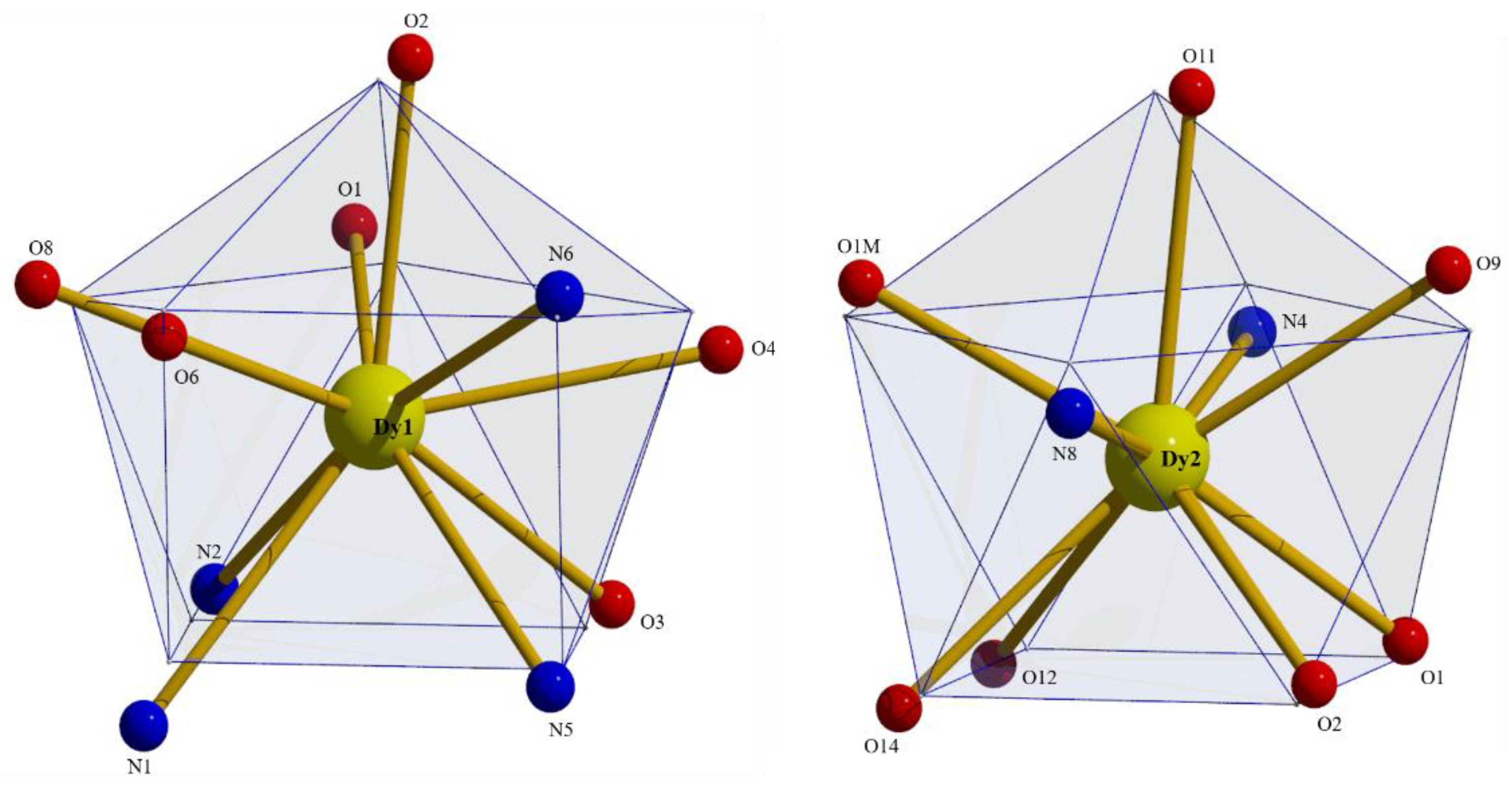
2.2. Description of Structures
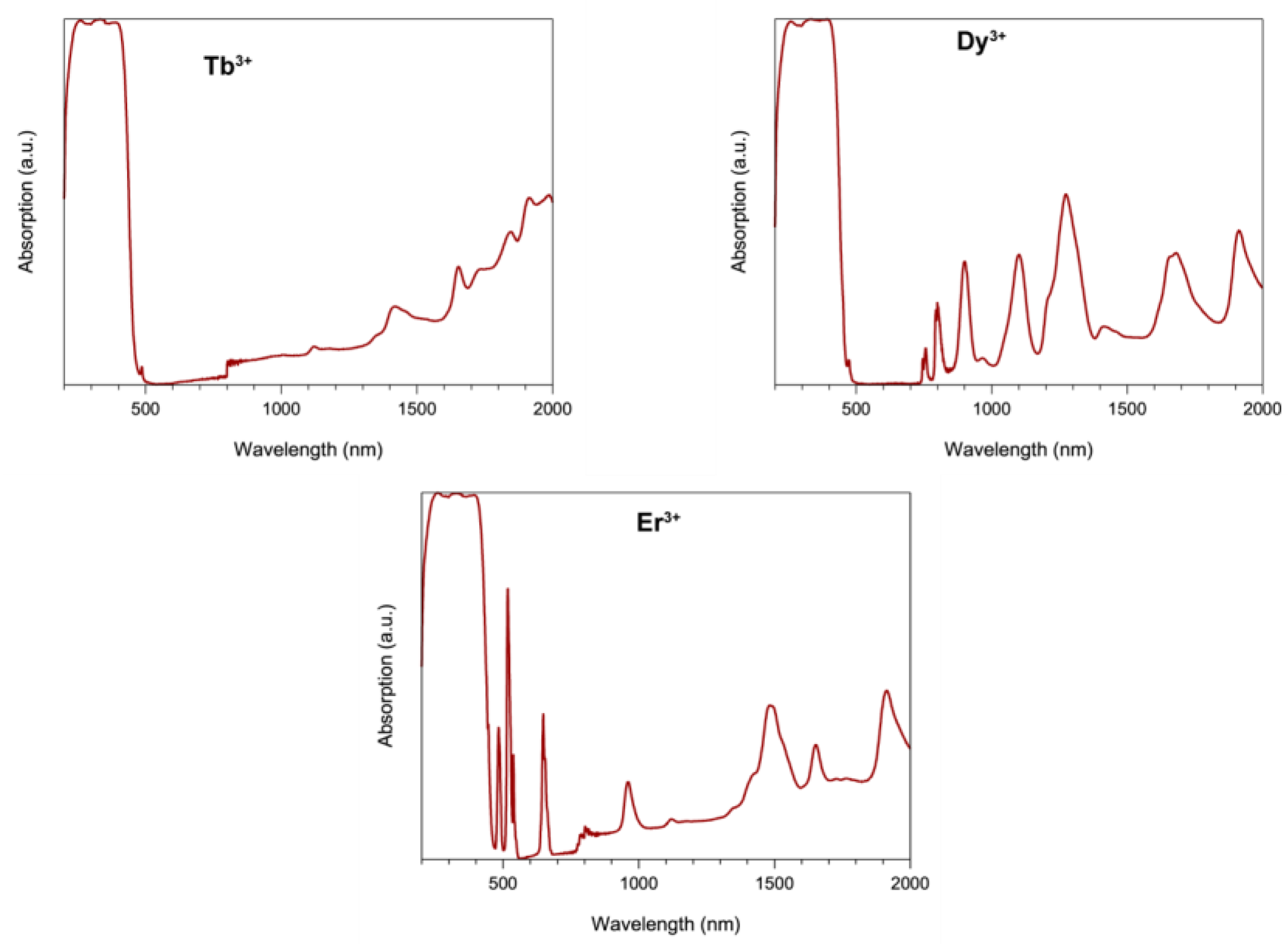
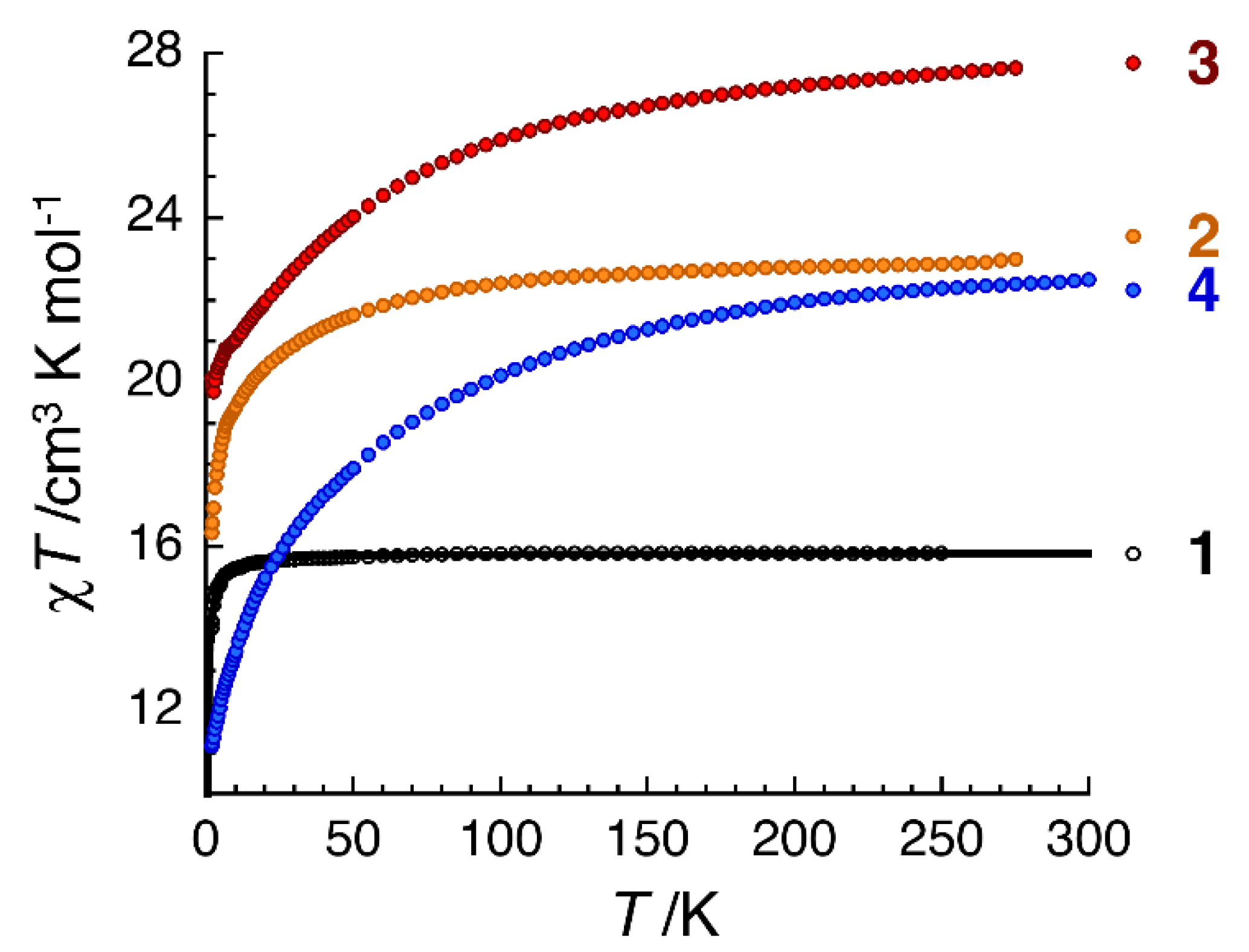
2.3. Dc Magnetic Susceptibility Studies
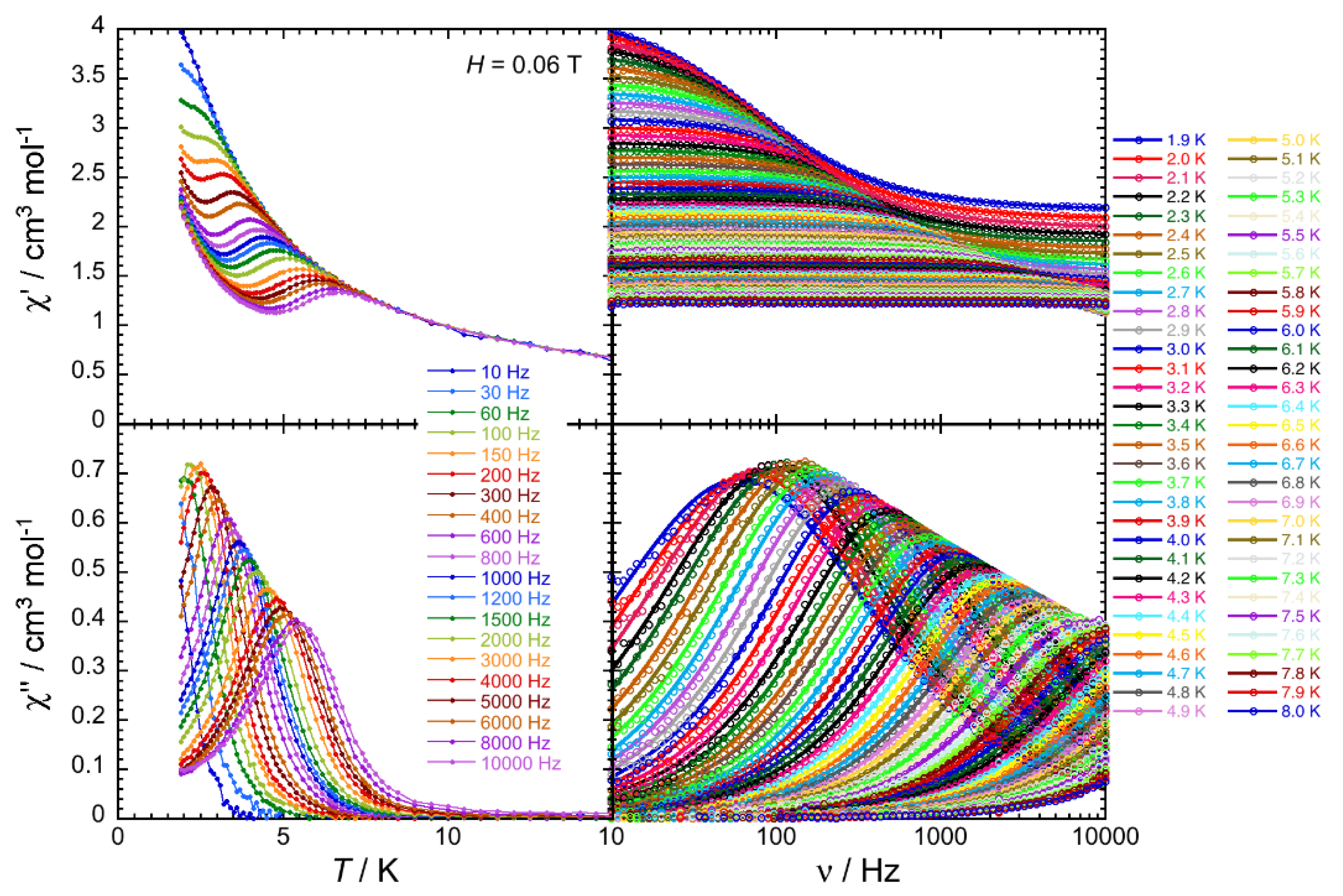
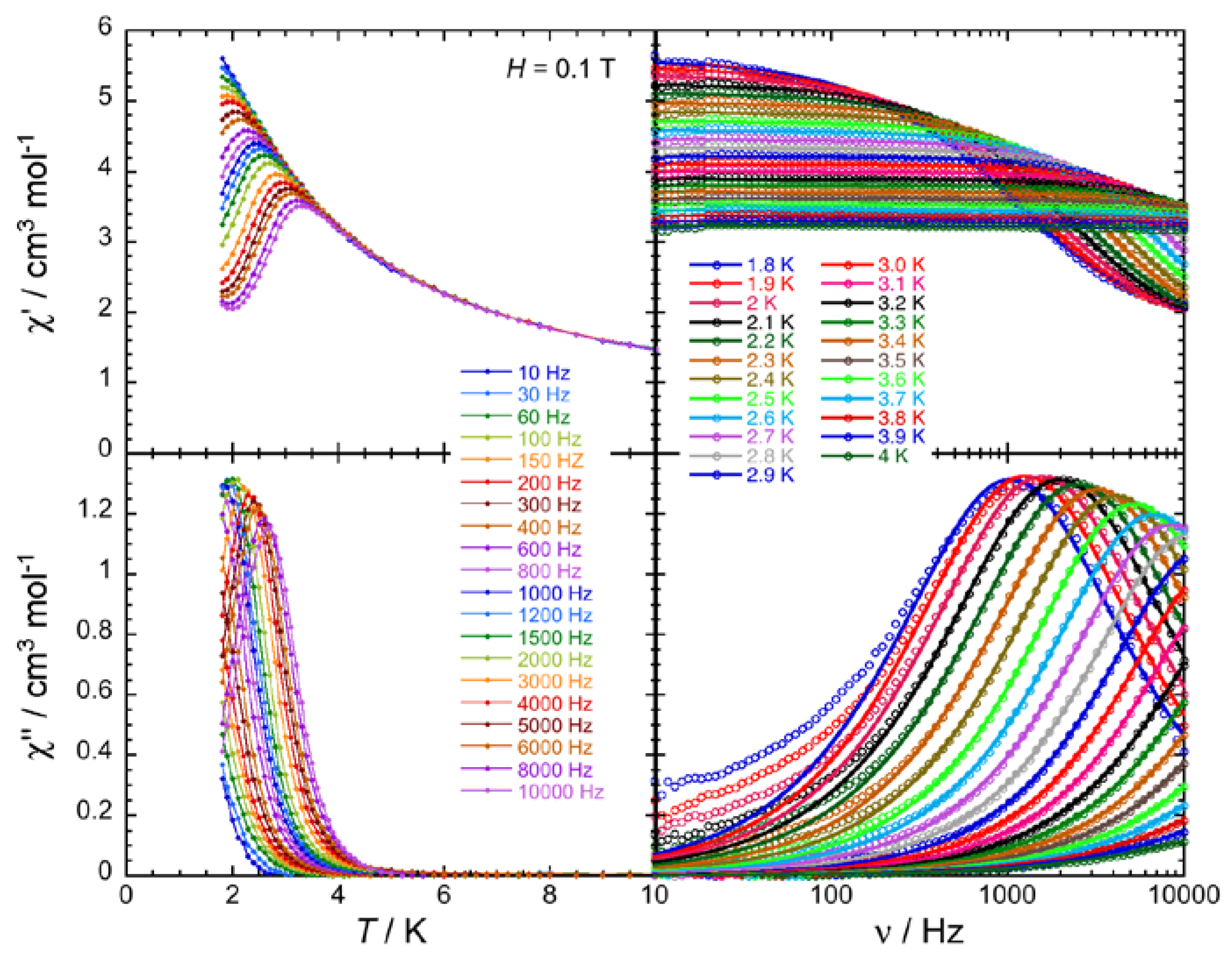
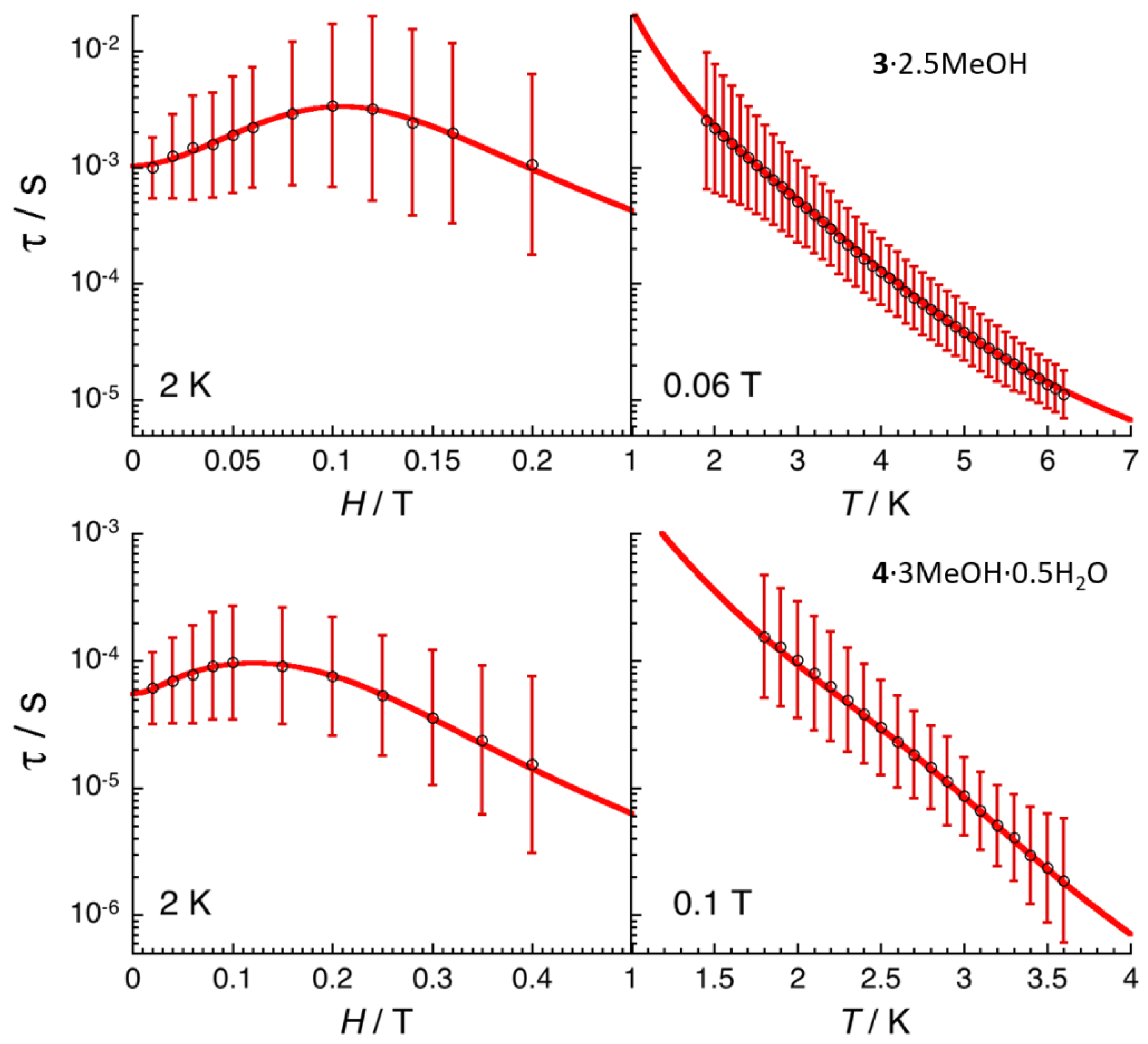
2.4. Ac Magnetic Susceptibilty Studies
3. Experimental Section
3.1. Materials, Physical and Spectroscopic Measurements
3.2. Synthesis of the Representative Complex [Gd2(NO3)4(L)2(H2O)]∙2MeOH∙2H2O (1∙2MeOH∙2H2O)
3.3. Syntheses of the Complexes [Τb2(ΝO3)4(L)2(H2O)]∙2ΜeOH∙1.5H2O (2∙2ΜeOH∙1.5H2O), [Dy2(ΝO3)4(L)2(MeOH)]∙ΜeOH∙2H2O (3∙2.5ΜeOH) and [Er2(ΝO3)4(L)2(H2O)]∙3ΜeOH∙0.5H2O (4∙3MeOH∙0.5H2O)
3.4. Single-Crystal X-ray Crystallography
4. Concluding Comments and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cador, O.; Le Guennic, B.; Pointillart, F. Electro-activity and magnetic switching in lanthanide-based single-molecule magnets. Inorg. Chem. Front. 2019, 6, 3398–3417. [Google Scholar] [CrossRef] [Green Version]
- Bagai, R.; Christou, G. The drosophila of single-molecule magnetism: [Mn12O12(O2CR)16(H2O)4]. Chem. Soc. Rev. 2009, 38, 1011–1026. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Maniaki, D.; Pilichos, E.; Perlepes, S.P. Coordination Clusters of 3d-Metals That Behave as Single-Molecule Magnets (SMMs): Synthetic Routes and Strategies. Front. Chem. 2018, 6, 461. [Google Scholar] [CrossRef]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.-Y.; Kaizu, Y. Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef]
- Briganti, M.; Fernandez Garcia, G.; Jung, J.; Sessoli, R.; Le Guennic, B.; Totti, F. Covalency and magnetic anisotropy in lanthanide single molecule magnets: The DyDOTA archetype. Chem. Sci. 2019, 10, 7233–7245. [Google Scholar] [CrossRef] [PubMed]
- Canaj, A.B.; Singh, M.K.; Regincos Marti, E.; Damjanovic, M.; Wilson, C.; Céspedes, O.; Wernsdorfer, W.; Rajaraman, G.; Murrie, M. Boosting axiality in stable high-coordinate Dy(III) single-molecule magnets. Chem. Commun. 2019, 55, 5950–5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandropoulos, D.I.; Vingesh, K.R.; Xie, H.; Dunbar, K.R. Switching On Single-Molecule Magnet Properties of Homoleptic Sandwich Tris(pyrazolyl)borate Dysprosium(III) Cations via Intermolecular Dipolar Coupling. Dalton Trans. 2019, 48, 10610–10618. [Google Scholar] [CrossRef]
- Ortu, F.; Reta, D.; Ding, Y.-S.; Goodwin, C.A.P.; Gregson, M.P.; McInnes, E.J.L.; Winpenny, R.E.P.; Zheng, Y.-Z.; Liddle, S.T.; Mills, D.P.; et al. Studies of hysteresis and quantum tunneling of the magnetization in dysprosium(III) single molecule magnets. Dalton Trans. 2019, 48, 8541–8545. [Google Scholar] [CrossRef]
- Hilgar, J.D.; Bernbeck, M.G.; Rinehart, J.D. Million-fold relaxation time enhancement across a series of phosphino-supported erbium single-molecule magnets. J. Am. Chem. Soc. 2019, 141, 1913–1917. [Google Scholar] [CrossRef]
- Goodwin, C.A.P.; Reta, D.; Ortu, F.; Chilton, N.F.; Mills, D.P. Synthesis and Electronic Structures of Heavy Lanthanide Metallocenium Cations. J. Am. Chem. Soc. 2017, 139, 18714–18724. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem. Int. Ed. 2017, 56, 11445–11449. [Google Scholar] [CrossRef]
