
Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases

Abstract
: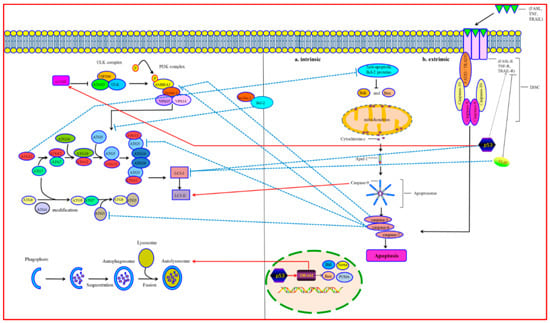
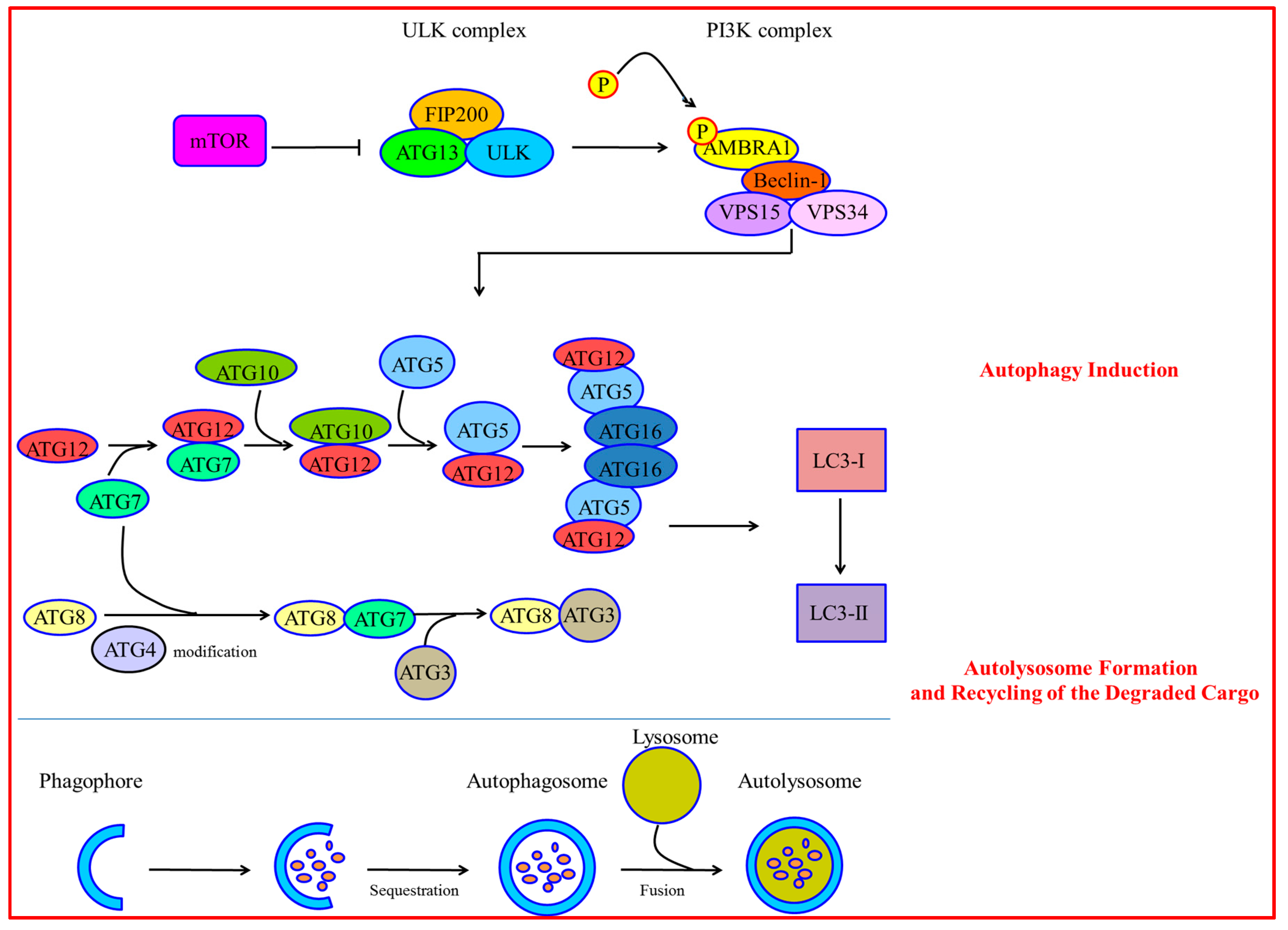
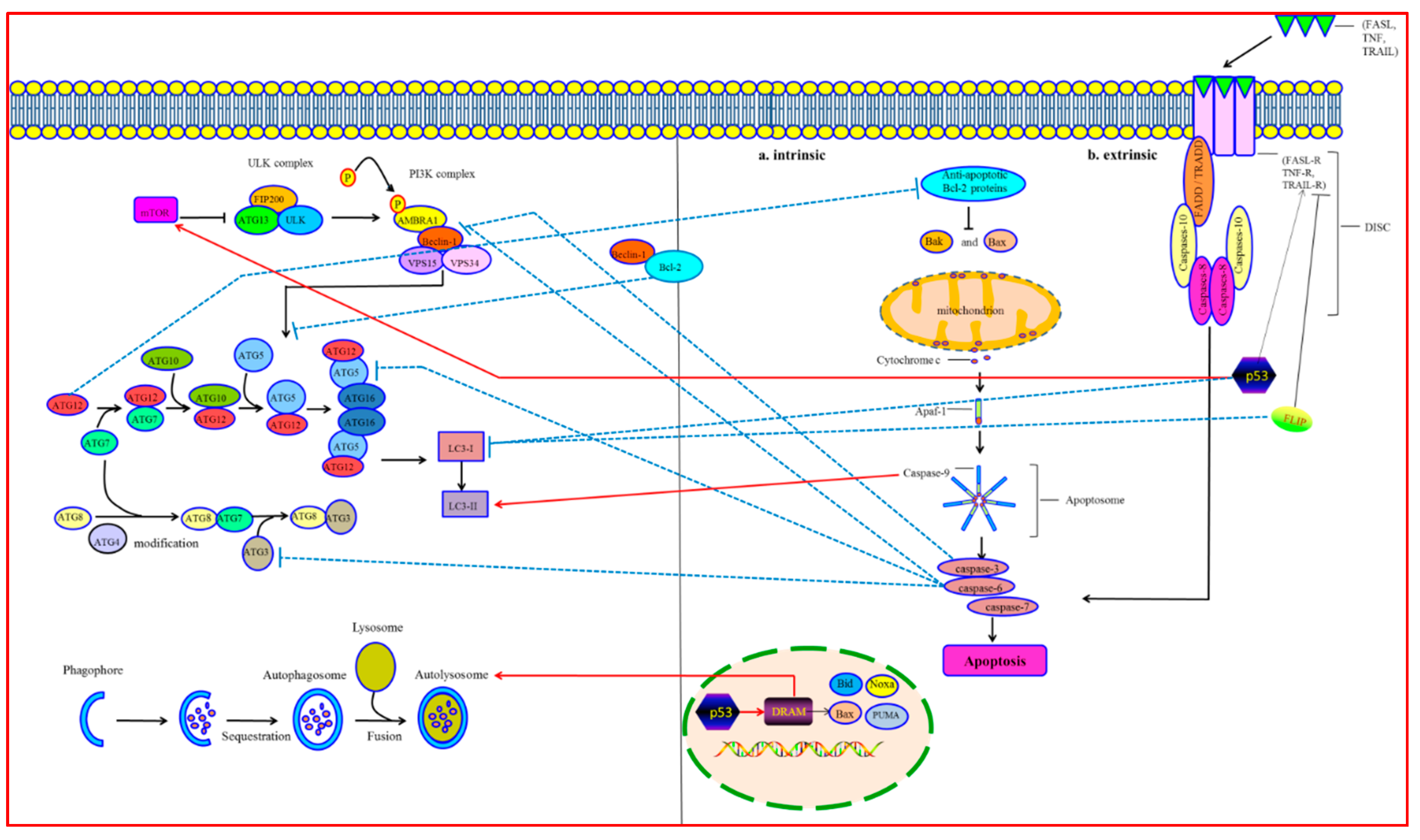
1. Molecular Mechanisms of Autophagy
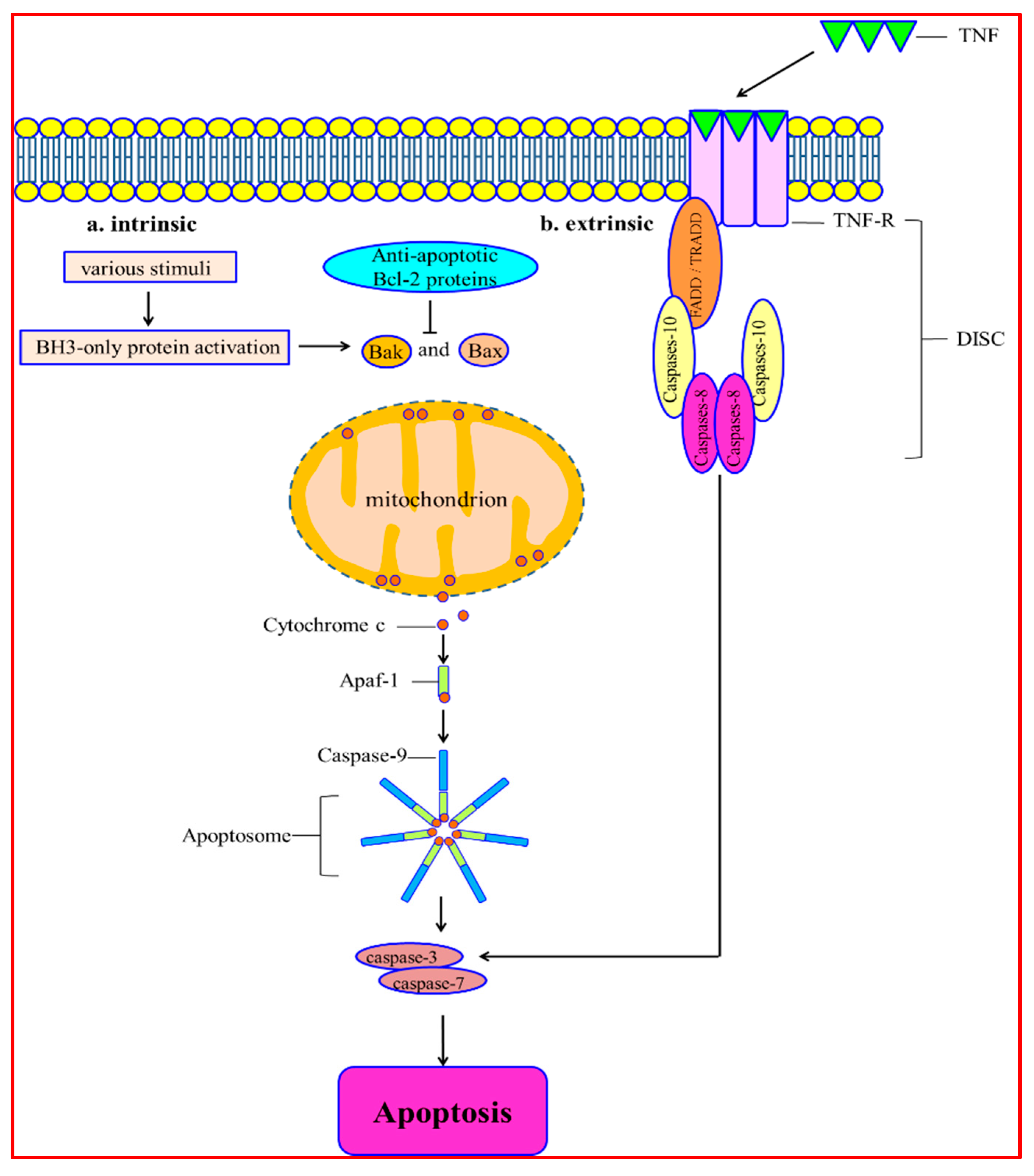
2. Molecular Mechanisms of Apoptosis
3. Crosstalk between Autophagy and Apoptosis
3.1. Bcl-2/Beclin-1
3.2. Atgs
3.3. Caspases
3.4. p53
3.5. FLIP
3.6. Mitoptosis
3.7. Mitophagy
4. The Relationship between Autophagy and Apoptosis in Cardiac Diseases
4.1. Ischemic Heart Disease
4.2. Pressure Overload–Induced Cardiac Disease
4.3. Diabetic Cardiomyopathy
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wang, T.; Zhu, H.; Zhang, P.; Han, R.; Liu, Y.; Ni, P.; Shen, H.; Xu, W.; Xu, H. HMGB1 modulates Lewis cell autophagy and promotes cell survival via RAGE-HMGB1-Erk1/2 positive feedback during nutrient depletion. Immunobiology 2015, 220, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, L.; Zhong, S.; Xian, R.; Yuan, B. Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp. Mol. Pathol. 2015, 98, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Jacobi, A.; Vater, C.; Zou, L.; Zou, X.; Stiehler, M. Icariin promotes angiogenic differentiation and prevents oxidative stress-induced autophagy in endothelial progenitor cells. Stem Cells 2015, 33, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Shi, H.; Ren, Y.; Guo, F.; Ni, W.; Qiao, J.; Wang, P.; Zhang, H.; Chen, C. Bovine viral diarrhea virus infection induces autophagy in MDBK cells. J. Microbiol. 2014, 52, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Sung, M.S.; Lee, E.G.; Yoo, H.G.; Cheon, Y.H.; Chae, H.J.; Yoo, W.H. A pathogenic role for ER stress-induced autophagy and ER chaperone GRP78/BiP in T lymphocyte systemic lupus erythematosus. J. Leukoc. Biol. 2015, 97, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Orogo, A.M.; Gustafsson, Å.B. Therapeutic targeting of autophagy: Potential and concerns in treating cardiovascular disease. Circ. Res. 2015, 116, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Tukaj, C. The significance of macroautophagy in health and disease. Folia Morphol. (Warsz) 2013, 72, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Mizushima, N. Role of ULK-FIP200 complex in mammalian autophagy: FIP200, a counterpart of yeast Atg17? Autophagy 2009, 5, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Di-Bartolomeo, S.; Piacentini, M.; Cecconi, F. Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy 2011, 7, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Klionsky, D.J. Defining the membrane precursor supporting the nucleation of the phagophore. Autophagy 2014, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy 2010, 6, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Tooze, S.A. Recycling endosomes contribute to autophagosome formation. Autophagy 2012, 8, 1682–1683. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Beier, V.; Franquelim, H.G.; Wollert, T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 2014, 156, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Ruivo, R.; Anne, C.; Sagné, C.; Gasnier, B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta 2009, 1793, 636–649. [Google Scholar] [CrossRef] [PubMed]
- D’souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R. Danon disease: Clinical features, evaluation, and management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, J.; Geng, J.; Nair, U.; Klionsky, D.J. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol. Biol. Cell 2006, 17, 5094–5104. [Google Scholar] [CrossRef] [PubMed]
- Rogalińska, M. Alterations in cell nuclei during apoptosis. Cell. Mol. Biol. Lett. 2002, 7, 995–1018. [Google Scholar] [PubMed]
- Chen, C.W.; Wu, M.S.; Huang, Y.J.; Lin, P.W.; Shih, C.J.; Lin, F.P.; Chang, C.Y. Iridovirus CARD Protein Inhibits Apoptosis through Intrinsic and Extrinsic Pathways. PLoS ONE 2015, 10, e0129071. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kwon, S.B.; Ham, S.H.; Jeong, E.S.; Choi, Y.K.; Choi, K.D.; Hong, J.T.; Jung, S.H.; Yoon, D.Y. H9 Inhibits Tumor Growth and Induces Apoptosis via Intrinsic and Extrinsic Signaling Pathway in Human Non-Small Cell Lung Cancer Xenografts. J. Microbiol. Biotechnol. 2015, 25, 648–657. [Google Scholar] [PubMed]
