
Exo⇔Endo Isomerism, MEP/DFT, XRD/HSA-Interactions of 2,5-Dimethoxybenzaldehyde: Thermal, 1BNA-Docking, Optical, and TD-DFT Studies

 , , , ,
, , , ,  , ,
, ,
Abstract
:
1. Introduction
2. Experimental Section
2.1. Computational Methodology
2.2. XRD and HSA
2.3. 2,5-Dimethoxybenzaldehyde Crystallization
2.4. BNA Docking
3. Results and Discussion
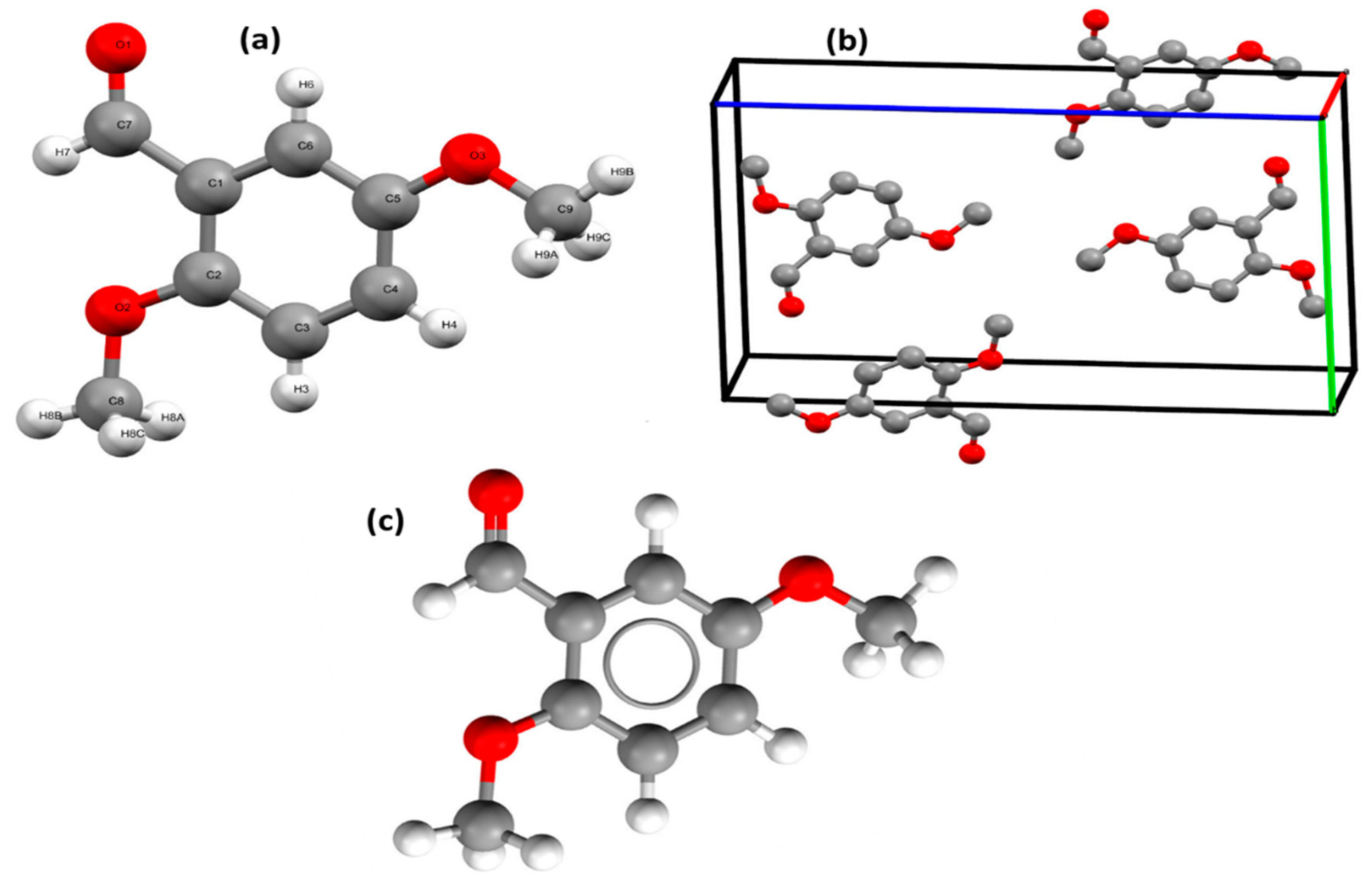
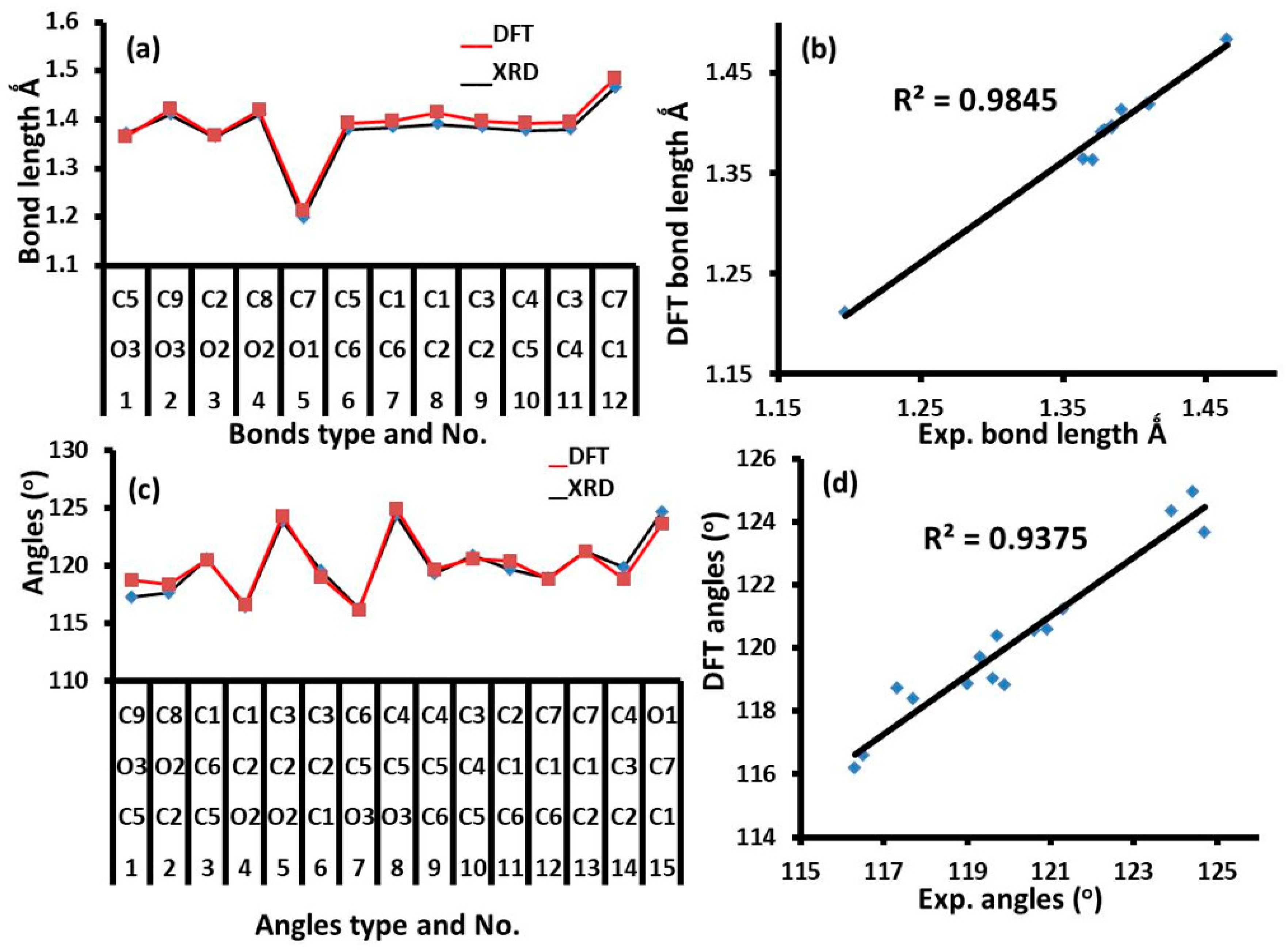
3.1. XRD and DFT Structure Analysis
3.2. B3LYP/6-311G(d,p) Structures
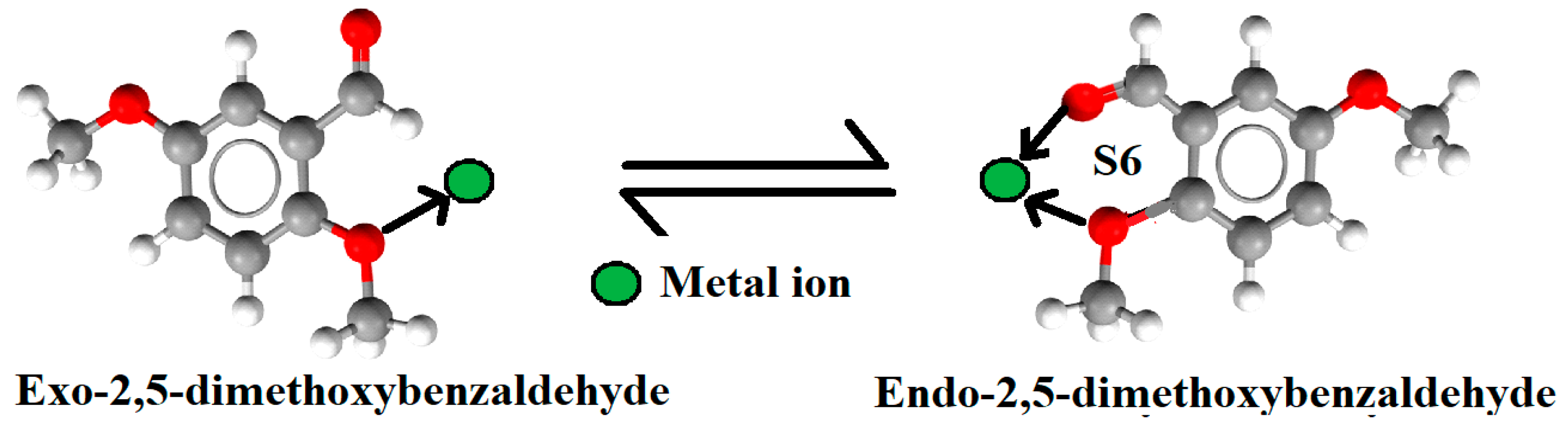
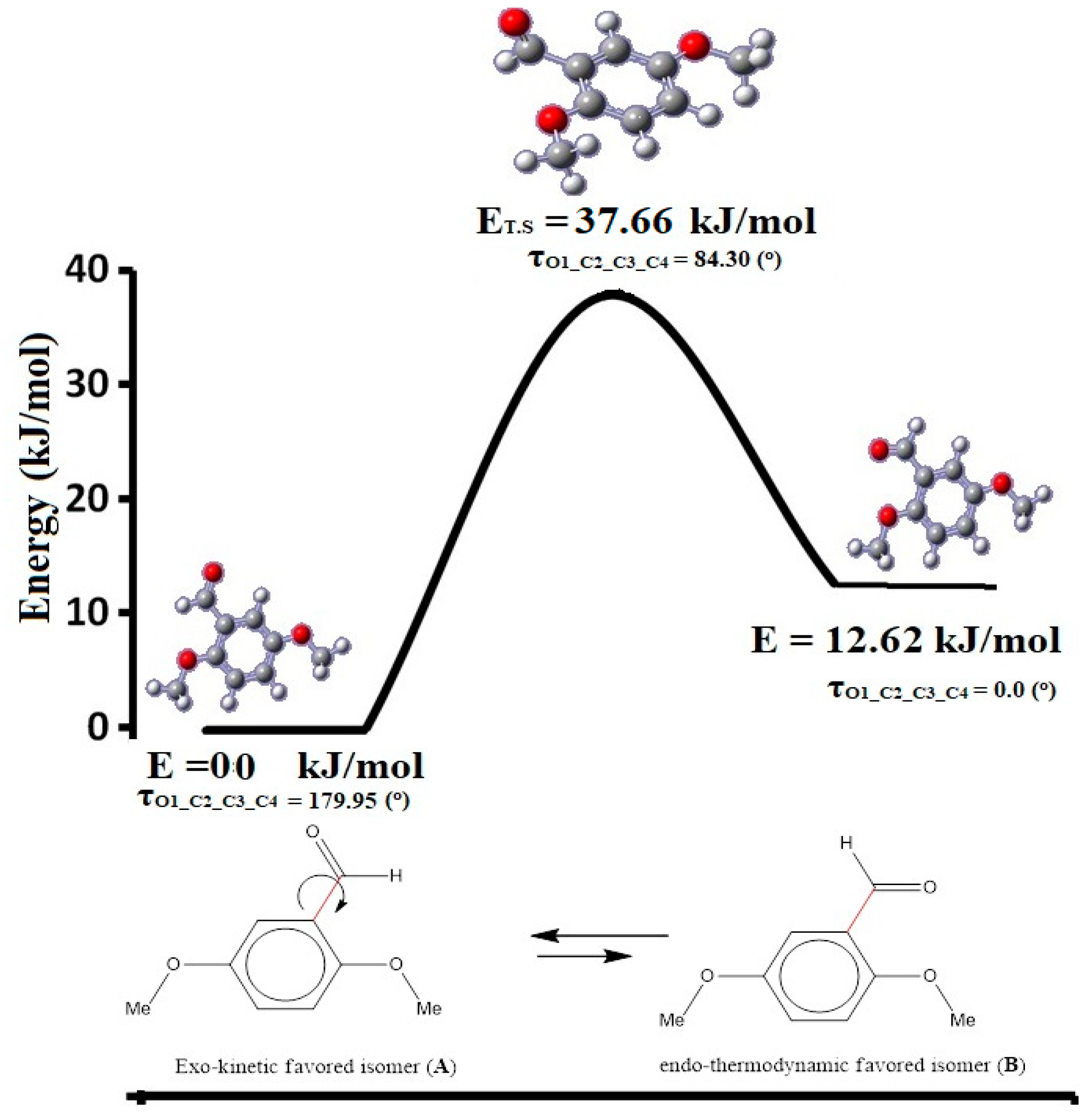
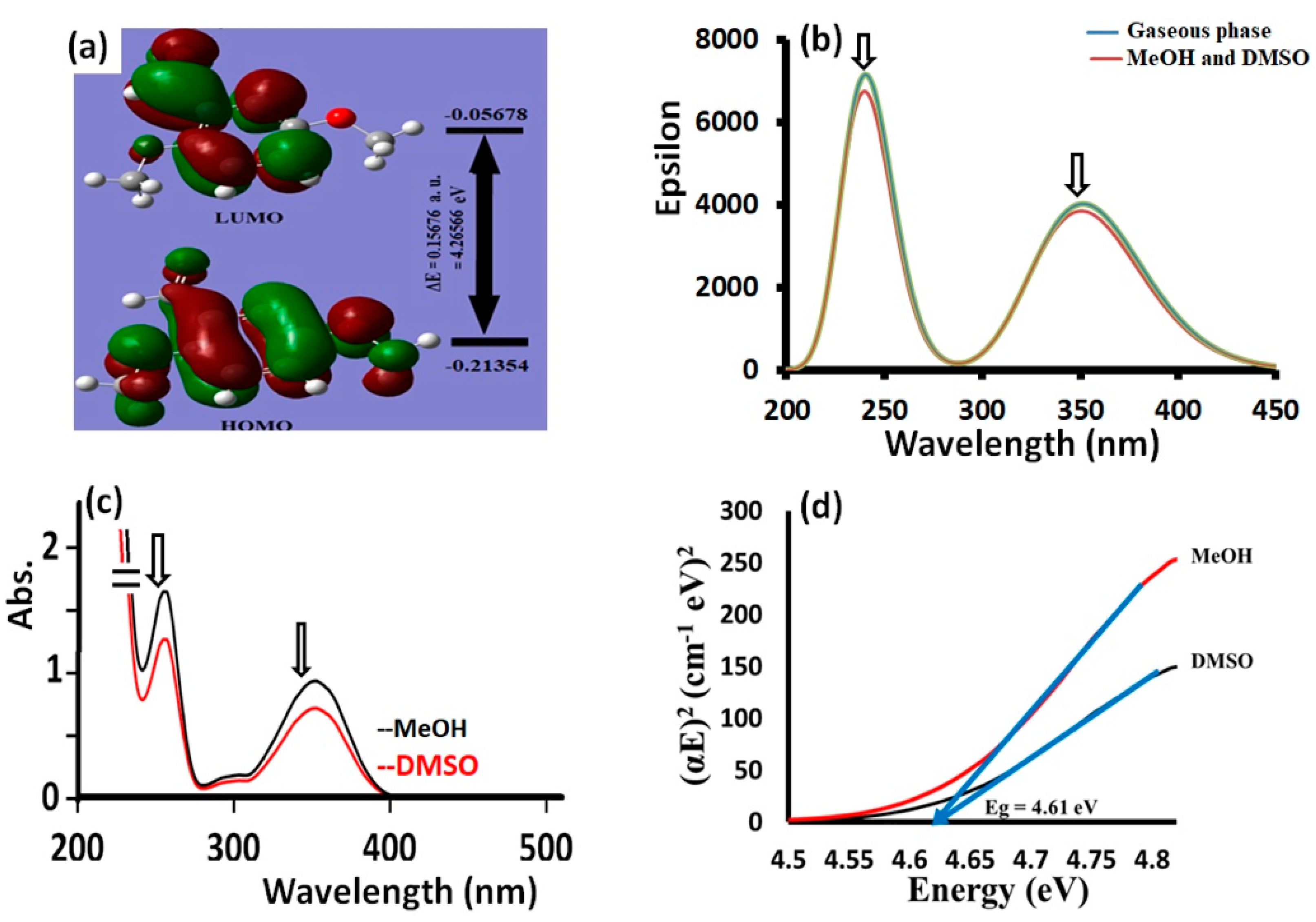
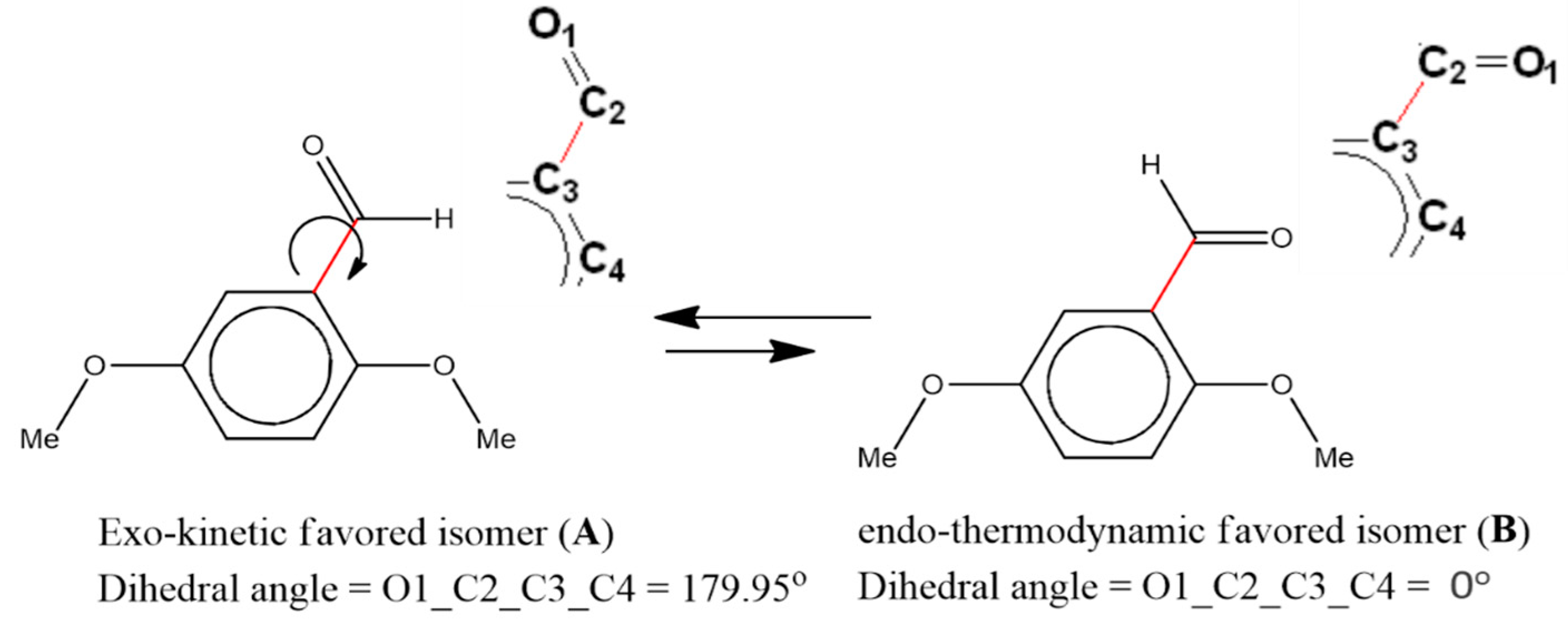
3.3. Exo⇔Endo Computational Isomerism
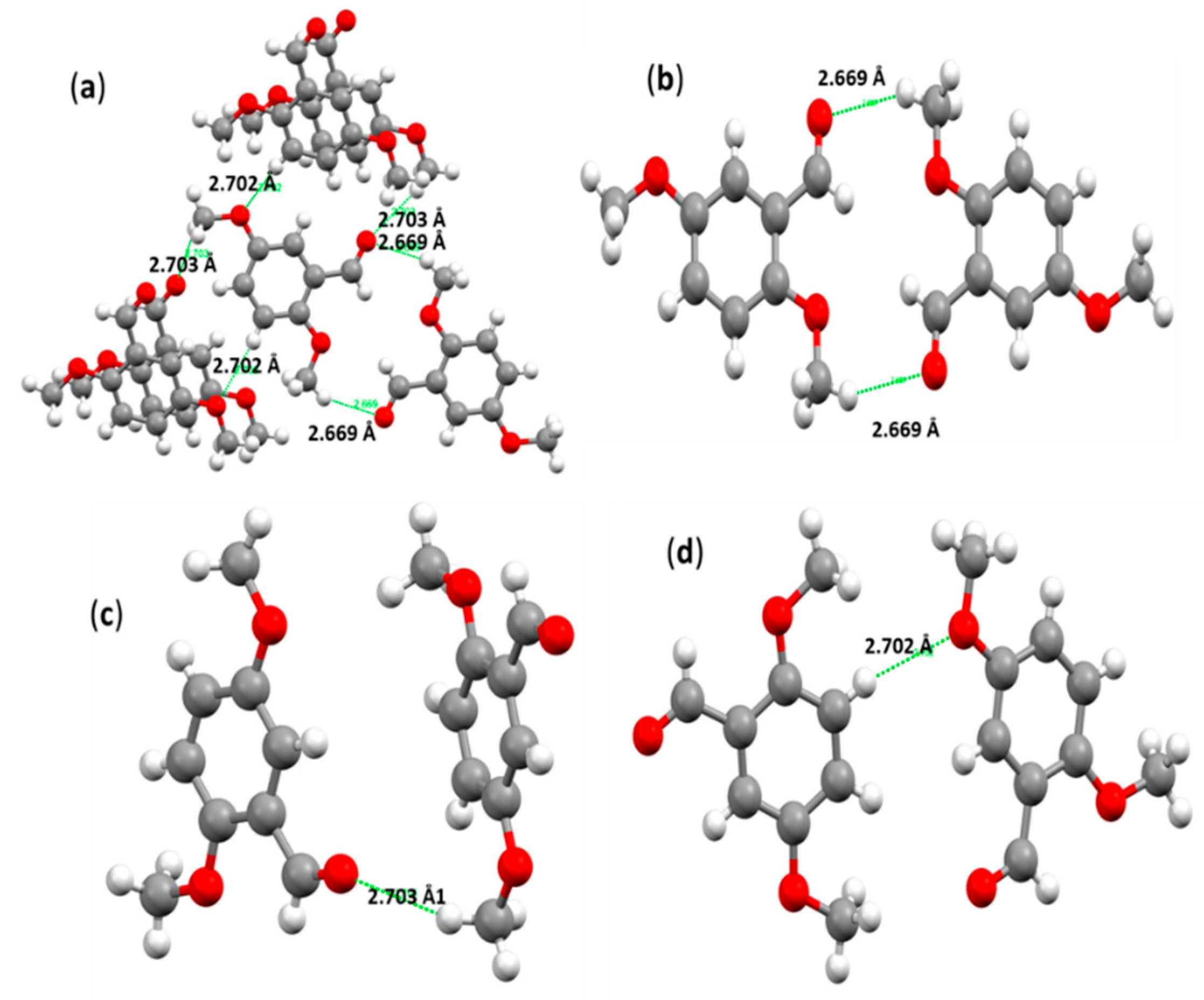
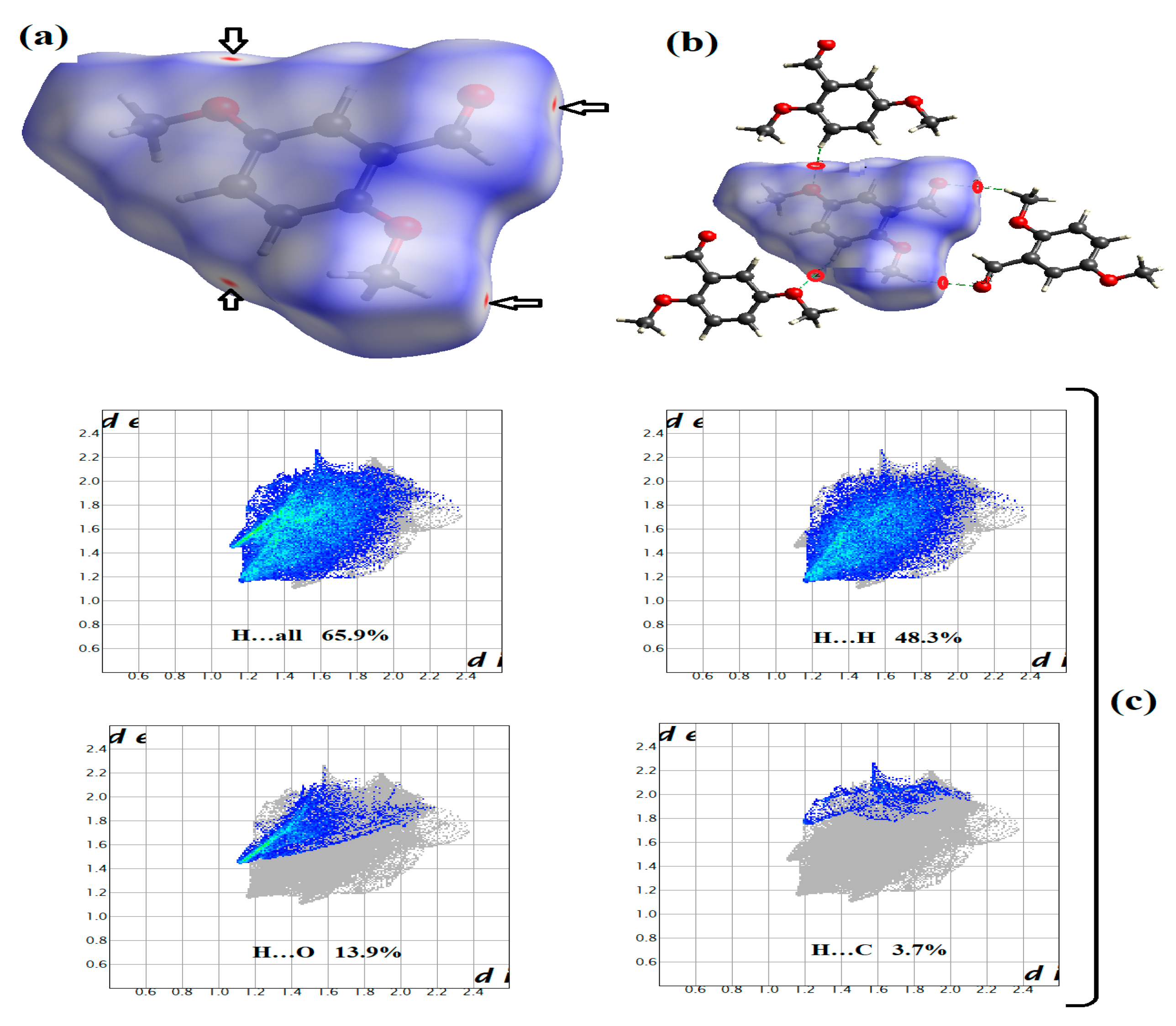
3.4. Crystal Interactions and HSA Investigation
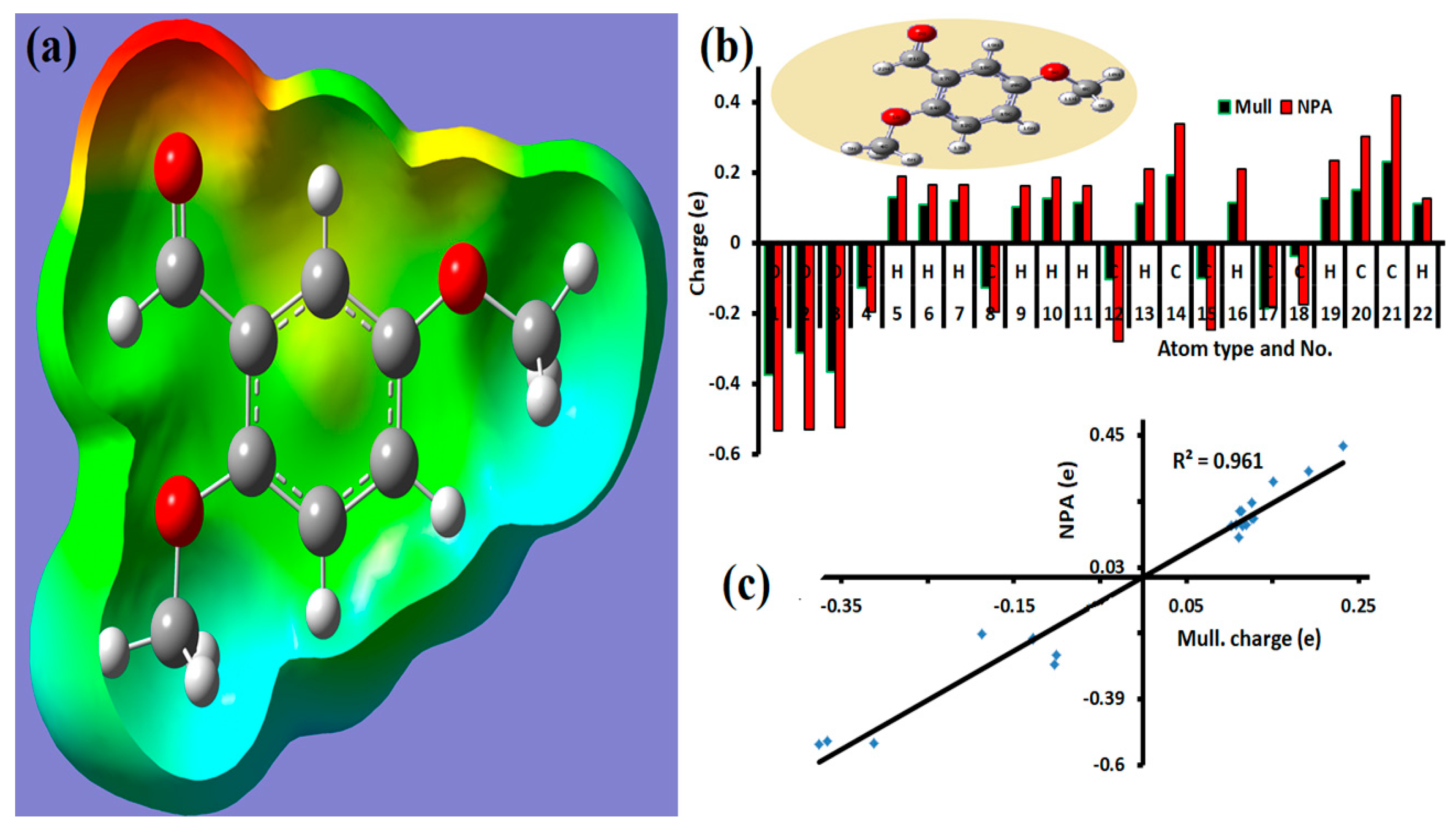
3.5. MEP Analysis and Atomic Charge Populations
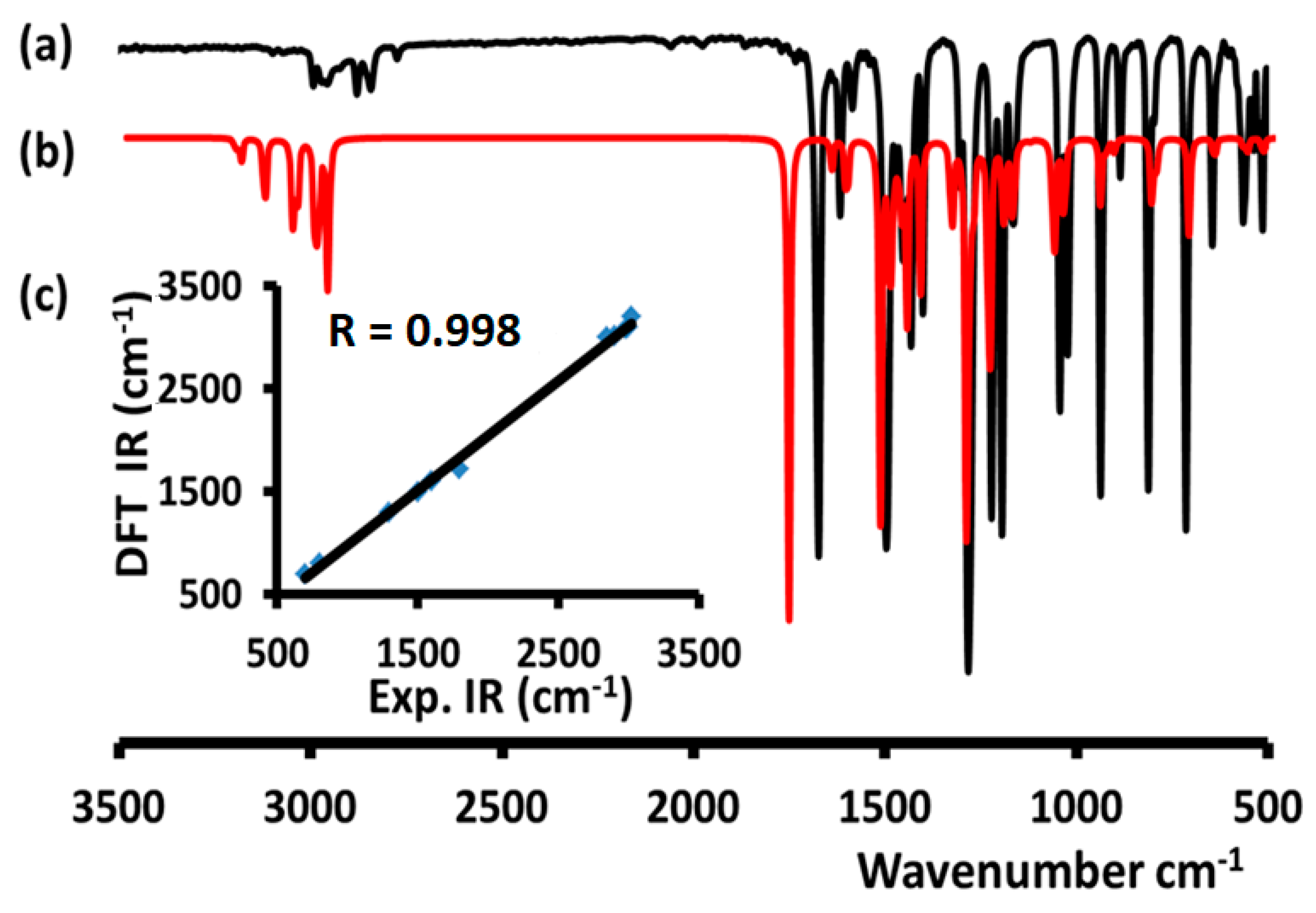
3.6. FTIR Investigations
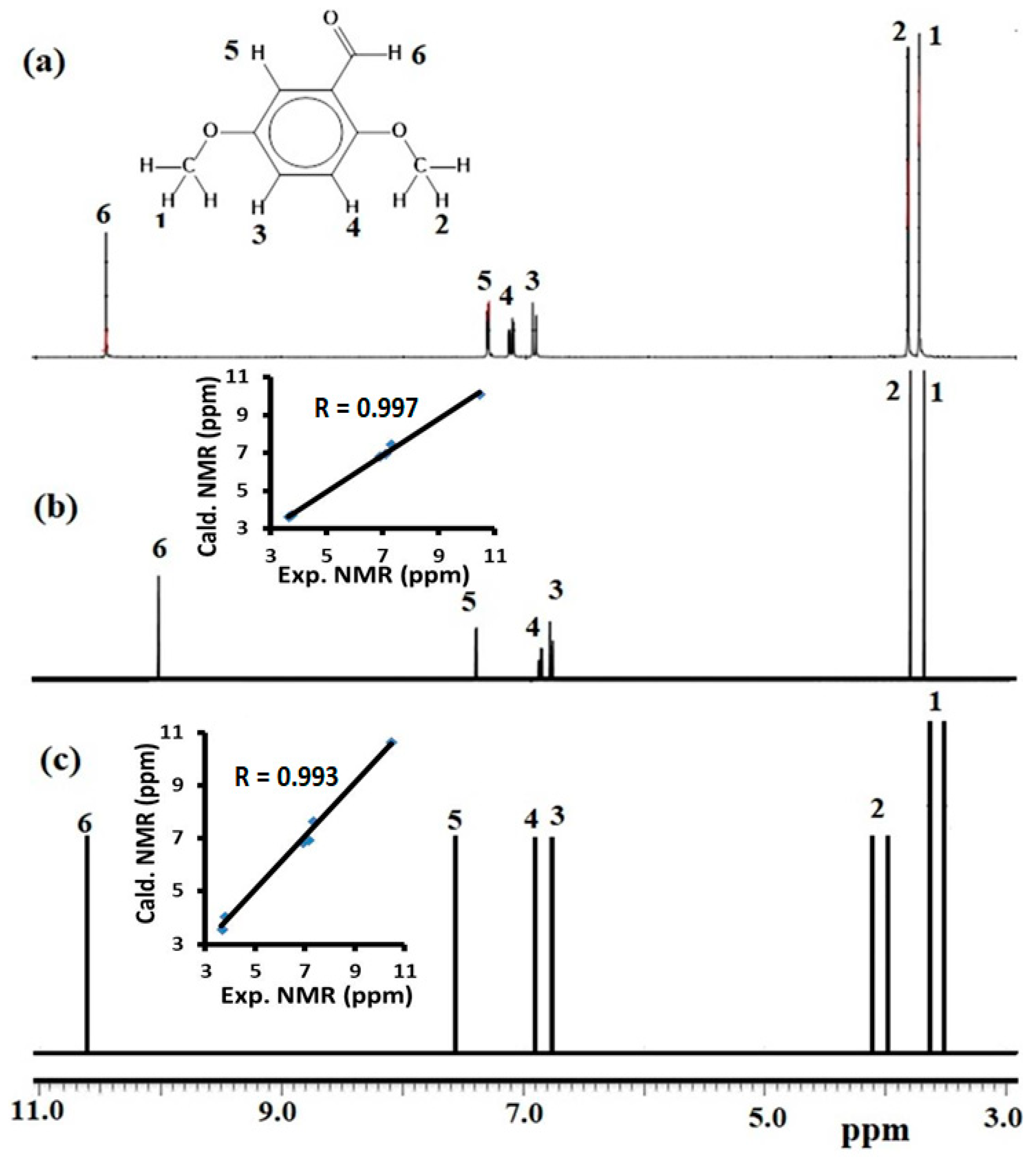
3.7. Computed and Experimental 1H NMR
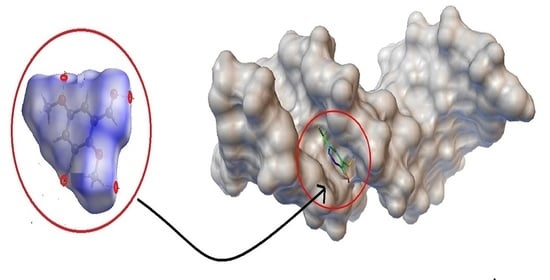
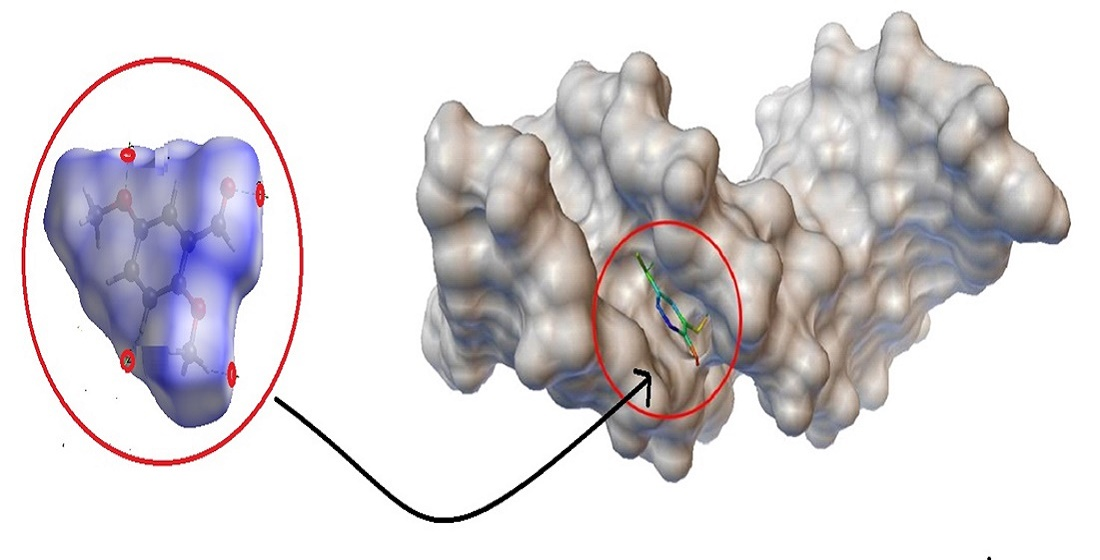
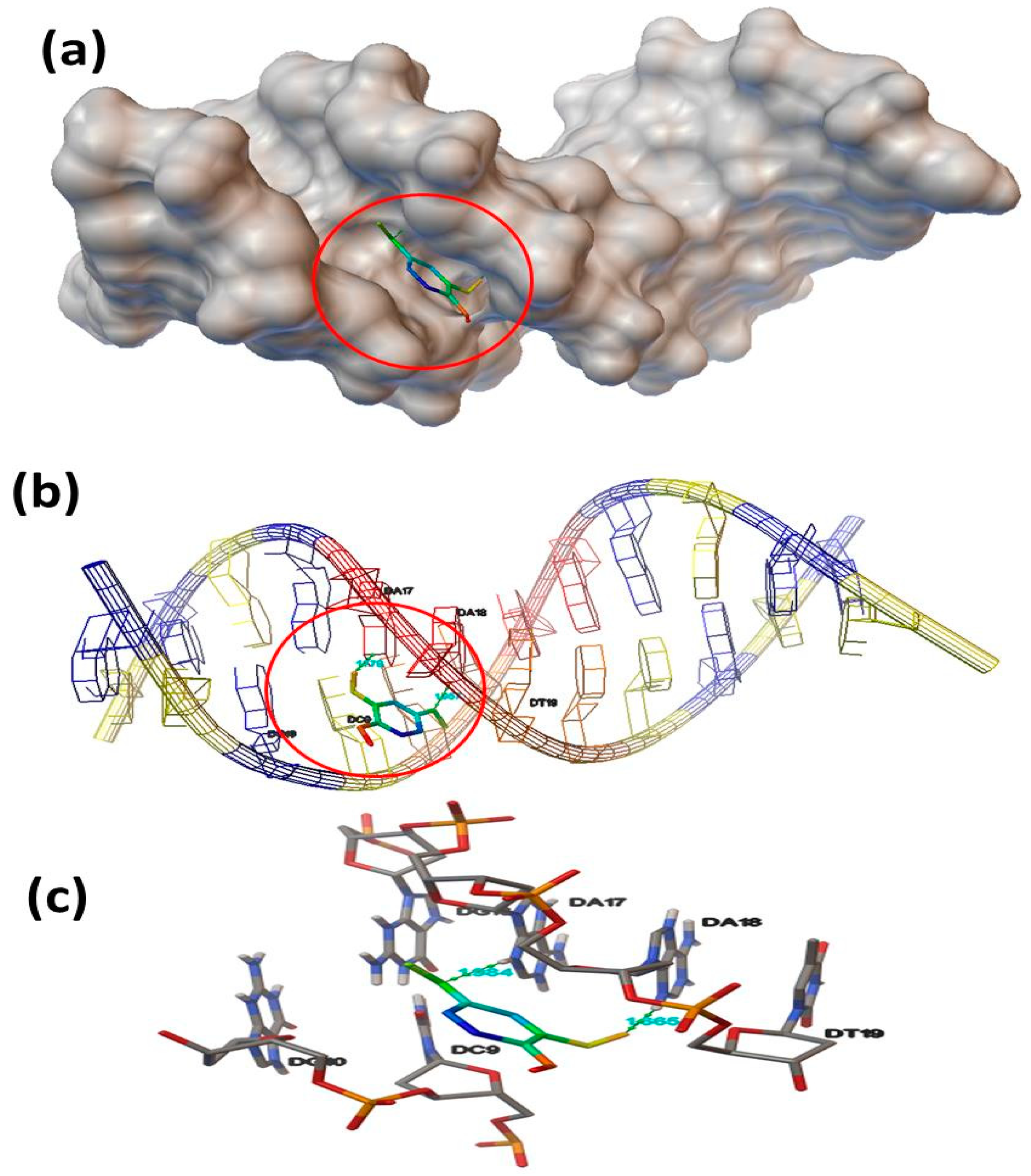
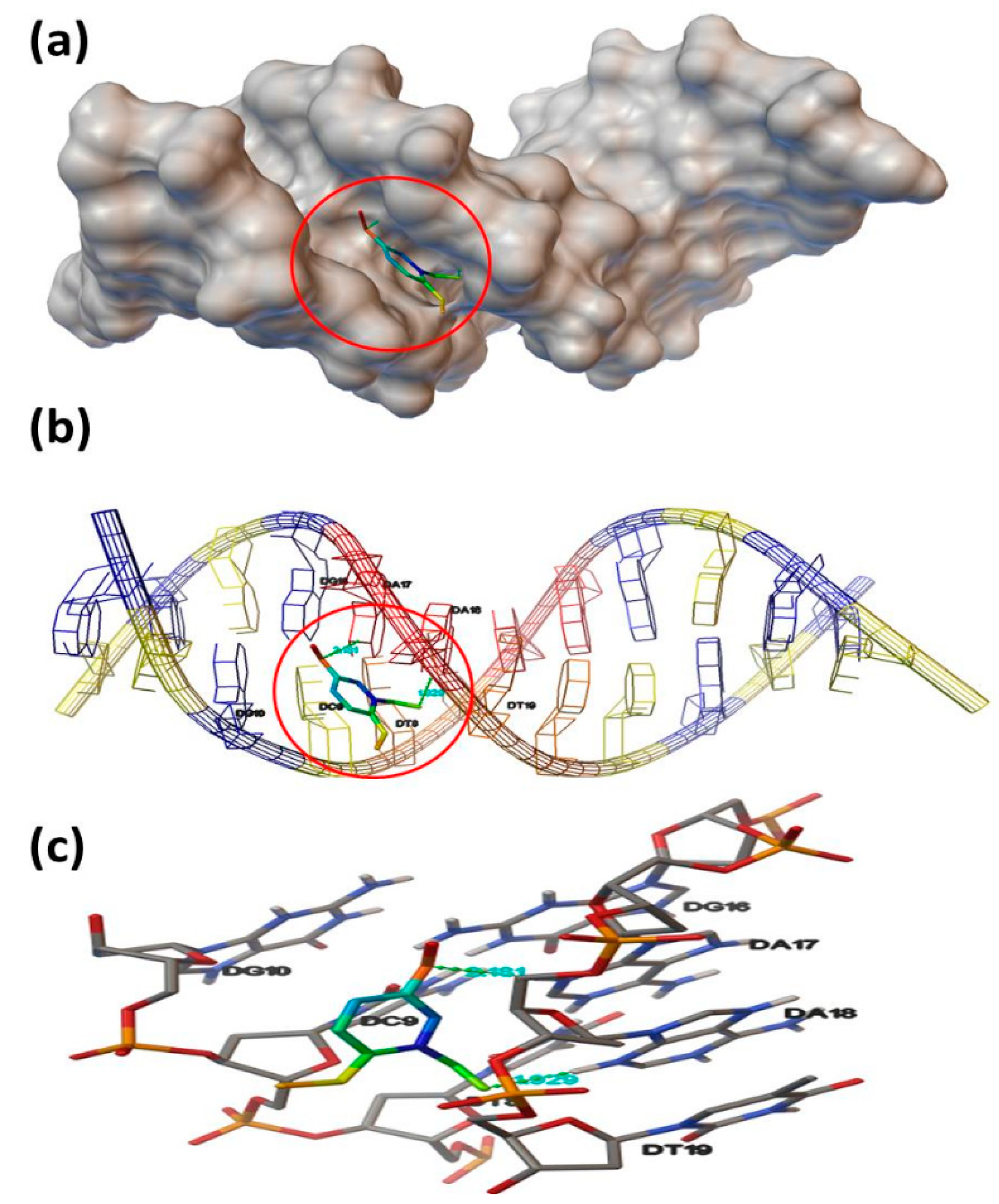
3.8. Molecular Docking
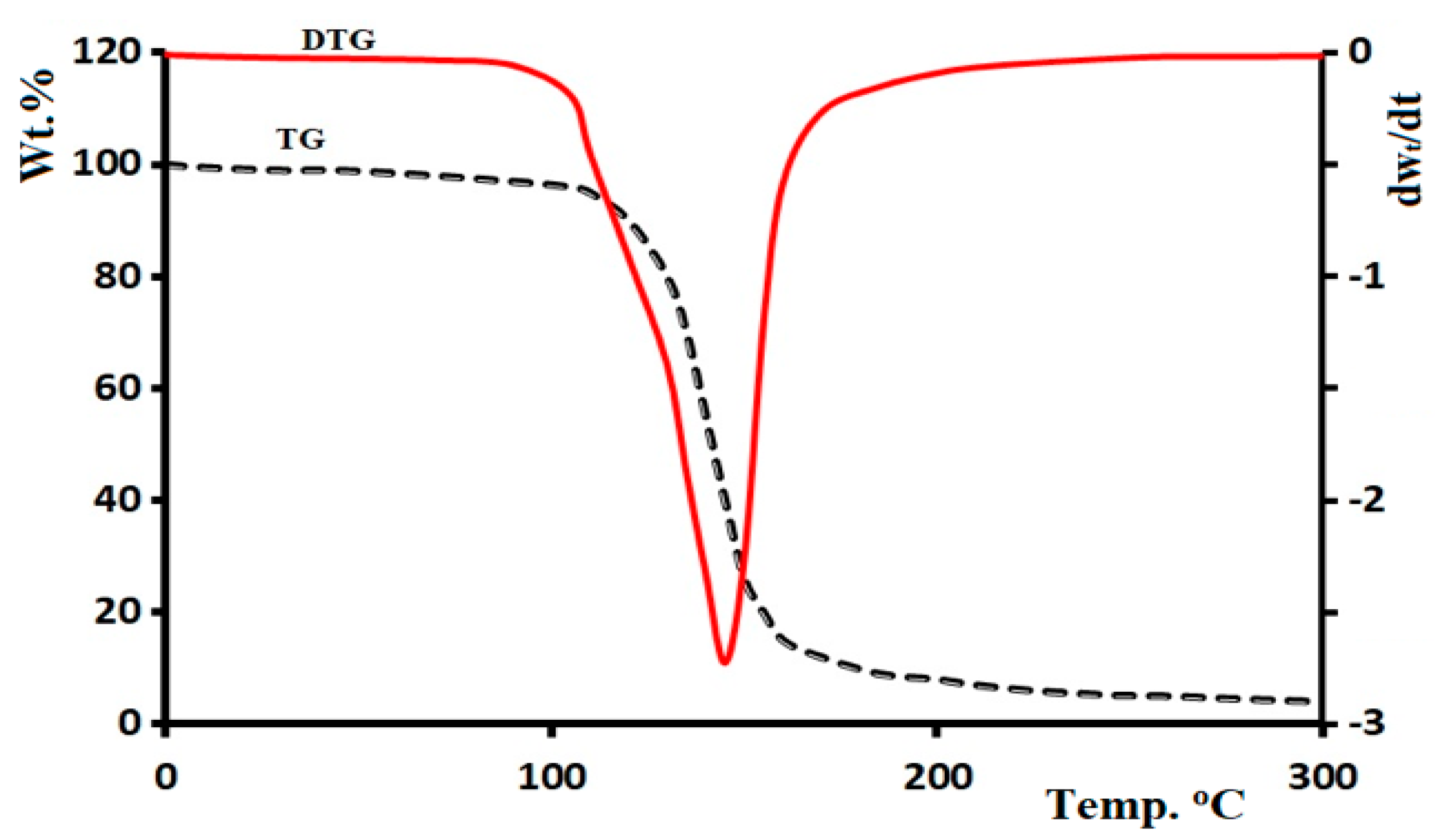
3.9. Thermal Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Avci, D.; Tamer, Ö.; Başoğlu, A.; Atalay, Y. 5-Methyl-2-thiophenecarboxaldehyde: Experimental and TD/DFT study. J. Mol. Struct. 2018, 1174, 52–59. [Google Scholar] [CrossRef]