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular magnetic hysteresis at 60 Kelvin in dysprosocenium. Nature 2017, 548, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Harriman, K.L.M.; Brosmer, J.L.; Ungur, L.; Diaconescu, P.L.; Murugesu, M. Pursuit of Record Breaking Energy Barriers: A Study of Magnetic Axiality in Diamide Ligated DyIII Single-Molecule Magnets. J. Am. Chem. Soc. 2017, 139, 1420–1423. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Rajeshkumar, T.; Rajaraman, G.; Murugavel, R. Is a strong axial crystal-field the only essential condition for a large magnetic anisotropy barrier? The case of non-Kramers Ho(III) versus Tb(III). Dalton Trans. 2018, 47, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.-C.; Jia, J.-H.; Liu, J.-L.; Vieru, V.; Ungur, L.; Chibotaru, L.F.; Lan, Y.; Wernsdorfer, W.; Gao, S.; et al. A Stable Pentagonal-Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization Reversal Barrier over 1000 K. J. Am. Chem. Soc. 2016, 138, 5441–5450. [Google Scholar] [CrossRef] [PubMed]
- Ζhang, H.; Νakanishi, R.; Κatoh, Κ.; Breedlove, B.K.; Kitagawa, Y.; Yamashita, M. Low coordinated mononuclear erbium(III) single-molecule magnets with C3v symmetry: A method by altering single-molecule magnet properties by incorporating hard and soft donors. Dalton Trans. 2018, 47, 302–305. [Google Scholar] [CrossRef]
- Ungur, L.; Chibotaru, L.F. Strategies toward High-Temperature Lanthanide-Based Single-Molecule Magnets. Inorg. Chem. 2016, 55, 10043–10056. [Google Scholar] [CrossRef] [PubMed]
- Harriman, K.L.M.; Errulat, D.; Murugesu, M. Magnetic Axiality: Design Principles from Molecules to Materials. Trends Chem. 2019, 1, 425–439. [Google Scholar] [CrossRef]
- Liddle, S.T.; Van Slageren, J. Improving f-element single molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Guo, Y.-N.; Tang, J. Recent advances in dysprosium-based single molecule magnets: Structural overview and synthetic strategies. Coord. Chem. Rev. 2013, 257, 1728–1763. [Google Scholar] [CrossRef]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Murugavel, R. Enriching lanthanide single-ion magnetism through symmetry and axiality. Chem. Commun. 2018, 54, 3685–3696. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2011, 2, 2078–2085. [Google Scholar] [CrossRef]
- Chilton, N.F.; Collison, D.; McInnes, E.J.L.; Winpenny, R.E.P.; Soncini, A. An electrostatic model for the determination of magnetic anisotropy in dysprosium complexes. Nat. Commun. 2013, 4, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Chen, Y.-C.; Tong, M.-L. Symmetry strategies for high performance lanthanide-based single-molecule magnets. Chem. Soc. Rev. 2018, 47, 2431–2453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, L.; Tang, J. Lanthanide single molecule magnets: Progress and perspective. Dalton Trans. 2015, 44, 3923–3929. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; Cador, O.; Le Guennic, B.; Quahab, L. Uncommon lanthanide ions in purely 4f Single Molecule Magnets. Coord. Chem. Rev. 2017, 346, 150–175. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Woodruff, D.N.; Bendix, J.; Clérac, R. Experimental Aspects of Lanthanide Single-Molecule Magnet Physics. In Lanthanides and Actinides in Molecular Magnetism; Layfield, R.A., Murugesu, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 125–152. [Google Scholar]
