
Focus on Chemistry of the 10-Dioxane-nido-7,8-dicarba-undecahydrido Undecaborate Zwitterion; Exceptionally Easy Abstraction of Hydrogen Bridge and Double-Action Pathways Observed in Ring Cleavage Reactions with OH− as Nucleophile †

 ,
, 
Abstract
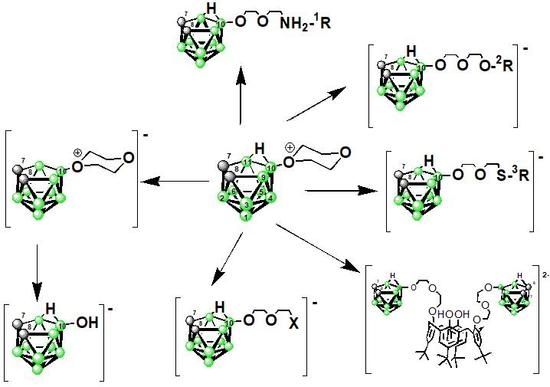
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Reaction Pathways of 1 with Nucleophiles
2.2. Crystallography
3. Materials and Methods
3.1. General
3.2. Instrumental Techniques
3.3. Synthetic Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hawthorne, M.F.; Young, D.C.; Garrett, P.M.; Owen, D.A.; Schwerin, S.G.; Tebbe, F.N.; Wegner, P.A. Preparation and characterization of (3)-1,2- and (3)-1,7-dicarbadodecahydroundecaborate(-1) Ions. J. Am. Chem. Soc. 1968, 90, 862–865. [Google Scholar] [CrossRef]
- Plešek, J.; Heřmánek, S.; Štíbr, B. Potassium Dodecahydro-7,8-Dicarba-Nido-Undecaborate(1-), K[7,8-C2B9H12], Intermediates, Stock Solution, and Anhydrous Salt. Inorg. Synth. 1983, 22, 231–234. [Google Scholar]
- Grimes, R.N. Eleven-Vertex Carboranes. In Carboranes, 3rd ed.; Academic Press Ltd-Elsevier Science Ltd.: London, UK, 2016; pp. 179–247. [Google Scholar]
- Frank, R.; Adhikari, A.K.; Auer, H.; Hey-Hawkins, E. Electrophile-Induced Nucleophilic Substitution of the nido-Dicarbaundecaborate Anion nido-7,8-C2B9H12-by Conjugated Heterodienes. Chem. Eur. J. 2014, 20, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Young, D.C.; Howe, D.V.; Hawthorne, M.F. Ligand derivatives of (3)-1, 2-dicarbadodecahydroundecaborate (-1). J. Am Chem. Soc. 1969, 91, 859–862. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Kalinin, V.N.; Zhigareva, G.G. Oxidation of dicarbadodecahydroundecaborate anions by mercuric-chloride in tetrahydrofuran and pyridine. Bull. Acad. Sci. USSR 1979, 28, 2198–2199. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Abramova, E.N.; Lobanova, I.A.; Sivaev, I.B.; Bragin, V.I.; Petrovskii, P.V.; Tsupreva, V.N.; Sorokina, O.V.; Bregadze, V.I. Synthesis of functional derivatives of 7,8-dicarba-nido-undecaborate anion by ring-opening of its cyclic oxonium derivatives. Collect. Czech. Chem. Commun. 2007, 72, 1676–1688. [Google Scholar] [CrossRef]
- Řezáčová, P.; Řezáčová, P.; Pokorná, J.; Brynda, J.; Kožíšek, M.; Cígler, P.; Lepšík, M.; Fanfrlík, J.; Řezáč, J.; Šašková, K.G.; et al. From nonpeptide toward noncarbon protease inhibitors: Metallacarboranes as specific and potent inhibitors of HIV protease. J. Med. Chem. 2009, 52, 7132–7141. [Google Scholar]
- Semioshkin, A.A.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of polyhedral boron hydrides and their synthetic applications. Dalton Trans. 2008, 977–992. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Kulikova, N.Y.; Nizhnik, E.A.; Vichuzhanin, M.V.; Starikova, Z.A.; Semioshkin, A.A.; Bregadze, V.I. Practical synthesis of 1,4-dioxane derivative of the closo-dodecaborate anion and its ring opening with acetylenic alkoxides. J. Organomet. Chem. 2008, 693, 519–525. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Cyclic Oxonium Derivatives as an Efficient Synthetic Tool for the Modification of Polyhedral Boron Hydrides. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press-Taylor & Francis Group: Boca Raton, FL, USA, 2012; pp. 623–637. [Google Scholar]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press, Elsevier: London, UK, 2016; pp. 1–1041. [Google Scholar]