- Da Silva, M.A.R.; Santos, A.F.L. Experimental thermochemical study of 3-acetyl-2-methyl-5-phenylthiophene. J. Chem. Thermodyn. 2010, 42, 128–133. [Google Scholar] [CrossRef]
- Brugman, S.J.T.; Engwerda, A.H.J.; Kalkman, E.; De Ronde, E.; Tinnemans, P.; Vlieg, E. The crystal structures of four dimethoxybenzaldehyde isomers. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 38–42. [Google Scholar] [CrossRef]
- Ma, C.; Liu, S.; Zhang, S.; Xu, T.; Yu, X.; Gao, Y.; Zhai, C.; Li, C.; Lei, C.; Fan, S.; et al. Evidence and perspective for the role of the NLRP3 inflammasome signaling pathway in ischemic stroke and its therapeutic potential (Review). Int. J. Mol. Med. 2018, 42, 2979–2990. [Google Scholar] [CrossRef] [Green Version]
- Atta, A.K.; Kim, S.-B.; Cho, D.-G. Catalytic Oxidative Conversion of Aldehydes to Carboxylic Esters and Acids under Mild Conditions. Bull. Korean Chem. Soc. 2011, 32, 2070–2072. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, D.M.; Charlton, A. Methods for direct generation of a-alkyl-substituted aldehydes. Tetrahedron 2014, 70, 2207. [Google Scholar] [CrossRef]
- Ding, Y.-Q.; Cui, Y.; Li, T.-D. New Views on the Reaction of Primary Amine and Aldehyde from DFT Study. J. Phys. Chem. A 2015, 119, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, V.; Santhanam, R.; Rani, T.; Rosi, H.; Mohan, S. Conformational, vibrational, NMR and DFT studies of N-methylacetanilide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 104, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Sevvanthi, S.; Muthu, S.; Raja, M. Molecular docking, vibrational spectroscopy studies of (RS)-2-(tert-butylamino)-1-(3-chlorophenyl)propan-1-one: A potential adrenaline uptake inhibitor. J. Mol. Struct. 