- Lada, Ζ.G.; Κatsoulakou, Ε.; Perlepes, S.P. Synthesis and Chemistry of Single-molecule Magnets. In Single-Molecule Magnets: Molecular Architectures and Building Blocks for Spintronics; Holynska, M., Ed.; Wiley-VCH: Weinheim, Germany, 2019; pp. 245–313. [Google Scholar]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. Strong exchange and magnetic blocking in N23--radical-bridged lanthanide complexes. Nat. Chem. 2011, 3, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhang, P. Polynuclear Lanthanide Single Molecule Magnets. In Lanthanides and Actinides in Molecular Magnetism; Layfield, R.A., Murugesu, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 61–88. [Google Scholar]
- Blagg, R.J.; Ungur, L.; Tuna, F.; Speak, J.; Comar, P.; Collison, D.; Wernsdorfer, W.; McInnes, E.J.L.; Chibotaru, L.F.; Winpenny, R.E.P. Magnetic relaxation pathways in lanthanide single-molecule magnets. Nat. Chem. 2013, 5, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.U.; Al-Harrasi, A.; Rawson, J.M.; Cavey, E.L.; Regier, J.; Alexandropoulos, D.; Pilkington, M.; Thompson, L.K. Slow magnetic relaxation in Dy2 and Dy4 complexes of a versatile, trifunctional polydentate N, O-ligand. Dalton Trans. 2019, 48, 14269–14278. [Google Scholar] [CrossRef]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic hysteresis up to 80 Kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, S.A. Lanthanide and Actinide Chemistry; Wiley: Chichester, UK, 2006. [Google Scholar]
- Lucas, J.; Lucas, P.; LeMercier, T.; Rollat, A.; Davenport, W.G. Rare Earths: Science, Technology, Production and Use; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Gould, C.; Randall McClain, R.; Yu, J.; Groshens, T.J.; Furche, F.; Harvey, B.G.; Long, J.R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(III) and Dysprosium(III). J. Am. Chem. Soc. 2019, 141, 12967–12973. [Google Scholar] [CrossRef] [PubMed]
- Habib, F.; Murugesu, M. Lessons learned from dinuclear lanthanide nano-magnets. Chem. Soc. Rev. 2013, 42, 3278–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aromi, G.; Aguilà, D.; Gamez, P.; Luis, F.; Roubeau, O. Design of magnetic coordination complexes for quantum computing. Chem. Soc. Rev. 2012, 41, 537–546. [Google Scholar] [CrossRef]
- Aguilà, D.; Barrios, L.A.; Velasco, V.; Roubeau, O.; Repollés, A.; Alonso, P.J.; Sesé, J.; Teat, S.J.; Luis, F.; Aromi, G. Heterodimetallic [LnLn’] Lanthanide Complexes: Toward a Chemical Design of Two-Qubit Molecular Spin Quantum Gates. J. Am. Chem. Soc. 2014, 136, 14215–14222. [Google Scholar] [CrossRef] [Green Version]
- Aguilà, D.; Barrios, L.A.; Luis, F.; Repollés, A.; Roubeau, O.; Teat, S.J.; Aromi, G. Synthesis and Properties of a Family of Unsymmetric Dinuclear Complexes of LnIII (Ln = Eu, Gd, Tb). Inorg. Chem. 2010, 49, 6784–6786. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Brunet, G.; Holmberg, R.J.; Habib, F.; Korobkov, I.; Murugesu, M. Terminal solvent effects of the anisotropy barriers of Dy2 systems. Dalton Trans. 2016, 45, 16709–16715. [Google Scholar] [CrossRef]
- Guo, Y.-N.; Xu, G.-F.; Wernsdorfer, W.; Ungur, L.; Guo, Y.; Tang, J.; Zhang, H.-J.; Chibotaru, L.F.; Powell, A.K. Strong Axiality and Ising Exchange Interaction Suppress Zero-Field Tunneling of Magnetization of an Asymmetric Dy2 Single-Molecule Magnet. J. Am. Chem. Soc. 2011, 133, 11948–11951. [Google Scholar] [CrossRef]
