
Sensitive Assay for the Lactonase Activity of Serum Paraoxonase 1 (PON1) by Harnessing the Fluorescence Turn-On Characteristics of Bioorthogonally Synthesized and Geometrically Controlled Chemical Probes


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis of the Fluorescent Chemical Probes 16b
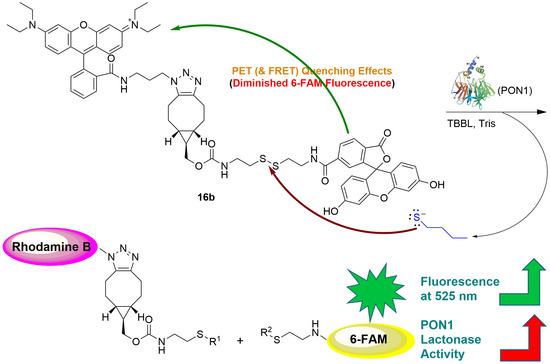
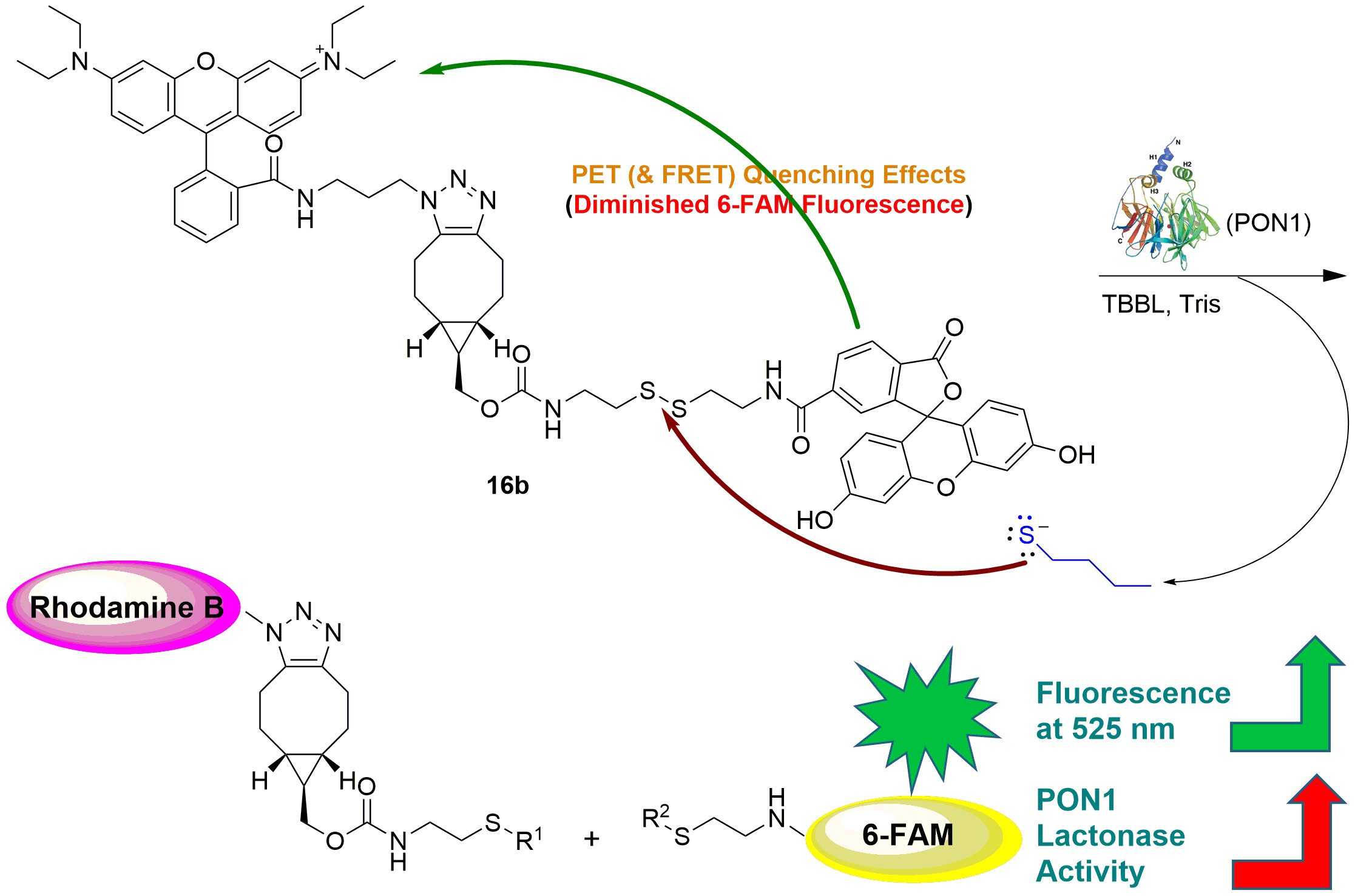
2.2. Contact Quenching and Fluorogenic Properties of the Fluorescent Chemical Probes 16a and 16b
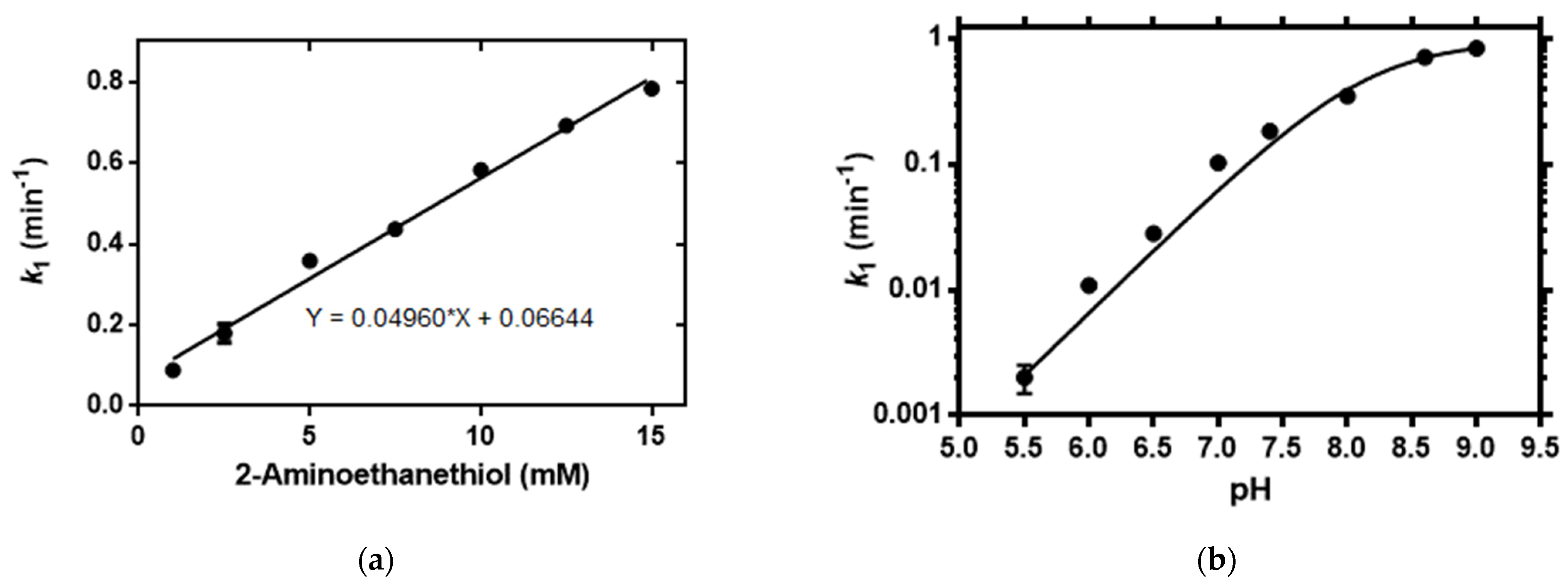
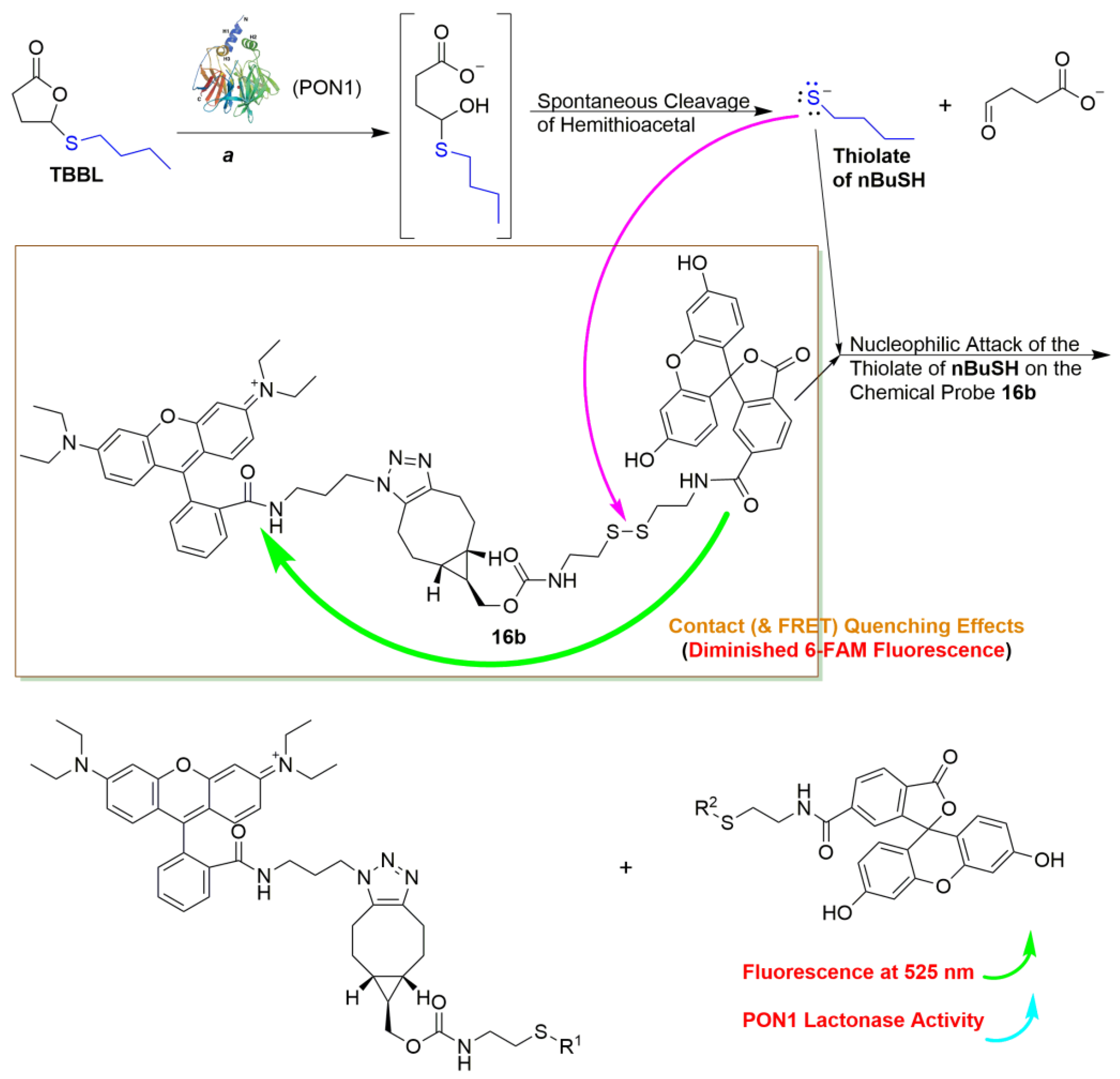
2.3. Mechanistic Studies of the Fluorogenic Reaction of 16b with 2-AET
2.4. Development of the 16b-Based Fluorescence Turn-On Assay for Measuring PON1 Lactonase Activity
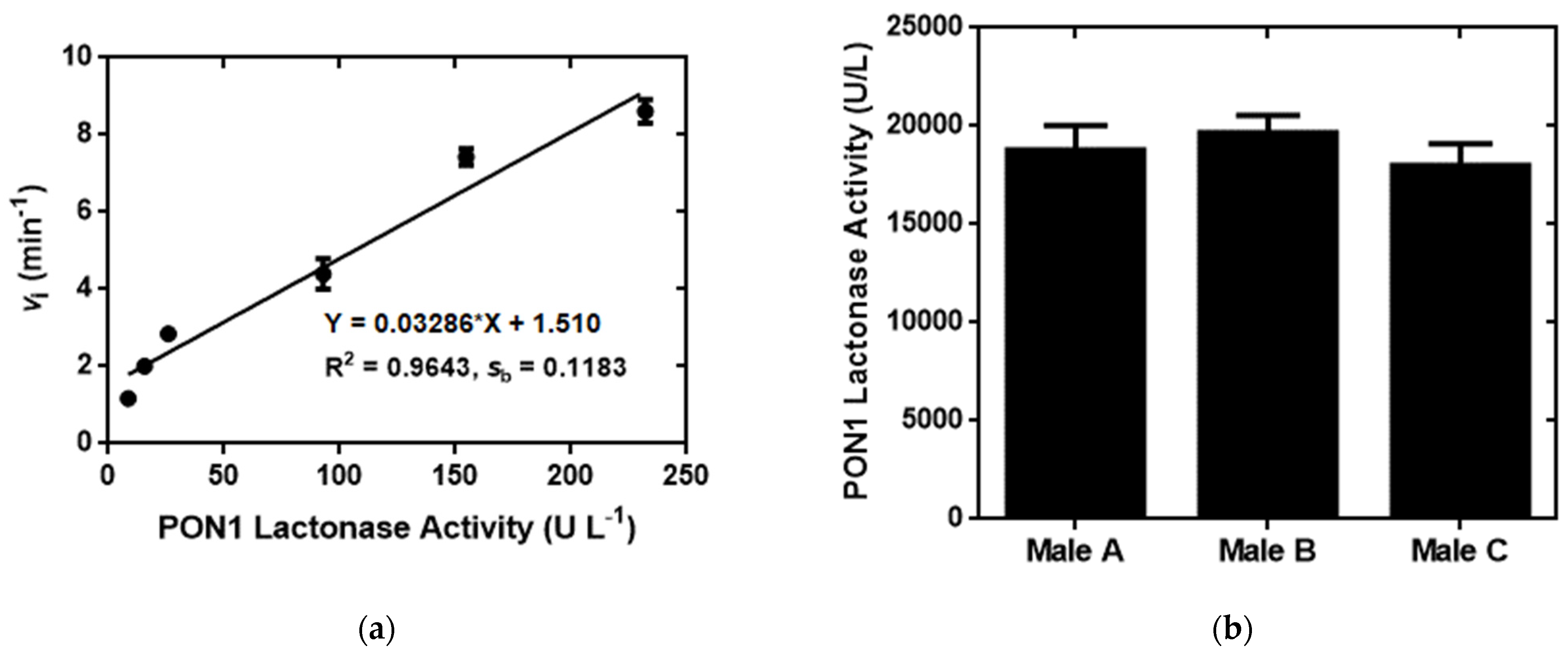
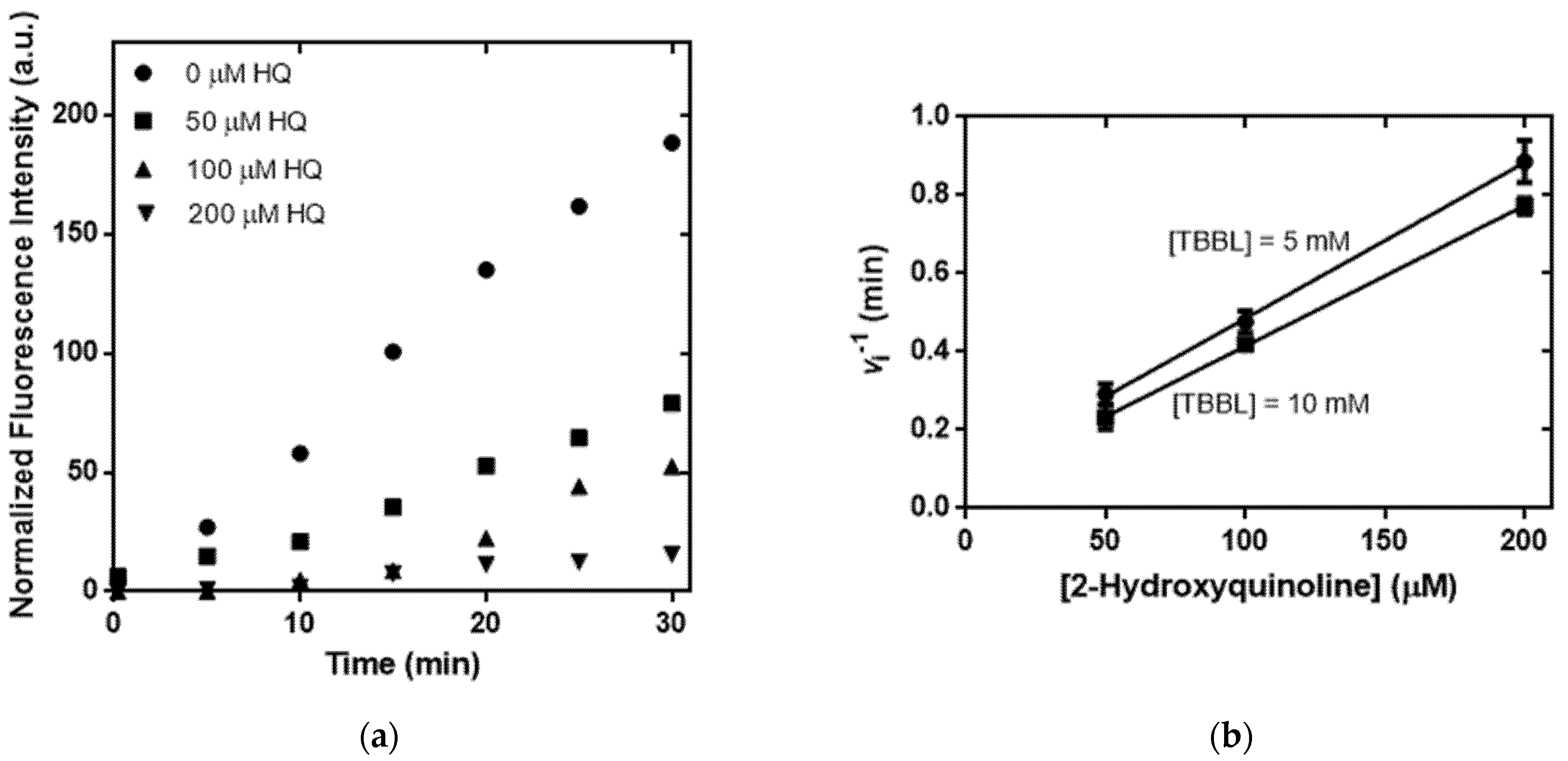
2.5. Sensitive and Accurate Determination of PON1 Lactonase Activity in Human Serum by the Fluorescence Turn-On Assay in the Presence of 16b
2.6. Kinetic Analysis of PON1 Inhibition to Corroborate the Requirement of PON1 Lactonase Activity for Turning on the 6-FAM Fluorescence in the 16b-Based Assay
3. Materials and Methods
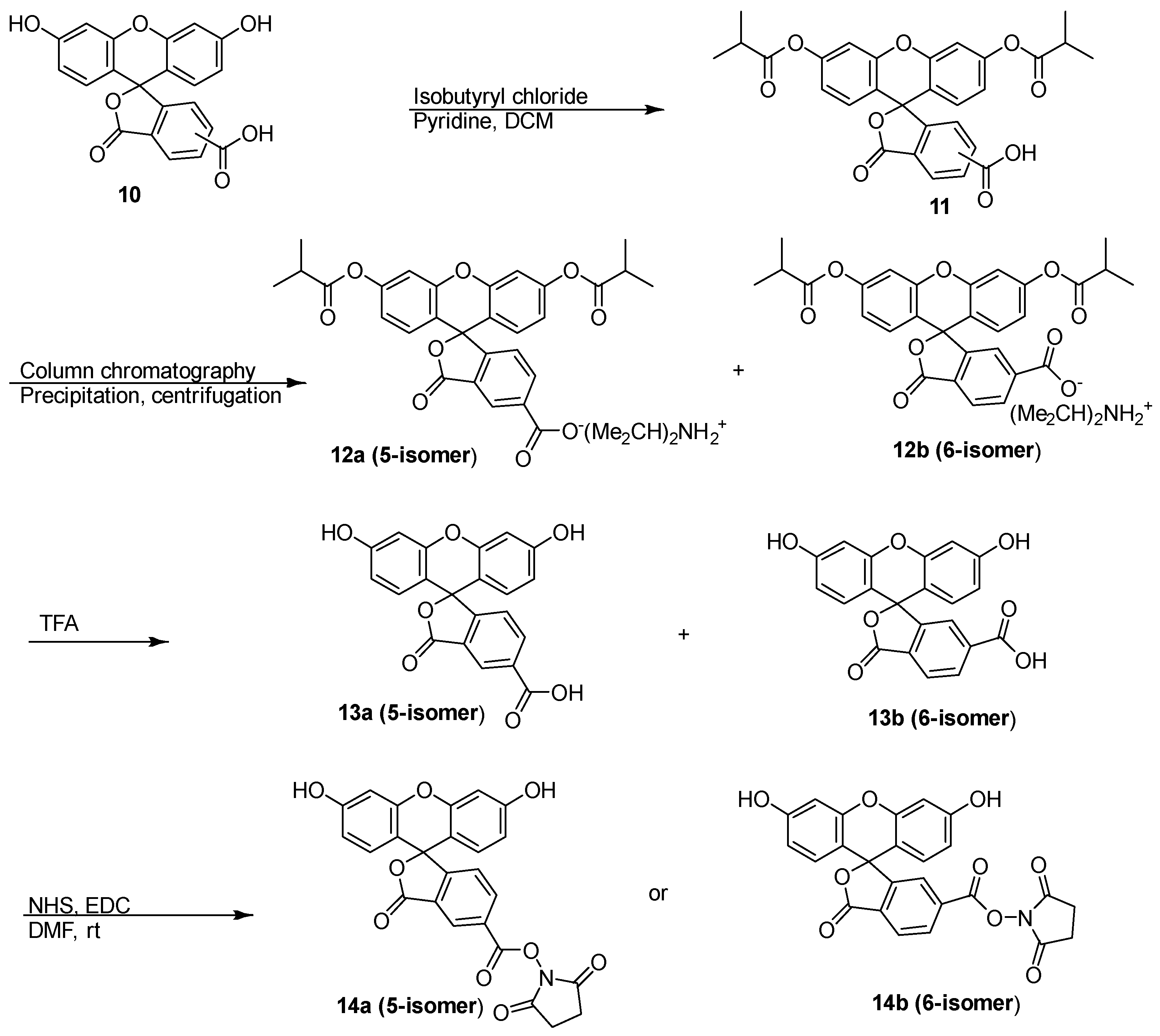
3.1. Synthesis of Azido-Rhodamine B (9), Separation and Regeneration of 5-FAM (13a) and 6-FAM (13b) for Synthesis of the Corresponding Geometric Isomer-Pure N-Hydroxysuccinimide Ester Derivatives (14), and Synthesis of the Rhodamine B-Carboxyfluorescein (FAM) Paired Fluorescent Chemical Probes 16
3.1.1. Synthesis of Azido-Rhodamine B (9)
3.1.2. Separation and Regeneration of 5-FAM (13a) and 6-FAM (13b) for the Synthesis of the Corresponding Geometric Isomer-Pure N-Hydroxysuccinimide (NHS) Ester Derivatives (14)
3′,6′-bis(isobutyryloxy)-5(6)-carboxyfluorescein (11), Diisopropylammonium 3′,6′-bis(isobutyryloxy)-5-carboxyfluorescein (12a) and Diisopropylammonium 3′,6′-bis(isobutyryloxy)-6-carboxyfluorescein (12b)