- Shin, E.J.; Schram, K.; Zheng, X.L.; Sweeney, G. Leptin attenuates hypoxia/reoxygenation-induced activation of the intrinsic pathway of apoptosis in rat H9c2 cells. J. Cell. Physiol. 2009, 221, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Mancini, F.; Moretti, F. Mitochondrial MDM4 (MDMX): An unpredicted role in the p53-mediated intrinsic apoptotic pathway. Cell Cycle 2009, 8, 3854–3859. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.; Sinha-Hikim, A.P.; Datta, S.; Ferrini, M.G.; Brown, D.; Kovacheva, E.L.; Gonzalez-Cadavid, N.F.; Sinha-Hikim, I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis 2008, 13, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, M.; Ekoff, M.; Labi, V.; Strasser, A.; Huang, D.; Nilsson, G. Pro-apoptotic Bax is the major and Bak an auxiliary effector in cytokine deprivation-induced mast cell apoptosis. Cell Death Dis. 2010, 1, e43. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Puthalakath, H. Bcl-2 family proteins: The sentinels of the mitochondrial apoptosis pathway. IUBMB Life 2008, 60, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, S.; Rojas-Rivera, D.; Lisbona, F.; Parra, V.; Court, F.A.; Villegas, R.; Cheng, E.H.; Korsmeyer, S.J.; Lavandero, S.; Hetz, C. A BAX/BAK and cyclophilin D-independent intrinsic apoptosis pathway. PLoS ONE 2012, 7, e37782. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, J.; Li, Y.; Qin, D.; Li, P. Mitochondrial fission controls DNA fragmentation by regulating endonuclease G. Free Radic. Biol. Med. 2010, 49, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N. Systems biology of death receptor networks: Live and let die. Cell Death Dis. 2014, 5, e1259. [Google Scholar] [CrossRef] [PubMed]
- Sayers, T.J. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol. Immunother. 2011, 60, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N. Regulation of death receptor-induced apoptosis induced via CD95/FAS and other death receptors. Mol. Biol. (Mosk) 2011, 45, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Pennarun, B.; Meijer, A.; de-Vries, E.G.; Kleibeuker, J.H.; Kruyt, F.; de-Jong, S. Playing the DISC: Turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim. Biophys. Acta 2010, 1805, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.; Tharakan, B.; Bhat, G.K. Caspases—An update. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 151, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Kilbride, S.M.; Prehn, J.H. Central roles of apoptotic proteins in mitochondrial function. Oncogene 2013, 32, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; He, L. Beclin 1 biology and its role in heart disease. Curr. Cardiol. Rev. 2015, 11, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le-Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: Impact on the aging process. Ageing Res. Rev. 2013, 12, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Van-Delft, M.F.; Huang, D.C. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006, 16, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Bovellan, M.; Fritzsche, M.; Stevens, C.; Charras, G. Death-associated protein kinase (DAPK) and signal transduction: Blebbing in programmed cell death. FEBS J. 2010, 277, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Huebener, P.; Gwak, G.Y.; Schwabe, R.F. Comment on: HMGB1-dependent and -independent autophagy. Autophagy 2015, 11, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Lane, J.D. Atg4D at the interface between autophagy and apoptosis. Autophagy 2009, 5, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ishii, J.; Asai, E.; Ohsumi, Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 2012, 8, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schüchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Dash, R.; Das, S.K.; Azab, B.; Su, Z.Z.; Lee, S.G.; Grant, S.; Yacoub, A.; Dent, P.; Curiel, D.T.; et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res. 2010, 70, 3667–3676. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.; Hock, A.K.; Giampazolias, E.; Oberst, A.; Green, D.R.; Debnath, J.; Ryan, K.M.; Vousden, K.H.; Tait, S.W. Ubiquitination and proteasomal degradation of Atg12 regulates its proapoptotic activity. Autophagy 2014, 10, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Al-Harbi, S.; Almasan, A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods Mol. Biol. 2015, 1219, 1–9. [Google Scholar] [PubMed]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Oshima, S.; Nibe, Y.; Kobayashi, M.; Maeyashiki, C.; Nemoto, Y.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; et al. RIPK3 regulates p62-LC3 complex formation via the caspase-8-dependent cleavage of p62. Biochem. Biophys. Res. Commun. 2015, 456, 298–304. [Google Scholar]
