
The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes


Abstract
:
1 Introduction
1.1. Hypertrophy in Chondrogenesis of MSCs in Vitro
1.2. Hypertrophy in Articular Chondrocytes during OA Progression


2. Signaling Pathways in Hypertrophy

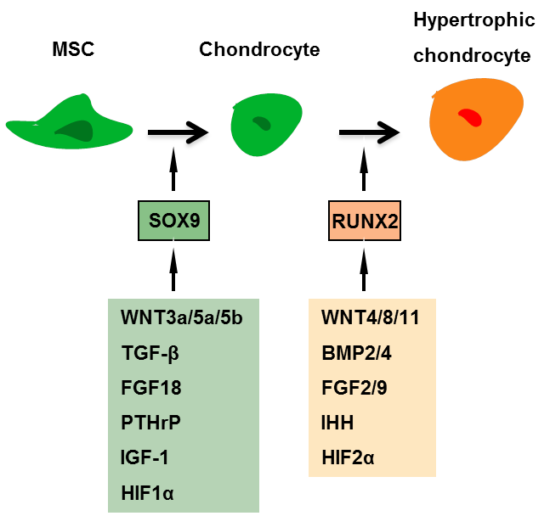{kind=link}
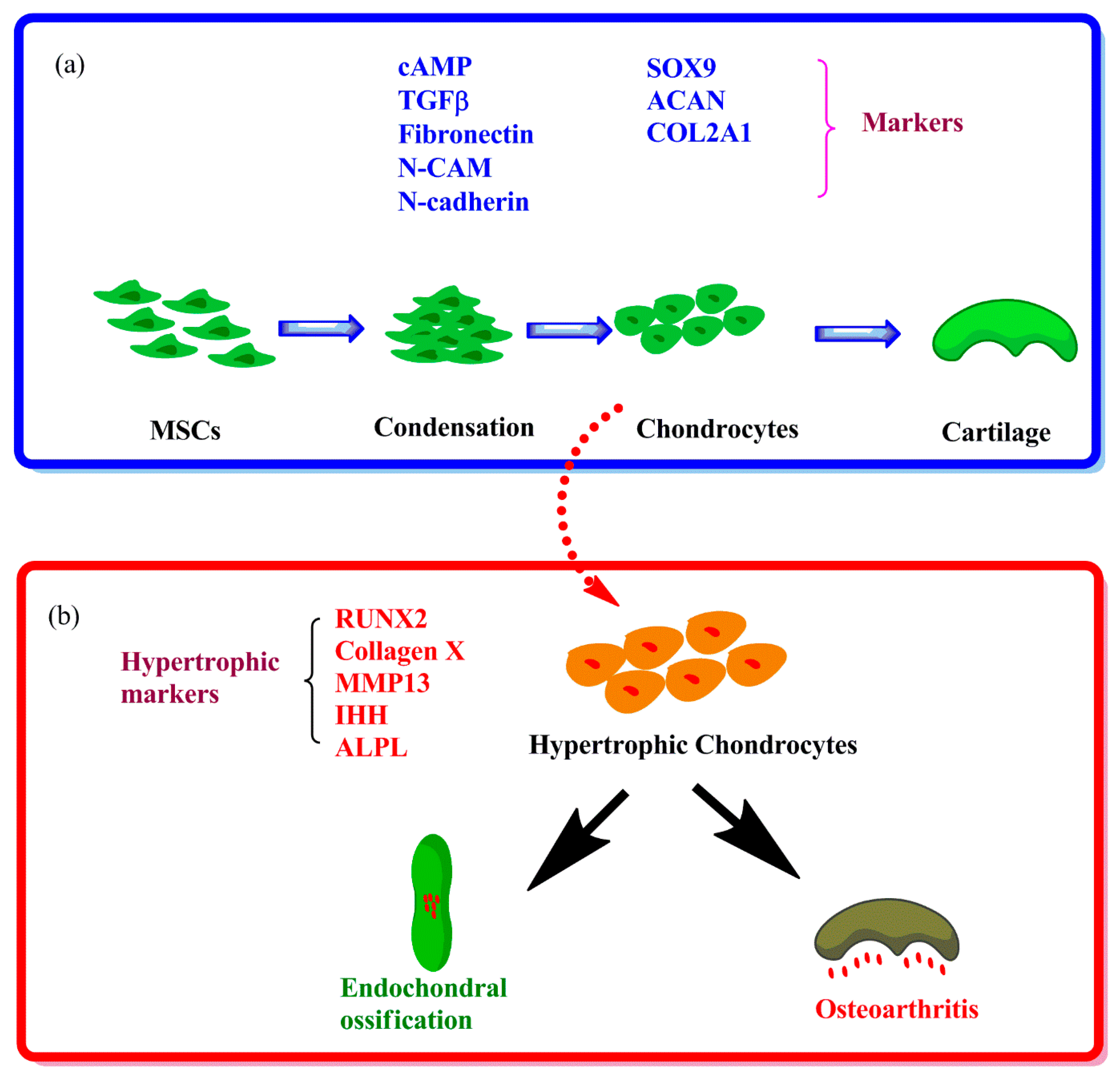{kind=link}
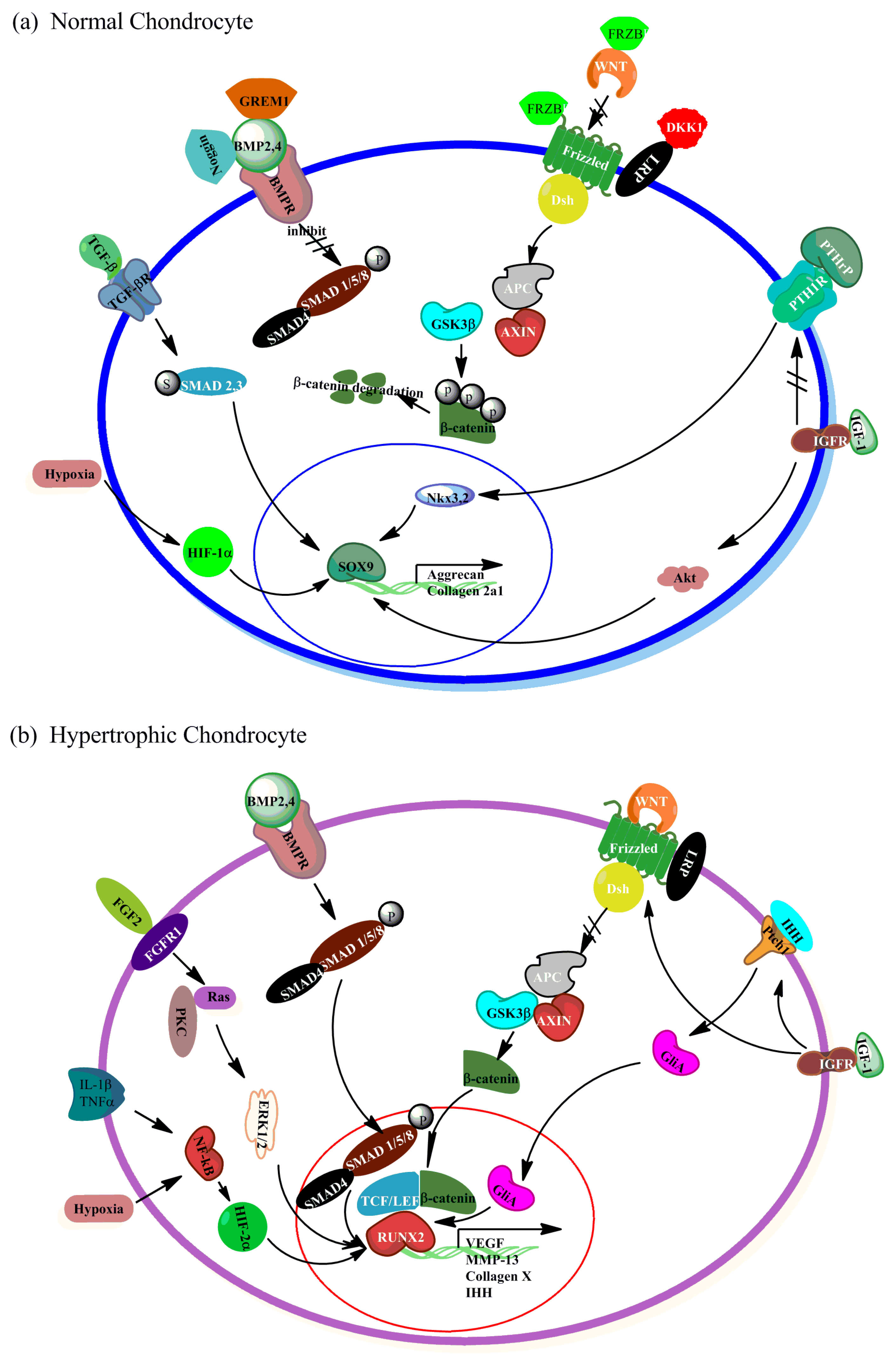{kind=link}
| Signal | Subtypes | Main Functions |
|---|---|---|
| WNT | WNT3a | Promotes chondrogenic differentiation; delays chondrocyte hypertrophy |
| WNT4 | Blocks chondrogenic differentiation; promotes chondrocyte hypertrophy | |
| WNT5a | Promotes chondrogenic differentiation; delays chondrocyte hypertrophy | |
| WNT5b | Promotes chondrogenic differentiation; delays chondrocyte hypertrophy | |
| WNT8 | Blocks chondrogenic differentiation; promotes chondrocyte hypertrophy | |
| WNT9a | Blocks both chondrogenic differentiation and chondrocyte hypertrophy | |
| WNT11 | Promotes chondrogenic differentiation; stimulates RUNX2 and IHH expression | |
| WNT16 | Upregulation is accompanied by the downregulation of FRZB | |
| BMP/TGF-β | BMP2 | Induces chondrocyte hypertrophy |
| BMP4 | Induces chondrocyte hypertrophy | |
| BMP7 | Maintain chondrogenic potential and prevents chondrocyte hypertrophy; | |
| TGF-β | Promotes chondrogenic differentiation; inhibits chondrocyte hypertrophy | |
| PTHrP | Blocks hypertrophy by stimulating Nkx3.2 and prevent RUNX2 expression | |
| IHH | Promotes chondrocyte hypertrophy; Stimulates proliferating chondrocytes to produce PTHrP | |
| FGF | FGF2 | Promotes expression of RUNX2 |
| FGF8 | Catabolic mediator with a pathological role in rat and rabbit articular cartilage | |
| FGF9 | Promotes chondrocyte hypertrophy | |
| FGF18 | Promotes chondrocyte proliferation and differentiation in the early stages of cartilage development | |
| IGF | IGF-1 | Promotes chondrocyte proliferation and maturation; augments chondrocyte hypertrophy |
| HIF | HIF-1α | Potentiates BMP2-induced SOX9 expression and cartilage formation, while inhibiting RUNX2 expression and endochondral ossification |
| HIF-2α | Increases expression of collagen X, MMP13 and VEGF |
2.1. WNT Signaling
2.2. Bone Morphogenetic Protein (BMP)/Transforming Growth Factor-β (TGFβ) Signaling
2.2.1. BMP Signaling
2.2.2. TGF-β Signaling
2.3. The Crosstalk between BMP/TGFβ and WNT Signaling in Regulating Hypertrophy
2.4. Parathyroid Hormone-Related Peptide (PTHrP)/Indian Hedgehog (IHH) Signaling
2.5. Fibroblast Growth Factor (FGF) Signaling
2.6. Insulin Like Growth Factor (IGF) Signaling
2.7. Hypoxia-Inducible Factor (HIF) Signaling
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MSCs | Mesenchymal stem cells |
| OA | Osteoarthritis |
| WNT | Wingless-type MMTV integration site |
| BMP | Bone morphogenetic protein |
| TGFβ | Transforming growth factor-β |
| PTHrP | Parathyroid hormone-related peptide |
| IHH | Indian hedgehog |
| FGF | Fibroblast growth factor |
| IGF | Insulin like growth factor |
| HIF | Hypoxia-inducible factor |
| AC | Articular cartilage |
| SOX9 | Sex determining region Y box 9 |
| ACAN | Aggrecan |
| Col2A1 | Collagen type II |
| cAMP | Cyclic adenosine monophosphate |
| N-CAM | Neural cell adhesion molecule |
| ECM | extracellular matrix |
| RUNX2 | Runt-related transcription factor 2 |
| MMP | Matrix metalloproteinase |
| ALPL | Alkaline phosphatase |
| VEGF | Vascular endothelial growth factor |
| JNK | c-Jun N-terminal kinase |
| AXIN2 | Axis inhibitor 2 |
| APC | Adenomatous polyposis coli |
| GSK3β | Glycogen synthase kinase 3β |
| TCF | T cell-specific factor |
| LEF | lymphoid enhancer binding protein |
| LRP5/6 | Low density lipoprotein receptor related protein 5/6 |
| SFRP 1 | WNT antagonist secreted frizzled related protein 1 |
| GPCR | G-protein coupled receptor |
| PI3K | phosphoinositide 3-kinase (PI3K) |
| PKB/Akt | protein kinase B |
| NF-κB | Nuclear factor κ-light chain-enhancer of activated B cells |
| BMPR | BMP receptor |
| SMAD | Sma and Mad related proteins |