- Diaz-Ortega, I.F.; Herrera, J.M.; Aravena, D.; Ruiz, E.; Gupta, T.; Rajaraman, G.; Nojiri, H.; Colacio, E. Designing a Dy2 Single-Molecule Magnet with Two Well-Differentiated Relaxation Processes by Using a Nonsymmetric Bis-bidentate Bipyrimidine-N-Oxide Ligand: A Comparison with Mononuclear Counterparts. Inorg. Chem. 2018, 57, 6362–6375. [Google Scholar] [CrossRef] [Green Version]
- Aguilà, D.; Barrios, L.A.; Velasco, V.; Arnedo, L.; Aliaga-Alcalde, N.; Menelaou, M.; Teat, S.J.; Roubeau, O.; Luis, F.; Aromi, G. Lanthanide Contraction within a Series of Asymmetric Dinuclear [Ln2] Complexes. Chem. Eur. J. 2013, 19, 5881–5891. [Google Scholar] [CrossRef]
- Grove, H.; Kelly, T.L.; Thompson, L.K.; Zhao, L.; Xu, Z.; Abedin, T.S.M.; Miller, D.O.; Goeta, A.E.; Wilson, C.; Howard, J.A.K. Copper(II) Complexes of a Series of Alkoxy Diazine Ligands: Mononuclear, Dinuclear and Tetranuclear Examples with Structural, Magnetic and DFT Studies. Inorg. Chem. 2004, 43, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Pilichos, E.; Spanakis, E.; Maniaki, E.-K.; Raptopoulou, C.P.; Psycharis, V.; Turnbull, M.M.; Perlepes, S.P. Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(II) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters. Magnetochemistry 2019, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Akkurt, M.; Khandar, A.A.; Tahir, M.N.; Yazdi, S.A.H.; Afkhami, F.A. Dibromido {N’-[1-(pyridin-2-yl)ethylidene]picolinohydrazide-κ2N’,O} cadmium. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, m842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.-Q.; Mao, X.-J.; Zhang, X.; Cai, H.-X.; Bie, H.-Y.; Xu, J.; Jia, L. Syntheses, Crystal Structures and Antitumor Activities of Three Ln(III) Complexes with 2-Acetylpyridine Picolinohydrazone. Chin. J. Inorg. Chem. 2015, 31, 1–8. [Google Scholar]
- Κitamura, F.; Sawaguchi, Κ.; Mori, A.; Takagi, S.; Suzuki, Τ.; Kobayashi, A.; Kato, Μ.; Nakajima, K. Coordination Structure Conversion of Hydrazone-Palladium(II) Complexes in the Solid State and in Solution. Inorg. Chem. 2015, 54, 8436–8448. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Bauzá, A.; Gurbanov, A.V.; Zubkov, F.I.; Maniukiewicz, W.; Rodriguez-Dieguez, A.; López-Torres, E.; Frontera, A. The role of unconventional stacking interactions in the supramolecular assemblies of Hg(II) coordination compounds. CrystEngComm 2016, 18, 9056–9066. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Stilinovic, V.; Bauzá, A.; Frontera, A.; Bartyzel, A.; Ruiz-Pérez, C.; Kirillov, A.M. Inorganic-organic hybrid materials based on PbBr2 and pyridine-hydrazone blocks-Structural and theoretical study. RSC Adv. 2016, 6, 60385–60393. [Google Scholar] [CrossRef] [Green Version]
- Dawe, L.N.; Abedin, T.S.M.; Kelly, T.K.; Thompson, L.K.; Miller, D.O.; Zhao, L.; Wilson, C.; Leech, M.A.; Howard, J.A.K. Self-assembled polymetallic square grids ([2×2] M4, [3×3] M9) and trigonal bipyramidal clysters (M5)-structural and magnetic properties. J. Mater. Chem. 2016, 16, 2645–2659. [Google Scholar] [CrossRef]