2018, 1173, 251–260. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Ghabbour, H.A.; Atef, S.; Zarrouk, A.; Warad, I.; Gu, Y.; Liu, Y. Quantum chemical insight into the molecular structure of L-chemosensor 1,3-dimethyl-5-(thien-2-ylmethylene)-pyrimidine-2,4,6-(1H,3H,5H)-trione: Naked-eye colorimetric detection of copper(II) anions. J. Theor. Comput. Chem. 2018, 17, 1850005. [Google Scholar] [CrossRef]
- Barakat, A.; Soliman, S.M.; Ghabbour, H.A.; Ali, M.; Al-Majid, A.M.; Zarrouk, A.; Warad, I. Intermolecular interactions in crystal structure, Hirshfeld surface, characterization, DFT and thermal analysis of 5-((5-bromo-1 H -indol-3-yl)methylene)-1,3-dimethylpyrimidine-2,4,6(1 H, 3 H, 5 H )-trione indole. J. Mol. Struct. 2017, 1137, 354–361. [Google Scholar] [CrossRef]
- Zi, Y.; Zhu, M.; Li, X.; Xu, Y.; Wei, H.; Li, D.; Mu, C. Effects of carboxyl and aldehyde groups on the antibacterial activity of oxidized amylose. Carbohydr. Polym. 2018, 192, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Barnsley, J.E.; Wagner, P.; Officer, D.L.; Gordon, K.C. Aldehyde isomers of porphyrin: A spectroscopic and computational study. J. Mol. Struct. 2018, 1173, 665–670. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Guedes, I.A.; Camila, S.D.M.; Dardenne, L.E. Receptor–ligand molecular docking. Biophys. Rev. 2014, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, P.L.; Arya, D.P. Natural product DNA major groove binders. Nat. Prod. Rep. 2011, 29, 134–143. [Google Scholar] [CrossRef]