| ALK | Activin-like kinase |
| ADAMTS | A disintegrin like and metalloproteinase with thrombospondin type I motif |
| Nkx3.2 | NK3 homeobox 2 |
| Gli | GLI-Kruppel family members |
| PTH1R | PTHrP receptor 1 |
| PKCδ | Protein kinase C delta |
| ERKs | extracellular-signal-regulated kinases |
| MEK | Mitogen-activated protein kinase kinase |
| FGFR3 | The FGF receptor 3 |
| IGF1R | IGF-I receptor |
| rAAV | Recombinant adeno-associated virus |
| WISP3 | WNT induced secreted protein 3 |
| ROS | Reactive oxygen species |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| DMM | Medial meniscus |
| T3 | triiodothyronine |
| DKK1 | Dickkopf |
| FRZB | Frizzled-related protein |
| GREM1 | Gremlin 1 |
References
- Studer, D.; Millan, C.; Öztürk, E.; Maniura-Weber, K.; Zenobi-Wong, M. Molecular and biophysical mechanisms regulating hypertrophic differentiation in chondrocytes and mesenchymal stem cells. Eur. Cell Mater. 2012, 24, 118–135. [Google Scholar] [PubMed]
- Hall, B.K.; Miyake, T. All for one and one for all: Condensations and the initiation of skeletal development. Bioessays 2000, 22, 138–147. [Google Scholar] [CrossRef]
- Mrugala, D.; Dossat, N.; Ringe, J.; Delorme, B.; Coffy, A.; Bony, C.; Charbord, P.; Häupl, T.; Daures, J.P.; Noël, D.; et al. Gene expression profile of multipotent mesenchymal stromal cells: Identification of pathways common to TGFβ3/BMP2-induced chondrogenesis. Cloning Stem Cells 2009, 11, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Leijten, J.C.; Emons, J.; Sticht, C.; van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S. Gremlin 1, frizzled-related protein, and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheumatol. 2012, 64, 3302–3312. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Fosang, A.J.; Beier, F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pract. Res. Clin. Rheumatol. 2011, 25, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 2010, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Khan, I.M.; Richardson, K.; Nelson, L.; McCarthy, H.E.; Analbelsi, T.; Singhrao, S.K.; Dowthwaite, G.P.; Jones, R.E.; Baird, D.M.; et al. Identification and clonal characterisation of a progenitor cell sub-population in normal human articular cartilage. PLoS ONE 2010, 5, e13246. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Vuoristo, J.T.; Larson, B.L.; Prockop, D.J. In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 4397–4402. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheumatol. 2006, 54, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.B.; Tuan, R.S. Functional characterization of hypertrophy in chondrogenesis of human mesenchymal stem cells. Arthritis Rheumatol. 2008, 58, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Felson, D.T. Osteoarthritis. BMJ 2006, 332, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R. The Wnts. Genome Biol. 2002, 3, 1–15. [Google Scholar]
- Yano, F.; Kugimiya, F.; Ohba, S.; Ikeda, T.; Chikuda, H.; Ogasawara, T.; Ogata, N.; Takato, T.; Nakamura, K.; Kawaguchi, H.; et al. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem. Biophys. Res. Commun. 2005, 333, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, R.; Ohta, Y.; Yuasa, T.; Kondo, N.; Hoang, T.; Addya, S.; Fortina, P.; Pacifici, M.; Iwamoto, M.; Enomoto-Iwamoto, M. Roles of β-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab. Investig. 2011, 91, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.B.; Brockbank, S.M.; van Lent, P.L.; van Beuningen, H.M.; Geurts, J.; Takahashi, N.; van der Kraan, P.M.; van de Loo, F.A.; Schreurs, B.W.; Clements, K.; et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: Prominent role of Wnt-induced signaling protein 1. Arthritis Rheumatol. 2009, 60, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of β-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult β-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.F.; Soung do, Y.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J. Cell. Physiol. 2006, 208, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Lyons, J.P.; Mori-Akiyama, Y.; Yang, X.; Zhang, R.; Zhang, Z.; Deng, J.M.; Taketo, M.M.; Nakamura, T.; Behringer, R.R.; et al. Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 2004, 18, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, Y.; Otani, T.; Kanatani, N.; Koyama, E.; Kitagaki, J.; Komori, T.; Yamada, Y.; Costantini, F.; Wakisaka, S.; Pacifici, M.; et al. Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J. Biol. Chem. 2005, 280, 19185–19195. [Google Scholar] [CrossRef] [PubMed]