- Abedi, M.; Yesilel, O.Z.; Mahmoudi, G.; Bauzá, A.; Lofland, S.E.; Yerli, Y.; Kaminsky, W.; Garczarek, P.; Zareba, I.K.; Ienco, A.; et al. Tetranuclear Manganese(II) Complexes of Hydrazone and Carboxyhydrazone Ligands: Synthesis, Crystal Structures, Magnetic Properties, Hirshfeld Surface Analysis and DFT Calculations. Inorg. Chim. Acta 2016, 443, 101–109. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Mylonas-Margaritis, I.; Mayans, J.; Sakellakou, S.-M.; Raptopoulou, C.P.; Psycharis, V.; Escuer, A.; Perlepes, S.P. Using the Singly Deprotonated Triethanolamine to Prepare Dinuclear Lanthanide(III) Complexes: Synthesis, Structural Characterization and Magnetic Studies. Magnetochemistry 2017, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadis, Ν.C.; Granadeiro, C.Μ.; Mayans, J.; Raptopoulou, C.P.; Bekiari, V.; Cunha-Silva, L.; Psycharis, V.; Escuer, A.; Balula, S.S.; Konidaris, K.F.; et al. Mutlifunctionality in Two Families of Dinuclear Lanthanide(III) Complexes with a Tridentate Schiff-Base Ligand. Inorg. Chem. 2019, 58, 9581–9585. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, Ν.C.; Kalofolias, D.A.; Philippidis, A.; Tzani, S.; Raptopoulou, C.P.; Psycharis, V.; Milios, C.J.; Escuer, A.; Perlepes, S.P. A family of dinuclear lanthanide(III) complexes from the use of a tridentate Schiff base. Dalton Trans. 2015, 4, 10200–10209. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, N.C.; Mylonas-Margaritis, I.; Psycharis, V.; Raptopoulou, C.P.; Kalofolias, D.A.; Milios, C.J.; Klouras, N.; Perlepes, S.P. Dinuclear, tetrakis(acetatο)-bridged lanthanide(III) complexes from the use of 2-acetylpyridine hydrazοne. Inorg. Chem. Commun. 2015, 51, 99–102. [Google Scholar] [CrossRef]
- Nikolaou, H.; Terzis, A.; Raptopoulou, C.P.; Psycharis, V.; Bekiari, V.; Perlepes, S.P. Unique Dinuclear, Tetrakis(nitrato-O,O’)-Bridged Lanthanide(III) Complexes from the Use of Pyridine-2-Amidoxime: Synthesis, Structural Studies and Spectroscopic Characterization. J. Surf. Interfaces Mater. 2014, 2, 311–318. [Google Scholar] [CrossRef]
- Anastasiadis, Ν.C.; Granadeiro, C.Μ.; Klouras, N.; Cunha-Silva, L.; Raptopoulou, C.P.; Psycharis, V.; Bekiari, V.; Balula, S.S.; Escuer, A.; Perlepes, S.P. Dinuclear Lanthanide(III) Complexes by Metal-Ion-Assisted Hydration of Di-2-pyridyl ketone Azine. Inorg. Chem. 2013, 52, 4145–4147. [Google Scholar] [CrossRef]
- Bekiari, V.; Thiakou, Κ.A.; Raptopoulou, C.P.; Perlepes, S.P.; Lianos, P. Structure and photophysical behavior of 2,2-bipyrimidine/lanthanide ion complexes in various environments. J. Lumin. 2008, 128, 481–488. [Google Scholar] [CrossRef]
- Messimeri, A.; Papadimitriou, C.; Raptopoulou, C.P.; Escuer, A.; Perlepes, S.P.; Boudalis, A.K. The benzoate/nitrate/2,2’:6’,2’’-terpyridine ‘blend’ in lanthanide(III) chemistry. Relevance to the separation of lanthanides and actinides by solvent extraction. Inorg. Chem. Commun. 2007, 10, 800–804. [Google Scholar] [CrossRef]
- Τhiakou, Κ.A.; Nastopoulos, V.; Terzis, A.; Raptopoulou, C.P.; Perlepes, S.P. Di-2-pyridyl ketone in lanthanide(III) chemistry: Mononuclear and dinuclear erbium(III) complexes. Polyhedron 2006, 25, 539–549. [Google Scholar] [CrossRef]
- Thiakou, K.A.; Bekiari, V.; Raptopoulou, C.P.; Psycharis, V.; Lianos, P.; Perlepes, S.P. Dinuclear lanthanide(III) complexes from the use of di-2-pyridyl ketone: Preparation, structural characterization and spectroscopic studies. Polyhedron 2006, 25, 2869–2879. [Google Scholar] [CrossRef]