- Rehman, S.U.; Sarwar, T.; Husain, M.A.; Ishqi, H.M.; Tabish, M. Studying non-covalent drug–DNA interactions. Arch. Biochem. Biophys. 2015, 576, 49. [Google Scholar] [CrossRef]
- Arjmand, F.; Parveen, S.; Afzal, M.; Shahid, M. Synthesis, characterization, biological studies (DNA binding, cleavage, antibacterial and topoisomerase I) and molecular docking of copper(II) benzimidazole complexes. J. Photochem. Photobiol. B Biol. 2012, 114, 15–26. [Google Scholar] [CrossRef]
- Barnes, E.C.; Petersson, G.A.; Montgomery, J.A.; Frisch, M.J.; Martin, J.M.L.; Martin, J.M.L. Unrestricted Coupled Cluster and Brueckner Doubles Variations of W1 Theory. J. Chem. Theory Comput. 2009, 5, 2687–2693. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 2.1; University of Western Australia: Perth, Australia, 2007. [Google Scholar]
- Sheldrick, G. A short history ofSHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, H.R.; Wing, R.M.; Takano, T.; Broka, C.A.; Tanaka, S.; Itakura, K.; Dickerson, R.E. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. USA 1981, 78, 2179–2183. [Google Scholar] [CrossRef] [Green Version]
- Titi, A.; Warad, I.; Almutairi, S.M.; Fettouhi, M.; Messali, M.; Aljuhani, A.; Touzani, R.; Zarrouk, A. One-pot liquid microwave-assisted green synthesis of neutral trans-Cl2Cu(NNOH)2: XRD/HSA-interactions, antifungal and antibacterial evaluations. Inorg. Chem. Commun. 2020, 122, 108292. [Google Scholar] [CrossRef]