- Enomoto-Iwamoto, M.; Kitagaki, J.; Koyama, E.; Tamamura, Y.; Wu, C.; Kanatani, N.; Koike, T.; Okada, H.; Komori, T.; Yoneda, T.; et al. The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev. Biol. 2002, 251, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Church, V.; Nohno, T.; Linker, C.; Marcelle, C.; Francis-West, P. Wnt regulation of chondrocyte differentiation. J. Cell Sci. 2002, 115, 4809–4818. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Tabin, C.J. Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell 2001, 104, 341–351. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, E.; Yang, M.; Lu, L. Overexpression of Wnt11 promotes chondrogenic differentiation of bone marrow-derived mesenchymal stem cells in synergism with TGF-β. Mol. Cell. Biochem. 2014, 390, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Dell’accio, F.; de Bari, C.; Eltawil, N.M.; Vanhummelen, P.; Pitzalis, C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheumatol. 2008, 58, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Drissi, M.H. WNT5A regulates chondrocyte differentiation through differential use of the CaN/NFAT and IKK/NF-κB pathways. Mol. Endocrinol. 2010, 24, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. Bone morphogenetic proteins and articular cartilage: To serve and protect or a wolf in sheep clothing’s? Osteoarthr. Cartil. 2010, 18, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Im, G.I.; Lee, J.M.; Kim, H.J. Wnt inhibitors enhance chondrogenesis of human mesenchymal stem cells in a long-term pellet culture. Biotechnol. Lett. 2011, 33, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Pasold, J.; Osterberg, A.; Peters, K.; Taipaleenmäki, H.; Säämänen, A.M.; Vollmar, B.; Müller-Hilke, B. Reduced expression of Sfrp1 during chondrogenesis and in articular chondrocytes correlates with osteoarthritis in STR/ort mice. Exp. Cell Res. 2013, 319, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.Y.; Little, C.B. The interaction of canonical bone morphogenetic protein- and Wnt-signaling pathways may play an important role in regulating cartilage degradation in osteoarthritis. Arthritis Res. Ther. 2012, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major Smad pathways in TGF-β superfamily signalling. Genes Cells 2002, 7, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Steinert, A.F.; Proffen, B.; Kunz, M.; Hendrich, C.; Ghivizzani, S.C.; Nöth, U.; Rethwilm, A.; Eulert, J.; Evans, C.H. Hypertrophy is induced during the in vitro chondrogenic differentiation of human mesenchymal stem cells by bone morphogenetic protein-2 and bone morphogenetic protein-4 gene transfer. Arthritis Res. Ther. 2009, 11, R148. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Zhang, M.; Xie, R.; Wang, M.; Jin, H.; Hou, W.; Tang, D.; Harris, S.E.; Mishina, Y.; O’Keefe, R.J. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 2011, 124, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, Y.; Lim, Y.B.; Tang, D.; Xie, R.; Chen, A.; Tai, P.; Harris, S.E.; Xing, L.; Qin, Y.X.; et al. BMP-2 modulates β-catenin signaling through stimulation of Lrp5 expression and inhibition of β-TrCP expression in osteoblasts. J. Cell. Biochem. 2009, 108, 896–905. [Google Scholar] [CrossRef]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Maeda, S.; Imamura, T. Coordinate regulation of cell growth and differentiation by TGF-β superfamily and Runx proteins. Oncogene 2004, 23, 4232–4237. [Google Scholar] [CrossRef] [PubMed]