- Dash, B.P.; Satapathy, R.; Swain, B.R.; Mahanta, C.S.; Jena, B.B.; Hosmane, N.S. Cobalt bis(dicarbollide) anion and its derivatives. J. Organomet. Chem. 2017, 849, 170–194. [Google Scholar] [CrossRef]
- Plešek, J.; Heřmánek, S.; Franken, A.; Císařová, I.; Nachtigal, C. Dimethyl sulfate induced nucleophilic substitution of the bis(1,2-dicarbollido)-3-cobalt(1-) ate ion. Syntheses, properties and structures of its 8,8′-mu-sulfato, 8-phenyl and 8-dioxane derivatives. Collect. Czech. Chem. Commun. 1997, 62, 47–56. [Google Scholar] [CrossRef]
- Matveev, E.Y.; Akimov, S.S.; Kubasov, A.S.; Nichugovskii, A.I.; Nartov, A.S.; Retivov, V.M.; Zhizhina, K.Y.; Kuznetsov, N.T. Reaction of the B10H9O2C4H8 (-) anion with C-nucleophiles. Russ. J. Inorg. Chem. 2017, 62, 808–813. [Google Scholar] [CrossRef]
- El Anwar, S.; Laila, Z.; Ramsubhag, R.; Tlais, S.; Safa, A.; Dudley, G.; Naoufal, D. Synthesis and characterization of click-decahydrodecaborate derivatives by the copper(I) catalyzed 3+2 azide-alkyne cycloaddition reaction. J. Organomet. Chem. 2018, 865, 89–94. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Semioshkin, A.A.; Brellochs, B.; Sjoberg, S.; Bregadze, V.I. Synthesis of oxonium derivatives of the dodecahydro-closo-dodecaborate anion B12H12 (2-). Tetramethylene oxonium derivative of B12H12 (2-) as a convenient precursor for the synthesis of functional compounds for boron neutron capture therapy. Polyhedron 2000, 19, 627–632. [Google Scholar] [CrossRef]
- Bernard, R.; Cornu, D.; Gruner, B.; Dozol, J.F.; Miele, P.; Bonnetot, B. Synthesis of B12H12 (2-) based extractants and their application for the treatment of nuclear wastes. J. Organomet. Chem. 2002, 657, 83–90. [Google Scholar] [CrossRef]
- Bernard, R.; Cornu, D.; Perrin, M.; Scharff, J.P.; Miele, P. Synthesis and X-ray structural characterisation of the tetramethylene oxonium derivative of the hydrodecaborate anion. A versatile route for derivative chemistry of B10H10 (2-). J. Organomet. Chem. 2004, 689, 2581–2585. [Google Scholar] [CrossRef]
- Grűner, B.; Císařová, I.; Čáslavský, J.; Bonnetot, B.; Cornu, D. Synthesis of 12-hydroxy and 12-dioxane derivatives of the closo-1-carbadodecaborate(1-) ion. Variations on the Plesek’s cobalt bis(dicarbollide) pattern. Collect. Czech. Chem. Commun. 2002, 67, 953–964. [Google Scholar] [CrossRef]
- Oleshkevich, E.; Teixidor, F.; Rosell, A.; Vinas, C. Merging Icosahedral Boron Clusters and Magnetic Nanoparticles: Aiming toward Multifunctional Nanohybrid Materials. Inorg. Chem. 2018, 57, 462–470. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Maguire, J.A.; Hosmane, N.S. Polyhedral boron clusters in materials science. N. J. Chem. 2011, 35, 1955–1972. [Google Scholar] [CrossRef]
- Llop, J.; Masalles, C.; Viñas, C.; Teixidor, F.; Sillanpää, R.; Kivekäs, R. The 3,3′-Co(1,2-C2B9H11)(2) (-) anion as a platform for new materials: Synthesis of its functionalized monosubstituted derivatives incorporating synthons for conducting organic polymers. Dalton Trans. 2003, 556–561. [Google Scholar] [CrossRef]
- Nuñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral boron clusters: A perfect tool for the enhancement of polymer features. Chem. Soc. Rev. 2016, 45, 5147–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rais, J.; Grűner, B. Extraction with Metal bis(Dicarbollide) Anions; Metal bis(Dicarbollide) Extractants and their Applications in Separation Chemistry. In Ion Exchange, Solvent Extraction, 1st ed.; Marcus, Y., SenGupta, A.K., Eds.; Marcel Dekker: New York, NY, USA, 2004; Volume 17, pp. 243–334. [Google Scholar]
- Grűner, B.; Rais, J.; Selucký, P.; Lučaníkova, M. Recent Progress in Extraction Agents Based on Cobalt Bis(Dicarbollides) for Partitioning of Radionuclides from High-Level Nuclear Waste. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; Crc Press-Taylor & Francis Group: Boca Raton, FL, USA, 2012; pp. 463–490. [Google Scholar]
- Hao, E.; Vicente, M.G.H. Expeditious synthesis of porphyrin-cobaltacarborane conjugates. Chem. Commun. 2005, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Hao, E.H.; Jensen, T.J.; Courtney, B.H.; Vicente, M.G.H. Synthesis and cellular studies of porphyrin-cobaltacarborane conjugates. Bioconjugate Chem. 2005, 16, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, A.B.; Plešek, J.; Kříž, O.; Lesnikowski, Z.J. A nucleoside conjugate containing a metallacarborane group and its incorporation into a DNA oligonucleotide. Angew. Chem. Int. Edit. 2003, 42, 5740–5743. [Google Scholar] [CrossRef] [PubMed]
- Lesnikowski, Z.J. Challenges and Opportunities for the Application of Boron Clusters in Drug Design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef] [PubMed]
- Gabel, D. Boron clusters in medicinal chemistry: Perspectives and problems. Pure Appl. Chem. 2015, 87, 173–179. [Google Scholar] [CrossRef]
- Semioshkin, A.; Bregadze, V.I.; Godovikov, I.; Ilinova, A.; Laskova, Z.J.; Starikova, Z.A. Synthesis and structure of 1-iodo-7-dioxonium-decahydro-closo-dodecaborate. J. Organomet. Chem. 2011, 696, 2760–3765. [Google Scholar] [CrossRef]
- Schaffran, T.; Lissel, F.; Samatanga, B.; Karlsson, G.; Burghardt, A.; Edwards, K.; Winterhalter, M.; Peschka-Suss, R.; Schubert, R.; Gabel, D. Dodecaborate cluster lipids with variable headgroups for boron neutron capture therapy: Synthesis, physical-chemical properties and toxicity. J. Organomet. Chem. 2009, 694, 1708–1712. [Google Scholar] [CrossRef]
- Cígler, P.; Kožíšek, M.; Rezáčova, P.; Brynda, J.; Otwinowski, Z.; Pokorná, J.; Plešek, J.; Grűner, B.; Dolečková-Marešová, L.; Máša, M.; et al. From nonpeptide toward noncarbon protease inhibitors: Metallacarboranes as specific and potent inhibitors of HIV protease. Proc. Natl. Acad. Sci. USA 2005, 102, 15394–15399. [Google Scholar] [CrossRef] [Green Version]
- Sivaev, I.B.; Bregadze, V.V. Polyhedral Boranes for Medical Applications: Current Status and Perspectives. Eur. J. Inorg. Chem. 2009, 1433–1450. [Google Scholar] [CrossRef]
- Řezáčová, P.; Cigler, P.; Matějíček, P.; Lepšík, M.; Pokorná, J.; Grűner, B.; Konvalinka, J. Medicinal Application of Carboranes Inhibition of HIV Protease. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press-Taylor & Francis Group: Boca Raton, FL, USA, 2012; pp. 41–70. [Google Scholar]