- Warad, I.; Awwadi, F.F.; Abd Al-Ghani, B.; Sawafta, A.; Shivalingegowda, N.; Lokanath, N.K.; Mubarak, M.S.; Ben Hadda, T.; Zarrouk, A.; Al-Rimawi, F.; et al. Ultrasound-assisted synthesis of two novel [CuBr(diamine)2. H2O]Br complexes: Solvatochromism, crystal structure, physicochemical, Hirshfeld surface thermal, DNA/binding, antitumor and antibacterial activities. Ultrason. Sonochem. 2018, 48, 1. [Google Scholar] [CrossRef]
- Warad, I.; Musameh, S.; Sawafta, A.; Brandão, P.; Tavares, C.J.; Zarrouk, A.; Amereih, S.; Al Ali, A.; Shariah, R. Ultrasonic synthesis of Oct. trans-Br2Cu(N ∩ N)2 Jahn-Teller distortion complex: XRD-properties, solvatochromism, thermal, kinetic and DNA-binding evaluations. Ultrason. Sonochemistry 2019, 52, 428–436. [Google Scholar] [CrossRef]
- Abu Saleemh, F.; Musameh, S.; Sawafta, A.; Brandao, P.; Tavares, C.J.; Ferdov, S.; Barakat, A.; Al Ali, A.; Hamani, H.; Warad, I. Diethylenetriamine/diamines/copper (II) complexes [Cu(dien)(NN)]Br 2: Synthesis, solvatochromism, thermal, electrochemistry, single crystal, Hirshfeld surface analysis and antibacterial activity. Arab. J. Chem. 2017, 10, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Aouad, M.R.; Messali, M.; Rezki, N.; Al-Zaqri, N.; Warad, I. Single proton intramigration in novel 4-phenyl-3-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-1H-1,2,4-triazole-5(4H)-thione: XRD-crystal interactions, physicochemical, thermal, Hirshfeld surface, DFT realization of thiol/thione tautomerism. J. Mol. Liq. 2018, 264, 621–630. [Google Scholar] [CrossRef]
- Tauc, J.; Menth, A. States in the gap. J. Non-Cryst. Solids 1972, 8, 569–585. [Google Scholar] [CrossRef]