- Leboy, P.; Grasso-Knight, G.; D’Angelo, M.; Volk, S.W.; Lian, J.V.; Drissi, H.; Stein, G.S.; Adams, S.L. Smad-Runx interactions during chondrocyte maturation. J. Bone Jt. Surg. Am. 2001, 83, S15–S22. [Google Scholar]
- Yu, Y.Y.; Lieu, S.; Lu, C.; Miclau, T.; Marcucio, R.S.; Colnot, C. Immunolocalization of BMPs, BMP antagonists, receptors, and effectors during fracture repair. Bone 2010, 46, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.; Emans, P.J.; Cremers, A.; Surtel, D.A.; Coolsen, M.M.; van Rhijn, L.W.; Welting, T.J. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil. 2013, 21, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Tsumaki, N.; Nakase, T.; Miyaji, T.; Kakiuchi, M.; Kimura, T.; Ochi, T.; Yoshikawa, H. Bone morphogenetic protein signals are required for cartilage formation and differently regulate joint development during skeletogenesis. J. Bone Miner. Res. 2002, 17, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Zou, X.; Li, H.; Mygind, T.; Zeng, Y.; Lü, N.; Bünger, C. Molecular mechanism of osteochondroprogenitor fate determination during bone formation. Adv. Exp. Med. Biol. 2006, 585, 431–441. [Google Scholar] [PubMed]
- Karl, A.; Olbrich, N.; Pfeifer, C.; Berner, A.; Zellner, J.; Kujat, R.; Angele, P.; Nerlich, M.; Mueller, M.B. Thyroid hormone-induced hypertrophy in mesenchymal stem cell chondrogenesis is mediated by bone morphogenetic protein-4. Tissue Eng. Part A 2014, 20, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Barry, F.; Boynton, R.E.; Liu, B.; Murphy, J.M. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: Differentiation-dependent gene expression of matrix components. Exp. Cell Res. 2001, 268, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Hennig, T.; Bock, R.; Steck, E.; Richter, W. Impact of growth factors and PTHrP on early and late chondrogenic differentiation of human mesenchymal stem cells. J. Cell. Physiol. 2010, 223, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Aswad, R.; Kanaan, R.A.; Rico, M.C.; Owen, T.A.; Barbe, M.F.; Safadi, F.F.; Popoff, S.N. Connective tissue growth factor (CTGF) acts as a downstream mediator of TGF-β1 to induce mesenchymal cell condensation. J. Cell. Physiol. 2007, 210, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Tuli, R.; Tuli, S.; Nandi, S.; Huang, X.; Manner, P.A.; Hozack, W.J.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. Transforming growth factor-β-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J. Biol. Chem. 2003, 278, 41227–41236. [Google Scholar] [CrossRef] [PubMed]
- Ballock, R.T.; Heydemann, A.; Wakefield, L.M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B. TGF-β 1 prevents hypertrophy of epiphyseal chondrocytes: Regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev. Biol. 1993, 158, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Tschan, T.; Böhme, K.; Conscience-Egli, M.; Zenke, G.; Winterhalter, K.H.; Bruckner, P. Autocrine or paracrine transforming growth factor-β modulates the phenotype of chick embryo sternal chondrocytes in serum-free agarose culture. J. Biol. Chem. 1993, 268, 5156–5161. [Google Scholar] [PubMed]
- Dieudonne, S.C.; Semeins, C.M.; Goei, S.W.; Vukicevic, S.; Nulend, J.K.; Sampath, T.K.; Helder, M.; Burger, E.H. Opposite effects of osteogenic protein and transforming growth factor β on chondrogenesis in cultured long bone rudiments. J. Bone Miner. Res. 1994, 9, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Bohme, K.; Winterhalter, K.H.; Bruckner, P. Terminal differentiation of chondrocytes in culture is a spontaneous process and is arrested by transforming growth factor-β 2 and basic fibroblast growth factor in synergy. Exp. Cell Res. 1995, 216, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Hellingman, C.A.; Davidson, E.N.; Koevoet, W.; Vitters, E.L.; van den Berg, W.B.; van Osch, G.J.; van der Kraan, P.M. Smad signaling determines chondrogenic differentiation of bone-marrow-derived mesenchymal stem cells: Inhibition of Smad1/5/8P prevents terminal differentiation and calcification. Tissue Eng. Part A 2011, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, J.; Wang, B.; Jin, H.; Wang, M.; Zhang, Y.; Yang, Y.; Im, H.J.; O’Keefe, R.; Chen, D. Deletion of the transforming growth factor β receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheumatol. 