5-Carboxyfluorescein (5-FAM, 13a)
6-Carboxyfluorescein (6-FAM, 13b)
5-FAM NHS Ester (14a)
6-FAM NHS Ester (14b)
3.1.3. Synthesis of the Rhodamine B-FAM Paired Fluorescent Chemical Probes (16)
The exo–BCN-5-FAM Conjugate (15a)
The exo–BCN-6-FAM Conjugate (15b)
The Rhodamine B-5-FAM Paired Fluorescent Chemical Probe (16a)
The Rhodamine B-6-FAM Paired Fluorescent Chemical Probe (16b)
3.2. Spectroscopic Measurements
3.3. Specificity and Mechanism of the Reactions of the Fluorescent Chemical Probe 16b with Thiols
3.4. Development of Fluorescence Turn-On Assay Based on 16b and Able to Measure PON1 Lactonase Activity in Serum and in the Presence of a PON1 Inhibitor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Moya, C.; Máñez, S. Paraoxonases: Metabolic role and pharmacological projection. Naunyn-Schmiedebergs Arch. Pharmakol. 2018, 391, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, K.; Mach, T.; Cibor, D.; Owczarek, D.; Sapa, J.; Papież, M.; Pytko-Polończyk, J.; Krzyściak, W. Correlation of Paraoxonase-1 with the Severity of Crohn’s Disease. Molecules 2018, 23, 2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camps, J.; Iftimie, S.; García-Heredia, A.; Castro, A.; Joven, J. Paraoxonases and infectious diseases. Clin. Biochem. 2017, 50, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, Inflammation, and Metabolic Disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneses, M.J.; Silvestre, R.; Sousa-Lima, I.; Macedo, M.P. Paraoxonase-1 as a Regulator of Glucose and Lipid Homeostasis: Impact on the Onset and Progression of Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigagli, E.; Lodovici, M. Circulating Oxidative Stress Biomarkers in Clinical Studies on Type 2 Diabetes and Its Complications. Oxidative Med. Cell. Longev. 2019, 2019, 5953685. [Google Scholar] [CrossRef]