- Banfi, D.; Patiny, L. www.nmrdb.org: Resurrecting and Processing NMR Spectra On-line. Chim. Int. J. Chem. 2008, 62, 280–281. [Google Scholar] [CrossRef]
- Shi, J.-H.; Lou, Y.-Y.; Zhou, K.-L.; Pan, D.-Q. Exploration of intermolecular interaction of calf thymus DNA with sulfosulfuron using multi-spectroscopic and molecular docking techniques. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 204, 209–216. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C9H10O3 |
|---|---|
| CCDC | 1860213 |
| Temperature | 293(2) K |
| Formula weight | 166.17 |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, p21/n |
| Volume | 639.55(5) Å3 |
| Unit cell dimensions | a = 3.9469 (9), b = 11.580 (3), c = 17.886 (4) Å |
| β | 91.442 (17)° |
| V | 817.2 (3) (Å)3 |
| Crystal size | 0.29 × 0.26 × 0.23 mm |
| Z | 4 |
| Absorption coefficient | 0.1 mm−1 |
| No. of reflections | 1837 |
| Refinement method | Full-matrix least-squares on F2 |
| R(int), (sin θ/λ)max | 0.115, 0.650 (Å−1) |
| S | 1.05 |
| R[F2 > 2σ(F2)], wR(F2) | 0.062, 0.190 |
| Largest diff. peak and hole | 0.14, −0.20 eÅ−3 |
| Bond No. | Bonds | Exp. XRD | DFT | Angle No. | Angles (°) | Exp. XRD | DFT | |||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | O3 | C5 | 1.371(2) | 1.3639 | 1 | C5 | O3 | C9 | 117.3(2) | 118.73 |
| 2 | O3 | C9 | 1.409(3) | 1.4207 | 2 | C2 | O2 | C8 | 117.7(2) | 118.38 |
| 3 | O2 | C2 | 1.364(2) | 1.3655 | 3 | C5 | C6 | C1 | 120.6(2) | 120.56 |
| 4 | O2 | C8 | 1.410(3) | 1.4189 | 4 | O2 | C2 | C1 | 116.5(2) | 116.62 |
| 5 | O1 | C7 | 1.197(3) | 1.2124 | 5 | O2 | C2 | C3 | 123.9(2) | 124.35 |
| 6 | C6 | C5 | 1.379(3) | 1.393 | 6 | C1 | C2 | C3 | 119.6(2) | 119.03 |