2013, 65, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-β/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rigueur, D.; Lyons, K.M. TGFβ signaling in cartilage development and maintenance. Birth Defects Res. Part C Embryo Today 2014, 102, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Zuscik, M.J.; Baden, J.F.; Wu, Q.; Sheu, T.J.; Schwarz, E.M.; Drissi, H.; O’Keefe, R.J.; Puzas, J.E.; Rosier, R.N. 5-azacytidine alters TGF-β and BMP signaling and induces maturation in articular chondrocytes. J. Cell. Biochem. 2004, 92, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; Darowish, M.; Zuscik, M.J.; Chen, D.; Schwarz, E.M.; Rosier, R.N.; Drissi, H.; O’Keefe, R.J. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 2006, 21, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, T.; Tsuda, M.; Taniguchi, N.; Tajima, Y.; Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 2005, 280, 8343–8350. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of Runx2 function by TGF-β through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Narcisi, R.; Quarto, R.; Ulivi, V.; Muraglia, A.; Molfetta, L.; Giannoni, P. TGF β-1 administration during ex vivo expansion of human articular chondrocytes in a serum-free medium redirects the cell phenotype toward hypertrophy. J. Cell. Physiol. 2012, 227, 3282–3290. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-β signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zou, Y.; Guo, X.M.; Tan, H.S.; Denslin, V.; Yeow, C.H.; Ren, X.F.; Liu, T.M.; Hui, J.H.; Lee, E.H. Temporal activation of β-catenin signaling in the chondrogenic process of mesenchymal stem cells affects the phenotype of the cartilage generated. Stem Cells Dev. 2012, 21, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Itasaki, N.; Hoppler, S. Crosstalk between Wnt and bone morphogenic protein signaling: A turbulent relationship. Dev. Dyn. 2010, 239, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, I.; Malizos, K.N.; Tsezou, A. Bone morphogenetic protein-2-induced Wnt/β-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther. 2012, 14, R82. [Google Scholar] [CrossRef] [PubMed]
- Provot, S.; Kempf, H.; Murtaugh, L.C.; Chung, U.I.; Kim, D.W.; Chyung, J.; Kronenberg, H.M.; Lassar, A.B. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development 2006, 133, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xie, R.; Hou, W.; Wang, B.; Shen, R.; Wang, X.; Wang, Q.; Zhu, T.; Jonason, J.H.; Chen, D. PTHrP prevents chondrocyte premature hypertrophy by inducing cyclin-D1-dependent Runx2 and Runx3 phosphorylation, ubiquitylation and proteasomal degradation. J. Cell Sci. 2009, 122, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, X.; Lefebvre, V.; de Crombrugghe, B. Phosphorylation of SOX9 by cyclic AMP-dependent protein kinase A enhances SOX9’s ability to transactivate a Col2a1 chondrocyte-specific enhancer. Mol. Cell. Biol. 2000, 20, 4149–4158. [Google Scholar] [CrossRef] [PubMed]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Cho, S.W.; Shin, J.O.; Lee, M.J.; Kim, K.S.; Jung, H.S. Ihh and Runx2/Runx3 signaling interact to coordinate early chondrogenesis: A mouse model. PLoS ONE 2013, 8, e55296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, K.; Densmore, M.; Nishimura, R.; Lanske, B. Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions. J. Biol. Chem. 2014, 289, 24898–24910. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Karp, S.J.; Schipani, E.; St.-Jacques, B.; Hunzelman, J.; Kronenberg, H.; McMahon, A.P. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development 2000, 127, 543–548. [Google Scholar] [PubMed]