- Pilichos, E.; Mylonas-Margaritis, I.; Kontos, A.P.; Psycharis, V.; Klouras, N.; Raptopoulou, C.P.; Perlepes, S.P. Coordination and metal ion-mediated transformation of a polydentate ligand containing oxime, hydrazine and picolinoyl functionalities. Inorg. Chem. Commun. 2018, 94, 48–52. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; Wiley: New York, NY, USA, 1986; pp. 254–257. [Google Scholar]
- Maniaki, D.; Mylonas-Margaritis, I.; Mayans, J.; Savvidou, A.; Raptopoulou, C.P.; Bekiari, V.; Psycharis, V.; Escuer, A.; Perlepes, S.P. Slow magnetic relaxation and luminescence properties in lanthanide(III)/anil complexes. Dalton Trans. 2018, 47, 11859–11872. [Google Scholar] [CrossRef] [PubMed]
- Carnall, W.T.; Fields, P.R.; Kajnak, K. Electronic Energy Levels in the Trivalent Lanthanide Aquo Ions. I. Pr3+, Nd3+, Pm3+, Sm3+, Dy3+, Ho3+, Er3+, and Tm3+. J. Chem. Phys. 1968, 49, 4424–4442. [Google Scholar] [CrossRef]
- Carnall, W.T.; Fields, P.R.; Rajnak, K. Electronic Energy Levels of the Trivalent Lanthanide Aquo Ions. III. Tb3+. J. Chem. Phys. 1968, 49, 4447–4449. [Google Scholar] [CrossRef]
- Karraker, D.G. The Hypersensitive Transitions of Nd3+, Ho3+ and Er3+ Ions. Inorg. Chem. 1968, 7, 473–479. [Google Scholar] [CrossRef]
- Karraker, D.G. Hypersensitive Transitions of Six-, Seven-, and Eight-Coordinate Neodymium, Holmium, and Erbium Chelates. Inorg. Chem. 1967, 6, 1863–1868. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Girera, J.; Alemany, P.; Alvarez, S. SHAPE, Continuous Shape Measures Calculation; Version 2.0; Universitat de Barcelona: Barcelona, Spain, 2010. [Google Scholar]
- Long, J.; Habib, F.; Lin, P.-H.; Korobkov, I.; Enright, G.; Ungur, L.; Wernsdorfer, W.; Chibotaru, L.F.; Murugesu, M. Single-Molecule Magnet Behavior for an Antiferromagnetically Superexchange-Coupled Dinuclear Dysprosium(III) Complex. J. Am. Chem. Soc. 2011, 133, 5319–5328. [Google Scholar] [CrossRef]
- Roy, L.E.; Hughbanks, T. Magnetic Coupling in Dinuclear Gd Complexes. J. Am. Chem. Soc. 2006, 128, 568–575. [Google Scholar] [CrossRef]
- Lecren, L.; Wernsdorfer, W.; Li, Y.-G.; Roubeau, O.; Miyasaka, H.; Clérac, R. Quantum Tunneling and Quantum Phase Interference in a [MnII2MnIII2] Single-Molecule Magnet. J. Am. Chem. Soc. 2005, 127, 11311–11317. [Google Scholar] [CrossRef]
- Van Vleck, I.H. Paramagnetic Relaxation Times for Titanium and Chrome Alum. Phys. Rev. 1940, 57, 426–447. [Google Scholar] [CrossRef]
- Mazarakioti, E.C.; Regier, J.; Cunha-Silva, L.; Wernsdorfer, W.; Pilkington, M.; Tang, J.; Stamatatos, T.C. Large Energy Barrier and Magnetization Hysteresis at 5 K for a Symmetric {Dy2} Complex with Spherical Tricapped Trigonal Prismatic DyIII ions. Inorg. Chem. 2017, 56, 3568–3578. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Reta, D.; Chilton, N.F. Uncertainty estimates for magnetic relaxation times and magnetic relaxation parameters. Phys. Chem. Chem. Phys. 2019, 21, 23567–23575. [Google Scholar] [CrossRef]
- Shrivastava, K.N. Theory of Spin–Lattice Relaxation. Phys. Stat. Sol. A 1983, 117, 437–458. [Google Scholar] [CrossRef]
- Abragam, A.; Bleaney, B. Electron Paramagnetic Resonance of Transition Ions; Dover: New York, NY, USA, 1986. [Google Scholar]
- Ding, M.; Hickey, A.K.; Pink, M.; Telser, J.; Tierney, D.L.; Amoza, M.; Rouzières, M.; Ozumzerzifon, T.J.; Hoffert, W.A.; Shores, M.P.; et al. Magnetization Slow Dynamics in Ferrocenium Complexes. Chem. Eur. J. 2019, 25, 10625–10632. [Google Scholar] [CrossRef] [PubMed]