| 7 | C6 | C1 | 1.384(3) | 1.3978 | 7 | O3 | C5 | C6 | 116.3(2) | 116.2 |
| 8 | C2 | C1 | 1.391(3) | 1.4138 | 8 | O3 | C5 | C4 | 124.4(2) | 124.95 |
| 9 | C2 | C3 | 1.384(3) | 1.3964 | 9 | C6 | C5 | C4 | 119.3(2) | 119.73 |
| 10 | C5 | C4 | 1.377(3) | 1.3919 | 10 | C5 | C4 | C3 | 120.9(2) | 120.58 |
| 11 | C4 | C3 | 1.379(3) | 1.3939 | 11 | C6 | C1 | C2 | 119.7(2) | 120.39 |
| 12 | C1 | C7 | 1.465(3) | 1.4838 | 12 | C6 | C1 | C7 | 119.0(2) | 118.88 |
| 13 | C2 | C1 | C7 | 121.3(2) | 121.25 | |||||
| 14 | C2 | C3 | C4 | 119.9(2) | 118.84 | |||||
| 15 | C1 | C7 | O1 | 124.7(2) | 123.68 | |||||
| Atom No. | Atom | Mull | NPA | Atom No. | Atom | Mull | NPA |
|---|---|---|---|---|---|---|---|
| 1 | O | −0.37563 | −0.53238 | 12 | C | −0.10245 | −0.27906 |
| 2 | O | −0.31227 | −0.53113 | 13 | H | 0.111993 | 0.2103 |
| 3 | O | −0.36585 | −0.5234 | 14 | C | 0.191826 | 0.33756 |
| 4 | C | −0.12813 | −0.19544 | 15 | C | −0.10028 | −0.24795 |
| 5 | H | 0.128088 | 0.1876 | 16 | H | 0.113917 | 0.20922 |
| 6 | H | 0.107874 | 0.16505 | 17 | C | −0.1867 | −0.18217 |
| 7 | H | 0.11955 | 0.16505 | 18 | C | −0.03829 | −0.17568 |
| 8 | C | −0.12758 | −0.19749 | 19 | H | 0.126519 | 0.23468 |
| 9 | H | 0.102642 | 0.16179 | 20 | C | 0.151051 | 0.30193 |
| 10 | H | 0.126294 | 0.18724 | 21 | C | 0.23168 | 0.41763 |
| 11 | H | 0.115255 | 0.16178 | 22 | H | 0.110488 | 0.12487 |
| GRD | Value | |
|---|---|---|
| Global total energy | ET | −574.76611449 a.u |
| Low unoccupied molecular orbital | LUMO | −0.05678 a.u |
| High occupied molecular orbital | HOMO | −0.21354 a.u |