- Kawashima-Ohya, Y.; Satakeda, H.; Kuruta, Y.; Kawamoto, T.; Yan, W.; Akagawa, Y.; Hayakawa, T.; Noshiro, M.; Okada, Y.; Nakamura, S. Effects of parathyroid hormone (PTH) and PTH-related peptide on expressions of matrix metalloproteinase-2, -3, and -9 in growth plate chondrocyte cultures. Endocrinology 1998, 139, 2120–2127. [Google Scholar] [CrossRef] [PubMed]
- Lanske, B.; Amling, M.; Neff, L.; Guiducci, J.; Baron, R.; Kronenberg, H.M. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J. Clin. Investig. 1999, 104, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Kronenberg, H.M.; Chuang, P.T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Amizuka, N.; Henderson, J.E.; Hoshi, K.; Warshawsky, H.; Ozawa, H.; Goltzman, D.; Karaplis, A.C. Programmed cell death of chondrocytes and aberrant chondrogenesis in mice homozygous for parathyroid hormone-related peptide gene deletion. Endocrinology 1996, 137, 5055–5067. [Google Scholar] [PubMed]
- Weir, E.C.; Philbrick, W.M.; Amling, M.; Neff, L.A.; Baron, R.; Broadus, A.E. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc. Natl. Acad. Sci. USA 1996, 93, 10240–10345. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Leong, N.L.; Mung, J.C.; Hidaka, C.; Lu, H.H. Interaction between zonal populations of articular chondrocytes suppresses chondrocyte mineralization and this process is mediated by PTHrP. Osteoarthr. Cartil. 2008, 16, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Dickhut, A.; Rickert, M.; Richter, W. Human articular chondrocytes secrete parathyroid hormone-related protein and inhibit hypertrophy of mesenchymal stem cells in coculture during chondrogenesis. Arthritis Rheumatol. 2010, 62, 2696–2706. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Aulmann, A.; Dexheimer, V.; Grossner, T.; Richter, W. Intermittent PTHrP(1–34) exposure augments chondrogenesis and reduces hypertrophy of mesenchymal stromal cells. Stem Cells Dev. 2014, 23, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Kujat, R.; Prantl, L.; Nerlich, M.; Tuan, R.S.; Angele, P. Effect of parathyroid hormone-related protein in an in vitro hypertrophy model for mesenchymal stem cell chondrogenesis. Int. Orthop. 2013, 37, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, H.J.; Im, G.I. PTHrP promotes chondrogenesis and suppresses hypertrophy from both bone marrow-derived and adipose tissue-derived MSCs. Biochem. Biophys. Res. Commun. 2008, 373, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Kafienah, W.; Mistry, S.; Dickinson, S.C.; Sims, T.J.; Learmonth, I.; Hollander, A.P. Three-dimensional cartilage tissue engineering using adult stem cells from osteoarthritis patients. Arthritis Rheumatol. 2007, 56, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Im, G.I. PTHrP isoforms have differing effect on chondrogenic differentiation and hypertrophy of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2012, 421, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Zhai, D.Y.; Tous, E.; Rai, R.; Mauck, R.L.; Burdick, J.A. Enhanced MSC chondrogenesis following delivery of TGF-β3 from alginate microspheres within hyaluronic acid hydrogels in vitro and in vivo. Biomaterials 2011, 32, 6425–6434. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Mak, K.K.; Taketo, M.M.; Yang, Y. The Wnt/β-catenin pathway interacts differentially with PTHrP signaling to control chondrocyte hypertrophy and final maturation. PLoS ONE 2009, 4, e6067. [Google Scholar] [CrossRef] [PubMed]
- Ellman, M.B.; Yan, D.; Ahmadinia, K.; Chen, D.; An, H.S.; Im, H.J. Fibroblast growth factor control of cartilage homeostasis. J. Cell. Biochem. 2013, 114, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Blanc, A.; Filion, D.; Wang, H.; Plut, P.; Pfeffer, G.; Buschmann, M.D.; Henderson, J.E. Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis. J. Biol. Chem. 2005, 280, 20509–20515. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheumatol. 2009, 60, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.H.; Yu, K.; Lavine, K.J.; Ornitz, D.M. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Biol. 2007, 307, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Lewandoski, M.; Sun, X.; Martin, G.R. Fgf8 signalling from the AER is essential for normal limb development. Nat. Genet. 2000, 26, 460–463. [Google Scholar] [PubMed]