- Ferrín, G.; Rodríguez-Perálvarez, M.; Aguilar-Melero, P.; Ranchal, I.; Llamoza, C.; Linares, C.I.; González-Rubio, S.; Muntané, J.; Briceño, J.; López-Cillero, P.; et al. Plasma Protein Biomarkers of Hepatocellular Carcinoma in HCV-Infected Alcoholic Patients with Cirrhosis. PLoS ONE 2015, 10, e0118527. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, Y.; Su, Y.; Ye, X.; Liu, W.; Yang, Q.; Wang, J.; Fu, X.; Gong, Y.; Sun, H. Identification a novel clinical biomarker in early diagnosis of human non-small cell lung cancer. Glycoconj. J. 2019, 36, 57–68. [Google Scholar] [CrossRef]
- Shah, A.K.; Hartel, G.; Brown, I.; Winterford, C.; Na, R.; Cao, K.-A.L.; Spicer, B.A.; Dunstone, M.A.; Phillips, W.A.; Lord, R.V.; et al. Evaluation of Serum Glycoprotein Biomarker Candidates for Detection of Esophageal Adenocarcinoma and Surveillance of Barrett’s Esophagus. Mol. Cell. Proteom. 2018, 17, 2324–2334. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Ou, Q.; Chen, F.; Bi, J.; Li, W.; Ma, J.; Wang, R.; Huang, X. Evaluation of the prognostic value of paraoxonase 1 in the recurrence and metastasis of hepatocellular carcinoma and establishment of a liver-specific predictive model of survival. J. Transl. Med. 2018, 16, 327. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S.; Primo-Parmo, S.L.; La Du, B.N. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J. Clin. Investig. 1998, 101, 1581–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, C.G.; Batuca, J.R.; Marinho, A.T.; Caixas, U.; Monteiro, E.C.; Antunes, A.M.M.; Pereira, S.A. Quantification of the arylesterase activity of paraoxonase-1 in human blood. Anal. Methods 2013, 6, 289–294. [Google Scholar] [CrossRef]