| Energy difference | ΔEgap | 0.15676 a.u 4.26565 eV |
| Electron affinity | A | 1.545063 eV |
| Ionization potential | I | 5.810721 eV |
| Global hardness | ƞ | 2.13505 eV |
| Global softness | σ | 0.468372 eV |
| Chemical potential | μ | −3.67789 eV |
| Electronegativity | χ | 3.67789 eV |
| Electrophilicity | ω | 3.16781 eV |
| Dipole moment | u | 6.4226 D |
Sample Availability: Samples of the compounds are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Zaqri, N.; Suleiman, M.; Al-Ali, A.; Alkanad, K.; Kumara, K.; Lokanath, N.K.; Zarrouk, A.; Alsalme, A.; Alharthi, F.A.; Al-Taleb, A.; et al. Exo⇔Endo Isomerism, MEP/DFT, XRD/HSA-Interactions of 2,5-Dimethoxybenzaldehyde: Thermal, 1BNA-Docking, Optical, and TD-DFT Studies. Molecules 2020, 25, 5970. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245970
Al-Zaqri N, Suleiman M, Al-Ali A, Alkanad K, Kumara K, Lokanath NK, Zarrouk A, Alsalme A, Alharthi FA, Al-Taleb A, et al. Exo⇔Endo Isomerism, MEP/DFT, XRD/HSA-Interactions of 2,5-Dimethoxybenzaldehyde: Thermal, 1BNA-Docking, Optical, and TD-DFT Studies. Molecules. 2020; 25(24):5970. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245970
Chicago/Turabian StyleAl-Zaqri, Nabil, Mohammed Suleiman, Anas Al-Ali, Khaled Alkanad, Karthik Kumara, Neartur K. Lokanath, Abdelkader Zarrouk, Ali Alsalme, Fahad A. Alharthi, Afnan Al-Taleb, and et al. 2020. "Exo⇔Endo Isomerism, MEP/DFT, XRD/HSA-Interactions of 2,5-Dimethoxybenzaldehyde: Thermal, 1BNA-Docking, Optical, and TD-DFT Studies" Molecules 25, no. 24: 5970. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245970