- Montero, A.; Okada, Y.; Tomita, M.; Ito, M.; Tsurukami, H.; Nakamura, T.; Doetschman, T.; Coffin, J.D.; Hurley, M.M. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Investig. 2000, 105, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Chen, D.; Im, H.J. Fibroblast growth factor-2 promotes catabolism via FGFR1-Ras-Raf-MEK1/2-ERK1/2 axis that coordinates with the PKCdelta pathway in human articular chondrocytes. J. Cell. Biochem. 2012, 113, 2856–2865. [Google Scholar] [CrossRef] [PubMed]
- Uchii, M.; Tamura, T.; Suda, T.; Kakuni, M.; Tanaka, A.; Miki, I. Role of fibroblast growth factor 8 (FGF8) in animal models of osteoarthritis. Arthritis Res. Ther. 2008, 10, R90. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lavine, K.J.; Hung, I.H.; Ornitz, D.M. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev. Biol. 2007, 302, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Narayana, J.; Horton, W.A. FGFR3 biology and skeletal disease. Connect. Tissue Res. 2015, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shung, C.Y.; Ota, S.; Zhou, Z.Q.; Keene, D.R.; Hurlin, P.J. Disruption of a Sox9-β-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type II. Hum. Mol. Genet. 2012, 21, 4628–4644. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Mirando, A.J.; Deng, C.X.; Hsu, W. The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development. Sci. Signal. 2010, 3, ra40. [Google Scholar] [CrossRef] [PubMed]
- Minina, E.; Kreschel, C.; Naski, M.C.; Ornitz, D.M.; Vortkamp, A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 2002, 3, 439–449. [Google Scholar] [CrossRef]
- Yoon, B.S.; Pogue, R.; Ovchinnikov, D.A.; Yoshii, I.; Mishina, Y.; Behringer, R.R.; Lyons, K.M. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 2006, 133, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Colvin, J.S.; Bohne, B.A.; Harding, G.W.; McEwen, D.G.; Ornitz, D.M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 1996, 12, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.A.; Hegde, A.; Buckley, M.; Barnes, K.M.; Baron, J.; Nilsson, O. Spatial and temporal regulation of GH-IGF-related gene expression in growth plate cartilage. J. Endocrinol. 2007, 194, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B.; Wagner, J.; Zapf, J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J. Clin. Investig. 1994, 93, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.; Manthey, A.; Tyler, J. Insulin-like growth factor (IGF) receptor, IGF-I, interleukin-1 β (IL-1 β), and IL-6 mRNA expression in osteoarthritic and normal human cartilage. J. Histochem. Cytochem. 1996, 44, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, A.; Takata, S.; Matsui, Y.; Katoh, S.; Takahashi, M.; Yasui, N. Continuous infusion of insulin-like growth factor-I into the epiphysis of the tibia. Int. Orthop. 2008, 32, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, L.; Granero-Moltó, F.; O’Rear, L.; Myers, T.J.; Li, T.; Kregor, P.J.; Spagnoli, A. Subcellular localization of IRS-1 in IGF-I-mediated chondrogenic proliferation, differentiation and hypertrophy of bone marrow mesenchymal stem cells. Growth Factors 2009, 27, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Madry, H. Overexpression of human IGF-I via direct rAAV-mediated gene transfer improves the early repair of articular cartilage defects in vivo. Gene Ther. 2014, 21, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Bondy, C.A. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J. 1999, 13, 1985–1990. [Google Scholar] [PubMed]