- Prikaznov, A.V.; Las’kova, Y.N.; Semioshkin, A.A.; Sivaev, I.B.; Kisin, A.V.; Bregadze, V.I. Synthesis of boron-containing tyrosine derivatives based on the closo-decaborate and closo-dodecaborate anions. Russ. Chem. Bull. 2011, 60, 2550–2554. [Google Scholar] [CrossRef]
- Neumann, W.; Xu, S.; Sarosi, M.B.; Scholz, M.S.; Crews, B.C.; Ghebreselasie, K.; Banerjee, S.; Marnett, L.J.; Hey-Hawkins, E. nido-Dicarbaborate Induces Potent and Selective Inhibition of Cyclooxygenase-2. ChemMedChem 2016, 11, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Dabrowska, A.; Matuszewski, M.; Zwolinski, K.; Ignaczak, A.; Olejniczak, A.B. Insight into lipophilicity of deoxyribonucleoside-boron cluster conjugates. Eur. J. Pharm. Sci. 2018, 111, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Brynda, J.; Mader, P.; Šícha, V.; Fábry, M.; Poncová, K.; Bakardiev, M.; Grűner, B.; Cígler, P.; Řezáčová, P. Carborane-Based Carbonic Anhydrase Inhibitors. Angew. Chem.-Int. Edit. 2013, 52, 13760–13763. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Lee, S.S.; Knobler, C.B.; Hawthorne, M.F. Synthesis of Charge-compensated Dicarbollide Ligand Precursors and their Use in the Preparation of Novel Metallacarboranes. Inorg. Chem. 1991, 30, 2024–2031. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Sivaev, I.B.; Prikaznova, E.A.; Bregadze, V.I. Transition metal complexes with charge-compensated dicarbollide ligands. J. Organomet. Chem. 2014, 751, 221–250. [Google Scholar] [CrossRef]
- Tarres, M.; Viñas, C.; Gonzalez-Cardoso, P.; Hanninen, M.M.; Sillanpää, R.; Dorďovic, V.; Uchman, M.; Teixidor, F.; Matějíček, P. Aqueous Self-Assembly and Cation Selectivity of Cobaltabisdicarbollide Dianionic Dumbbells. Chem. Eur. J. 2014, 20, 6786–6794. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Kazakov, G.S.; Sivaev, I.B.; Bregadze, V.I. Synthesis of podands with nido-carboranyl groups as a basis for construction of crown ethers with an incorporated metallacarborane moiety. Russ. Chem. Bull. 2013, 62, 699–704. [Google Scholar] [CrossRef]
- Kazakov, G.S.; Stogniy, M.Y.; Sivaev, I.B.; Suponitsky, K.Y.; Godovikov, I.A.; Kirilin, A.D.; Bregadze, V.I. Synthesis of crown ethers with the incorporated cobalt bis(dicarbollide) fragment. J. Organomet. Chem. 2015, 798, 196–203. [Google Scholar] [CrossRef]
- Nekvinda, J.; Rozycka, D.; Rykowski, S.; Wyszko, E.; Fedoruk-Wyszomirska, A.; Gurda, D.; Orlicka-Plocka, M.; Giel-Pietraszuk, M.; Kiliszek, A.; Rypniewski, W.; et al. Synthesis of naphthalimide-carborane and metallacarborane conjugates: Anticancer activity, DNA binding ability. Bioorgan. Chem. 2020, 94, 16. [Google Scholar] [CrossRef]
- Grűner, B.; Brynda, J.; Das, V.; Šícha, V.; Štěpánková, J.; Nekvinda, J.; Holub, J.; Pospíšilová, K.; Fabry, M.; Pachl, P.; et al. Metallacarborane Sulfamides: Unconventional, Specific, and Highly Selective Inhibitors of Carbonic Anhydrase IX. J. Med. Chem. 2019, 62, 9560–9575. [Google Scholar] [CrossRef] [PubMed]
- El Anwar, S.; Assaf, K.I.; Begaj, B.; Samsonov, M.A.; Růžičková, Z.; Holub, J.; Bavol, D.; Nau, W.M.; Gabel, D.; Grűner, B. Versatile, one-pot introduction of nonahalogenated 2-ammonio-decaborate ions as boron cluster scaffolds into organic molecules; host-guest complexation with gamma-cyclodextrin. Chem. Commun. 2019, 55, 13669–13672. [Google Scholar] [CrossRef] [PubMed]