- Pescia, J. La relaxation des spins électronique avec le réseau. J. Phys. 1966, 27, 782–800. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SADABS Version 2.03; Bruker Analytical X-Ray Systems: Madison, WI, USA, 2000. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–4 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1∙2MeOH∙2H2O | 2∙2MeOH∙1.5H2O | 3∙2.5MeOH | 4∙3MeOH∙0.5H2O |
|---|---|---|---|---|
| Formula | C26H24Gd2N12O15∙2(CH4O)∙2(H2O) | (C26H24Tb2N12O15)2∙4(CH4O)∙3(H2O) | (C27H26Dy2N12O15)2∙5(CH4O) | (C26H24Er2N12O15)2∙6(CH4O)∙(H2O) |
| Formula weight | 1159.19 | 2307.04 | 1151.67 | 2368.45 |
| Crystal color | yellow | yellow | yellow | yellow |
| Crystal size, mm | 0.12 × 0.11 × 0.04 | 0.20 × 0.17 × 0.06 | 0.22 × 0.15 × 0.05 | 0.20 × 0.17 × 0.06 |
| Crystal system | triclinic | triclinic | triclinic | triclinic |
| Space group | Pī | Pī | Pī | Pī |
| Temperature, K | 120 | 120 | 120 | 120 |
| Radiation, Å | Mo Kα, 0.71073 | Mo Kα, 0.71073 | Mo Kα, 0.71073 | Mo Kα, 0.71073 |
| a, Å | 10.4504(10) | 10.3899(16) | 10.3194(8) | 10.3551(8) |
| b, Å | 12.3976(12) | 12.4185(18) | 12.1909(9) | 12.4468(10) |
| c, Å | 17.4313(17) | 17.609(3) | 17.8985(14) | 17.7769(13) |
| α, ° | 74.879(5) | 73.749(7) | 71.376(4) | 71.992(4) |
| β, ° | 85.147(5) | 84.738(7) | 84.075(4) | 83.975(4) |
| γ, ° | 68.821(4) | 69.114(7) | 71.252(4) | 69.411(4) |
| Volume, Å3 | 2032.8(3) | 2037.8(5) | 2020.6(3) | 2039.8(3) |
| Z | 2 | 1 | 1 | 1 |
| Calculated density, g·cm−3 | 1.894 | 1.880 | 1.913 | 1.928 |
| Absorption coefficient, mm−1 | 3.325 | 3.531 | 3.758 | 4.176 |
| θmin–θmax, ° | 1.918–29.713 | 2.098–28.438 | 2.084–25.505 | 2.101–30.489 |
| Reflections collected/unique | 47128/10876 | 30434/9998 | 120082/7400 | 44288/12236 |
| Completeness to 2θ | 0.940 | 0.974 | 0.984 | 0.996 |
| Rint | 0.0374 | 0.0692 | 0.0600 | 0.0296 |
| Refined parameters/restraints | 563/3 | 578/2 | 530/1 | 587/2 |
| R1[I > 2σ(I)] a, wR2 b (all data) | 0.0330, 0.0646 | 0.0545, 0.1100 | 0.0338, 0.0810 | 0.0241, 0.0554 |
| Goodness-of-fit on F2 | 1.059 | 1.069 | 1.107 | 1.036 |
| Interatomic Distances (Å) | ||||
|---|---|---|---|---|
| Ln = Gd (1∙2MeOH∙2H2O) | Ln = Tb (2∙2MeOH∙1.5H2O) | Ln = Dy (3∙2.5MeOH) | Ln = Er (4∙3MeOH∙0.5H2O) | |
| Ln1∙∙∙Ln2 | 4.000(1) | 3.969(1) | 3.945(1) | 3.933(1) |
| Ln1-O3 | 2.583(3) | 2.574(5) | 2.586(4) | 2.557(2) |
| Ln1-O4 | 2.500(3) | 2.475(5) | 2.468(4) | 2.448(2) |
| Ln1-O6 | 2.492(2) | 2.489(5) | 2.468(4) | 2.461(2) |
| Ln1-O8 | 2.543(3) | 2.511(5) | 2.485(4) | 2.489(2) |
| Ln1-O1 | 2.394(2) | 2.378(4) | 2.359(4) | 2.343(2) |
| Ln1-O2 | 2.389(2) | 2.380(5) | 2.364(3) | 2.347(2) |
| Ln1-N1 | 2.653(3) | 2.632(7) | 2.614(5) | 2.610(2) |
| Ln1-N2 | 2.573(3) | 2.551(6) | 2.523(4) | 2.517(2) |
| Ln1-N5 | 2.550(3) | 2.533(6) | 2.534(5) | 2.514(2) |
| Ln1-N6 | 2.547(3) | 2.534(6) | 2.512(4) | 2.506(2) |
| Ln2-O9 | 2.488(3) | 2.470(5) | 2.462(4) | 2.451(2) |
| Ln2-O11 | 2.458(3) | 2.465(5) | 2.436(4) | 2.443(2) |
| Ln2-O12 | 2.472(3) | 2.457(5) | 2.443(4) | 2.435(2) |
| Ln2-O14 | 2.472(3) | 2.452(5) | 2.439(4) | 2.418(2) |
| Ln2-O1 | 2.353(2) | 2.337(5) | 2.326(3) | 2.310(2) |
| Ln2-O2 | 2.383(2) | 2.361(4) | 2.348(4) | 2.344(2) |
| Ln2-N4 | 2.562(3) | 2.545(5) | 2.509(5) | 2.505(2) |
| Ln2-N8 | 2.528(3) | 2.511(6) | 2.503(5) | 2.478(2) |