- Repudi, S.R.; Patra, M.; Sen, M. WISP3-IGF1 interaction regulates chondrocyte hypertrophy. J. Cell Sci. 2013, 126, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, T.; Bijman, P.; Ahmed, S.F.; Farquharson, C. Insulin-like growth factor-I augments chondrocyte hypertrophy and reverses glucocorticoid-mediated growth retardation in fetal mice metatarsal cultures. Endocrinology 2004, 145, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Bohme, K.; Conscience-Egli, M.; Tschan, T.; Winterhalter, K.H.; Bruckner, P. Induction of proliferation or hypertrophy of chondrocytes in serum-free culture: The role of insulin-like growth factor-I, insulin, or thyroxine. J. Cell Biol. 1992, 116, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shao, Y.Y.; Ballock, R.T. Thyroid hormone-mediated growth and differentiation of growth plate chondrocytes involves IGF-1 modulation of β-catenin signaling. J. Bone Miner. Res. 2010, 25, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nishida, S.; Sakata, T.; Elalieh, H.Z.; Chang, W.; Halloran, B.P.; Doty, S.B.; Bikle, D.D. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology 2006, 147, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Silver, I.A. Measurement of pH and ionic composition of pericellular sites. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1975, 271, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Leijten, J.; Georgi, N.; Teixeira, L.M.; van Blitterswijk, C.A.; Post, J.N.; Karperien, M. Metabolic programming of mesenchymal stromal cells by oxygen tension directs chondrogenic cell fate. Proc. Natl. Acad. Sci. USA 2014, 111, 13954–13959. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Chang, C.C.; Shieh, M.J.; Wang, J.P.; Chen, Y.T.; Young, T.H.; Hung, S.C. Hypoxia enhances chondrogenesis and prevents terminal differentiation through PI3K/Akt/FoxO dependent anti-apoptotic effect. Sci. Rep. 2013, 3, 2683. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Pulsatelli, L.; Facchini, A. Signaling pathways in cartilage repair. Int. J. Mol. Sci. 2014, 15, 8667–8698. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Hu, N.; Liao, J.Y.; Lin, L.B.; Zhao, C.; Si, W.K.; Yang, Z.; Yi, S.X.; Fan, T.X.; Bao, W.; et al. HIF-1α as a regulator of BMP2-induced chondrogenic differentiation, osteogenic differentiation, and endochondral ossification in stem cells. Cell. Physiol. Biochem. 2015, 36, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, X.; Li, L.; Huang, H.; Xu, R.; Luyten, W. Insights on biology and pathology of HIF-1α/-2α, TGFβ/BMP, Wnt/β-catenin, and NF-κB pathways in osteoarthritis. Curr. Pharm. Des. 2012, 18, 3293–3312. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, H.; Ikeda, T.; Jeong, J.H.; Saito, T.; Yano, F.; Jung, Y.K.; Ohba, S.; Kawaguchi, H.; Chung, U.I.; Choi, J.Y. Analysis of the Runx2 promoter in osseous and non-osseous cells and identification of HIF2A as a potent transcription activator. Gene 2008, 416, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ryu, J.H.; Oh, H.; Jeon, J.; Kwak, J.S.; Kim, J.H.; Kim, H.A.; Chun, C.H.; Chun, J.S. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2α, is an essential catabolic regulator of osteoarthritis. Ann. Rheum. Dis. 2015, 74, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Shin, Y.; Huh, Y.H.; Yang, S.; Chun, C.-H.; Chun, J.-S. Hypoxia-inducible factor-2α regulates Fas-mediated chondrocyte apoptosis during osteoarthritic cartilage destruction. Cell Death Differ. 2012, 19, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Lafont, J.E.; Talma, S.; Murphy, C.L. Hypoxia-inducible factor 2α is essential for hypoxic induction of the human articular chondrocyte phenotype. Arthritis Rheumatol. 2007, 56, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. Int. J. Mol. Sci. 2015, 16, 19225-19247. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160819225
Zhong L, Huang X, Karperien M, Post JN. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. International Journal of Molecular Sciences. 2015; 16(8):19225-19247. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160819225
Chicago/Turabian StyleZhong, Leilei, Xiaobin Huang, Marcel Karperien, and Janine N. Post. 2015. "The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes" International Journal of Molecular Sciences 16, no. 8: 19225-19247. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160819225