- Furlong, C.E.; Marsillach, J.; Jarvik, G.P.; Costa, L.G. Paraoxonases-1, -2 and -3: What are their functions? Chem. Interact. 2016, 259, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.G.; Richter, R.J.; Keifer, M.; Broomfield, C.A.; Sowalla, J.; Furlong, C.E. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat. Genet. 1996, 14, 334–336. [Google Scholar] [CrossRef]
- Sorenson, R.C.; Bisgaier, C.L.; Aviram, M.; Hsu, C.; Billecke, S.; La Du, B.N. Human Serum Paraoxonase/Arylesterase’s Retained Hydrophobic N-Terminal Leader Sequence Associates with HDLs by Binding Phospholipids. Arter. Thromb. Vasc. Biol. 1999, 19, 2214–2225. [Google Scholar] [CrossRef] [Green Version]
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419. [Google Scholar] [CrossRef]
- Mackness, M.; Mackness, B. Human paraoxonase-1 (PON1): Gene structure and expression, promiscuous activities and multiple physiological roles. Gene 2015, 567, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.T.; Wadleigh, D.J.; Grijalva, V.; Ng, C.; Hama, S.; Gangopadhyay, A.; Shih, D.M.; Lusis, A.J.; Navab, M.; Fogelman, A.M. Human Paraoxonase-3 Is an HDL-Associated Enzyme With Biological Activity Similar to Paraoxonase-1 Protein but Is Not Regulated by Oxidized Lipids. Arter. Thromb. Vasc. Biol. 2001, 21, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.L.; La Du, B.N. Calcium binding by human and rabbit serum paraoxonases. Structural stability and enzymatic activity. Drug Metab. Dispos. 1998, 26, 653–660. [Google Scholar]
- Draganov, D.I.; Teiber, J.F.; Speelman, A.; Osawa, Y.; Sunahara, R.; La Du, B.N. Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res. 2005, 46, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Khersonsky, O.; Tawfik, D.S. Structure−Reactivity Studies of Serum Paraoxonase PON1 Suggest that Its Native Activity Is Lactonase. Biochemistry 2005, 44, 6371–6382. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Calcium-dependent Human Serum Homocysteine Thiolactone Hydrolase. A protective mechanism against protein N-homocysteinylation. J. Biol. Chem. 2000, 275, 3957–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackness, M.I.; Abbott, C.; Arrol, S.; Durrington, P.N. The role of high-density lipoprotein and lipid-soluble antioxidant vitamins in inhibiting low-density lipoprotein oxidation. Biochem. J. 1993, 294, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackness, M.; Arrol, S.; Abbott, C.; Durrington, P. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis 1993, 104, 129–135. [Google Scholar] [CrossRef]
- Gugliucci, A.; Caccavello, R.; Kotani, K.; Sakane, N.; Kimura, S. Enzymatic assessment of paraoxonase 1 activity on HDL subclasses: A practical zymogram method to assess HDL function. Clin. Chim. Acta 2013, 415, 162–168. [Google Scholar] [CrossRef]
- Teiber, J.F.; Draganov, D.I.; Du, B.N. Lactonase and lactonizing activities of human serum paraoxonase (PON1) and rabbit serum PON3. Biochem. Pharmacol. 2003, 66, 887–896. [Google Scholar] [CrossRef]
- Togawa, T.; Mukai, Y.; Ohata, K.; Suzuki, T.; Tanabe, S. Measurement of homocysteine thiolactone hydrolase activity using high-performance liquid chromatography with fluorescence detection and polymorphisms of paraoxonase in normal human serum. J. Chromatogr. B 2005, 819, 67–72. [Google Scholar] [CrossRef]
- Yeung, D.T.; Smith, J.R.; Sweeney, R.E.; Lenz, D.E.; Cerasoli, D.M. Direct detection of stereospecific soman hydrolysis by wild-type human serum paraoxonase. FEBS J. 2007, 274, 1183–1191. [Google Scholar] [CrossRef]
- Billecke, S.; Draganov, D.; Counsell, R.; Stetson, P.; Watson, C.; Hsu, C.; La Du, B.N. Human serum paraoxonase (PON1) isozymes Q and R hydrolyze lactones and cyclic carbonate esters. Drug Metab. Dispos. 2000, 28, 1335–1342. [Google Scholar]