| Ln2-O15/O1M | 2.402(3) | 2.383(5) | 2.383(4) | 2.347(2) |
| C6-N2 | 1.289(5) | 1.301(9) | 1.297(7) | 1.295(3) |
| N2-N3 | 1.411(4) | 1.406(7) | 1.403(6) | 1.411(3) |
| N3-C8 | 1.294(5) | 1.277(9) | 1.302(7) | 1.302(3) |
| C8-O1 | 1.308(4) | 1.309(8) | 1.299(6) | 1.307(3) |
| C19-N6 | 1.290(4) | 1.292(9) | 1.314(7) | 1.292(3) |
| N6-N7 | 1.398(4) | 1.386(8) | 1.396(6) | 1.402(3) |
| N7-C21 | 1.303(4) | 1.311(8) | 1.292(7) | 1.305(3) |
| C21-O2 | 1.313(4) | 1.304(8) | 1.309(6) | 1.310(3) |
| Ln-O-Ln Bond Angles (°) | ||||
| Ln1-O1-Ln2 | 114.8(1) | 114.7(2) | 114.7(1) | 115.4(1) |
| Ln1-O2-Ln2 | 113.9(1) | 113.7(2) | 113.7(1) | 113.9(1) |
| Compound a | Coordination Mode b,c | Nuclearity /Dimensionality | Coordination Geometry d | Ref. |
|---|---|---|---|---|
| [CdBr2(HL)] | 1.10011 | Mononuclear | tbp | [49] |
| [Ln(NO3)3(HL)(MeOH)2] (Ln = La, Ce) | 1.10011 | Mononuclear | cpa | [50] |
| [Nd(NO3)3(HL)(H2O)] | 1.10011 | Mononuclear | bsa | [50] |
| [PdCl2(HL)] | 1.01100 | Mononuclear | sp | [51] |
| [HgX2(HL)] (X = Cl, Br) | 1.10011 | Mononuclear | spy | [52] |
| [HgI2(HL)(H2O)] | 1.10011 | Mononuclear | oct | [52] |
| {[Pb3Br6(HL)2]}n | 1.10011 | 1D (metal-organic ribbon) | 7-coordinate e, oct | [53] |
| [PdCl(L) | 1.01011 | Mononuclear | sp | [51] |
| [PdCl(L) | 1.01100 | Mononuclear | sp | [51] |
| [Cu4(L)4(H2O)2](NO3)4 | 2.21011, 2.11111 | Rectangular [2 × 2] grid | spy, oct | [47] |
| [Mn4(CF3SO3)(L)4(H2O)3](CF3SO3)3 | 2.21011 | Square [2 × 2] grid | oct | [54] |
| [Mn5(L)6](ClO4)4 | 2.21011 | Trigonal bipyramidal topology | oct | [54] |
| [Mn4(N3)4(L)4] | 2.21011 | Square [2 × 2] grid | oct | [55] |
| [Cu4Br2(L)4]Br2 | 2.21011, 2.11111 | Rectangular [2 × 2] grid | spy, oct | [48] |
| [Ni4(NO3)2(L)4(H2O)](NO3)2 | 2.21011 | Square [2 × 2] grid | oct | [48] |
| [Co2(L)3](ClO4)3 | 2.01111 | Dinuclear helicate | oct | [48] |
| [Co(L)2](ClO4) | 1.10011 | Mononuclear | oct | [48] |
| [Ln2(ΝO3)4(L)2(H2O)] (Ln = Gd, Τb, Er) | 2.21011 | Dinuclear | sph, scsa | this work |
| [Dy2(NO3)4(L)2(MeOH)] | 2.21011 | Dinuclear | sph, scsa | this work |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniaki, D.; Perlepe, P.S.; Pilichos, E.; Christodoulou, S.; Rouzières, M.; Dechambenoit, P.; Clérac, R.; Perlepes, S.P. Asymmetric Dinuclear Lanthanide(III) Complexes from the Use of a Ligand Derived from 2-Acetylpyridine and Picolinoylhydrazide: Synthetic, Structural and Magnetic Studies. Molecules 2020, 25, 3153. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25143153
Maniaki D, Perlepe PS, Pilichos E, Christodoulou S, Rouzières M, Dechambenoit P, Clérac R, Perlepes SP. Asymmetric Dinuclear Lanthanide(III) Complexes from the Use of a Ligand Derived from 2-Acetylpyridine and Picolinoylhydrazide: Synthetic, Structural and Magnetic Studies. Molecules. 2020; 25(14):3153. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25143153
Chicago/Turabian StyleManiaki, Diamantoula, Panagiota S. Perlepe, Evangelos Pilichos, Sotirios Christodoulou, Mathieu Rouzières, Pierre Dechambenoit, Rodolphe Clérac, and Spyros P. Perlepes. 2020. "Asymmetric Dinuclear Lanthanide(III) Complexes from the Use of a Ligand Derived from 2-Acetylpyridine and Picolinoylhydrazide: Synthetic, Structural and Magnetic Studies" Molecules 25, no. 14: 3153. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25143153