- Peymann, T.; Kuck, K.; Gabel, D. Ring opening of tetrahydropyran attached to undecahydro-closo-dodecaborate(1-) by nucleophiles. Inorg. Chem. 1997, 36, 5138–5139. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Starikova, Z.A.; Sjoberg, S.; Bregadze, V.I. Synthesis of functional derivatives of the 3,3′-Co(1,2-C2B2H11)(2) (-) anion. J. Organomet. Chem. 2002, 649, 1–8. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press-Taylor & Francis Group: Boca Raton, FL, USA, 2016; pp. 5–89. [Google Scholar]
- Heřmánek, S. NMR as a tool for elucidation of structures and estimation of electron distribution in boranes and their derivatives. Inorg. Chim. Acta 1999, 289, 20–44. [Google Scholar] [CrossRef]
- Heřmánek, S. B-11 NMR-spectra of boranes, main-group heteroboranes, and substituted derivatives–Factors influencing chemical-shifts of skeletal atoms. Chem. Rev. 1992, 92, 325–362. [Google Scholar]
- Grimes, R.N. Metallacarboranes of the Transition and Lanthanide Elements. In Carboranes, 3rd ed.; Academic Press, Elsevier: London, UK, 2016; pp. 711–903. [Google Scholar]
- Holub, J.; Grűner, B.; Perekalin, D.S.; Golovanov, D.G.; Lyssenko, K.A.; Petrovskii, P.V.; Kudinov, A.R.; Štíbr, B. Synthesis and rearrangements of aminosubstituted ferra- and ruthenatricarbaboranes. Inorg. Chem. 2005, 44, 1655–1659. [Google Scholar] [CrossRef]
- Grűner, B.; Bačkovský, J.; Sillanpää, R.; Kivekäs, R.; Císařová, I.; Teixidor, F.; Viñas, C.; Štíbr, B. Amino-substituted ferra-bis(tricarbollides)—Metallatricarbaboranes designed for linear molecular constructions. Eur. J. Inorg. Chem. 2004, 1402–1410. [Google Scholar] [CrossRef]
- Grűner, B.; Lehtonen, A.; Kivekäs, R.; Sillanpää, R.; Holub, J.; Teixidor, F.; Viñas, C.; Štíbr, B. Unusual 9 -> 10 rearrangement of the substituted cage carbon in the ferratricarbollide series. Synthesis of the isomeric complexes 2-eta(5)-(C5H5)-10-X-closo-2,1,7,10-FeC3B8H10 (where X = H2N, MeHN, Me2N, and Bu(t)HN). Inorg. Chem. 2000, 39, 2577–2580. [Google Scholar] [CrossRef]
- Bakardjiev, M.; Holub, J.; Hnyk, D.; Císařová, I.; Londesborough, M.G.S.; Perekalin, D.S.; Štíbr, B. Structural dualism in the zwitterionic 7-RR′ NH-nido-7,8,9-C3B8H10 tricarbollide series: An example of absolute tautomerism. Angew. Chem. Int. Edit. 2005, 44, 6222–6226. [Google Scholar] [CrossRef]
- Štíbr, B. On Tautomerism. The Story of Absolute Tautomerism, an Unique Phenomenon in Chemistry. Chem. Listy 2008, 102, 902–905. [Google Scholar]
- Frank, R.; Auer, H.; Hey-Hawkins, E. Functionalisation of the nido-dicarbaborate anion nido-7,8-C2B9H12- by hydride abstraction. J. Organomet. Chem. 2013, 747, 217–224. [Google Scholar] [CrossRef]
- Plešek, J.; Grüner, B.; Heřmánek, S.; Báča, J.; Mareček, V.; Janchenová, J.; Lhotský, A.; Holub, K.; Selucký, P.; Rais, J.; et al. Synthesis of functionalized cobaltacarboranes based on the closo-(1,2-C2B9H11)(2)-3,3′-Co (-) ion bearing polydentate ligands for separation of M3+ cations from nuclear waste solutions. Electrochemical and liquid-liquid extraction study of selective transfer of M3+ metal cations to an organic phase. Molecular structure of the closo- (8-(2-CH3O-C5H4-O)- (CH2CH2O)(2)-1,2-C2B9H10)-(1’,2’-C2B9H11)-3,3′-Co Na determined by X-ray diffraction analysis. Polyhedron 2002, 21, 975–986. [Google Scholar]