- Khersonsky, O.; Tawfik, D.S. Chromogenic and Fluorogenic Assays for the Lactonase Activity of Serum Paraoxonases. ChemBioChem 2006, 7, 49–53. [Google Scholar] [CrossRef]
- Richter, R.J.; Jarvik, G.P.; Furlong, C.E. Paraoxonase 1 (PON1) status and substrate hydrolysis. Toxicol. Appl. Pharmacol. 2009, 235, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodveldt, C.; Tawfik, D.S. Shared Promiscuous Activities and Evolutionary Features in Various Members of the Amidohydrolase Superfamily. Biochemistry 2005, 44, 12728–12736. [Google Scholar] [CrossRef] [PubMed]
- Gaidukov, L.; Tawfik, D.S. The development of human sera tests for HDL-bound serum PON1 and its lipolactonase activity. J. Lipid Res. 2007, 48, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Yu, N.; Wang, C.; Zou, X.; Abulimite, Z.; Xia, Z. Evaluation of a new substrate for measurement of serum PON arylesterase activity. Talanta 2011, 88, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Carter, J.J.; Scott, J.E. A homogeneous cell-based assay for measurement of endogenous paraoxonase 1 activity. Anal. Biochem. 2010, 400, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garai-Ibabe, G.; Möller, M.; Pavlov, V. Ultrasensitive Assay for Detection of Serum Paraoxonase by Modulating the Growth of Fluorescent Semiconductor Nanoparticles. Anal. Chem. 2012, 84, 8033–8037. [Google Scholar] [CrossRef] [PubMed]
- Soukharev, S.; Hammond, D.J. A fluorogenic substrate for detection of organophosphatase activity. Anal. Biochem. 2004, 327, 140–148. [Google Scholar] [CrossRef]