- Plešek, J.; Grűner, B.; Macháček, J.; Císařová, I.; Čáslavský, J. 8-Dioxane ferra(III) bis(dicarbollide): A paramagnetic functional molecule as versatile building block for introduction of a Fe(III) centre into organic molecules. J. Organomet. Chem. 2007, 692, 4801–4804. [Google Scholar] [CrossRef]
- Grűner, B.; Mikulášek, L.; Báča, J.; Císařová, I.; Böhmer, V.; Danila, C.; Reinoso-Garcia, M.M.; Verboom, W.; Reinhoudt, D.N.; Casnati, A.; et al. Cobalt bis(dicarbollides)(1-) covalently attached to the calix 4 arene platform: The first combination of organic bowl-shaped matrices and inorganic metallaborane cluster anions. Eur. J. Org. Chem. 2005, 2022–2039. [Google Scholar] [CrossRef]
- Farras, P.; Teixidor, F.; Sillanpää, R.; Viñas, C. A convenient synthetic route to useful monobranched polyethoxylated halogen terminated 3,3-Co(1,2-C2B9H11)(2) (-) synthons. Dalton Trans. 2010, 39, 1716–1718. [Google Scholar] [CrossRef]
- Hosmane, N.S.; Maguire, J.A.; Zhu, Y.; Takagaki, M. Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment, 1st ed.; World Scientific Co., Pte, Ltd.: Hackensack, NJ, USA, 2012. [Google Scholar]
- Šubrtová, V.; Petříček, V.; Hummel, L. Structure of the zwitterionic 10- 2-(2-Dimethylsulfonioethoxy undecahydro-7,8-dicarba-nido-uncecaborate)(1-). Acta Crystallogr. C 1989, 45, 1964–1966. [Google Scholar] [CrossRef]
- Shmal’ko, A.V.; Anufriev, S.A.; Anisimov, A.A.; Stogniy, M.Y.; Sivaev, I.B.; Bregadze, V.I. Synthesis of cobalt and nickel 6,6-diphenylbis(dicarbollides). Russ. Chem. Bull. 2019, 68, 1239–1247. [Google Scholar] [CrossRef]
- Grűner, B.; Plzák, Z. High-performance liquid chromatographic separations of boron- cluster compounds. J. Chromatogr. A 1997, 789, 497–517. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakardjiev, M.; El Anwar, S.; Bavol, D.; Růžičková, Z.; Grűner, B. Focus on Chemistry of the 10-Dioxane-nido-7,8-dicarba-undecahydrido Undecaborate Zwitterion; Exceptionally Easy Abstraction of Hydrogen Bridge and Double-Action Pathways Observed in Ring Cleavage Reactions with OH− as Nucleophile . Molecules 2020, 25, 814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040814
Bakardjiev M, El Anwar S, Bavol D, Růžičková Z, Grűner B. Focus on Chemistry of the 10-Dioxane-nido-7,8-dicarba-undecahydrido Undecaborate Zwitterion; Exceptionally Easy Abstraction of Hydrogen Bridge and Double-Action Pathways Observed in Ring Cleavage Reactions with OH− as Nucleophile . Molecules. 2020; 25(4):814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040814
Chicago/Turabian StyleBakardjiev, Mário, Suzan El Anwar, Dmytro Bavol, Zdeňka Růžičková, and Bohumír Grűner. 2020. "Focus on Chemistry of the 10-Dioxane-nido-7,8-dicarba-undecahydrido Undecaborate Zwitterion; Exceptionally Easy Abstraction of Hydrogen Bridge and Double-Action Pathways Observed in Ring Cleavage Reactions with OH− as Nucleophile " Molecules 25, no. 4: 814. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040814