- Gong, M.-M.; Dai, C.-Y.; Severance, S.; Hwang, C.-C.; Fang, B.-K.; Lin, H.-B.; Huang, C.-H.; Ong, C.-W.; Wang, J.-J.; Lee, P.-L.; et al. A Bioorthogonally Synthesized and Disulfide-Containing Fluorescence Turn-On Chemical Probe for Measurements of Butyrylcholinesterase Activity and Inhibition in the Presence of Physiological Glutathione. Catalysts 2020, 10, 1169. [Google Scholar] [CrossRef]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L.J.A.; Rutjes, F.P.J.T.; van Hest, J.C.M.; Lefeber, D.J.; Friedl, P.; van Delft, F.L. Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three-Dimensional Imaging of Living Cells. Angew. Chem. Int. Ed. 2010, 49, 9422–9425. [Google Scholar] [CrossRef]
- Johansson, M.K.; Fidder, H.; Dick, D.; Cook, R.M. Intramolecular Dimers: A New Strategy to Fluorescence Quenching in Dual-Labeled Oligonucleotide Probes. J. Am. Chem. Soc. 2002, 124, 6950–6956. [Google Scholar] [CrossRef]
- Marras, S.A.E.; Kramer, F.R.; Tyagi, S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002, 30, e122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takakusa, H.; Kikuchi, K.; Urano, Y.; Higuchi, A.T.; Nagano, T. Intramolecular Fluorescence Resonance Energy Transfer System with Coumarin Donor Included in β-Cyclodextrin. Anal. Chem. 2001, 73, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-C.; Chen, H.-Y.; Ko, N.C.; Hwang, C.-C.; Wu, M.H.; Wang, L.F.; Wang, Y.-M.; Chang, S.-N.; Wang, E.-C.; Wang, T.-P. Effective and site-specific phosphoramidation reaction for universally labeling nucleic acids. Anal. Biochem. 2014, 449, 118–128. [Google Scholar] [CrossRef]
- Serjeant, E.P.; Dempsey, B. Ionisation Constants of Organic Acids in Aqueous Solution; Pergamon Press, Inc.: New York, NY, USA, 1979. [Google Scholar]
- Jones, D.P.; Carlson, J.L.; Samiec, P.S.; Sternberg, P.; Mody, V.C.; Reed, R.L.; Brown, L.A.S. Glutathione measurement in human plasma: Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clin. Chim. Acta 1998, 275, 175–184. [Google Scholar] [CrossRef]
- Aharoni, A.; Gaidukov, L.; Yagur, S.; Toker, L.; Silman, I.; Tawfik, D.S. Directed evolution of mammalian paraoxonases PON1 and PON3 for bacterial expression and catalytic specialization. Proc. Natl. Acad. Sci. USA 2004, 101, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horatscheck, A.; Wagner, S.; Ortwein, J.; Kim, B.G.; Lisurek, M.; Beligny, S.; Schütz, A.; Rademann, J. Benzoylphosphonate-Based Photoactive Phosphopeptide Mimetics for Modulation of Protein Tyrosine Phosphatases and Highly Specific Labeling of SH2 Domains. Angew. Chem. Int. Ed. 2012, 51, 9441–9447. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, B.-K.; Dai, C.-Y.; Severance, S.; Hwang, C.-C.; Huang, C.-H.; Hou, S.-Y.; Yeh, B.-L.; Gong, M.-M.; Chou, Y.-H.; Wang, J.-J.; et al. Sensitive Assay for the Lactonase Activity of Serum Paraoxonase 1 (PON1) by Harnessing the Fluorescence Turn-On Characteristics of Bioorthogonally Synthesized and Geometrically Controlled Chemical Probes. Molecules 2022, 27, 2435. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082435
Fang B-K, Dai C-Y, Severance S, Hwang C-C, Huang C-H, Hou S-Y, Yeh B-L, Gong M-M, Chou Y-H, Wang J-J, et al. Sensitive Assay for the Lactonase Activity of Serum Paraoxonase 1 (PON1) by Harnessing the Fluorescence Turn-On Characteristics of Bioorthogonally Synthesized and Geometrically Controlled Chemical Probes. Molecules. 2022; 27(8):2435. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082435
Chicago/Turabian StyleFang, Bo-Kai, Chia-Yen Dai, Scott Severance, Chi-Ching Hwang, Chien-Hui Huang, Sin-Yu Hou, Bao-Lin Yeh, Ming-Mao Gong, Yun-Hao Chou, Jeh-Jeng Wang, and et al. 2022. "Sensitive Assay for the Lactonase Activity of Serum Paraoxonase 1 (PON1) by Harnessing the Fluorescence Turn-On Characteristics of Bioorthogonally Synthesized and Geometrically Controlled Chemical Probes" Molecules 27, no. 8: 2435. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27082435