- Lee, H.J.; Lee, E.K.; Seo, Y.E.; Shin, Y.H.; Kim, H.S.; Chun, Y.H.; Yoon, J.S.; Kim, H.H.; Han, M.Y.; Kim, C.K.; et al. Roles of Bcl-2 and caspase-9 and -3 in CD30-induced human eosinophil apoptosis. J. Microbiol. Immunol. Infect. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.M.; Vandenabeele, P.; Corazzari, M.; et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Savaraj, N.; Kuo, M.T.; Wangpaichitr, M.; Varona-Santos, J.; Wu, C.; Nguyen, D.M.; Feun, L. TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol. Cell. Biochem. 2013, 374, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Kumar, S. Caspase-2 protocols. Methods Mol. Biol. 2014, 1133, 71–87. [Google Scholar] [PubMed]
- Gaglia, G.; Lahav, G. Constant rate of p53 tetramerization in response to DNA damage controls the p53 response. Mol. Syst. Biol. 2014, 10, 753. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J. Carcinog. Mutagen. 2013. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.J.; Zong, W.X. The cellular decision between apoptosis and autophagy. Chin. J. Cancer 2013, 32, 121–129. [Google Scholar] [PubMed]
- Carpenter, D.; Henderson, G.; Hsiang, C.; Osorio, N.; Ben-Mohamed, L.; Jones, C.; Wechsler, S.L. Introducing point mutations into the Atgs of the putative open reading frames of the HSV-1 gene encoding the latency associated transcript (LAT) reduces its anti-apoptosis activity. Microb. Pathog. 2008, 44, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Debnath, J. Atg12-Atg3 and mitochondria. Autophagy 2011, 7, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. Atg12 conjugation to Atg3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande-Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; de-Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. A Complex between Atg7 and Caspase-9: A novel mechanism of cross-regulation between autophagy and apoptosis. J. Biol. Chem. 2014, 289, 6485–6497. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Lopez-Cruzan, M.; Morgan, W.W.; Herman, B. Loss of caspase-2-dependent apoptosis induces autophagy after mitochondrial oxidative stress in primary cultures of young adult cortical neurons. J. Biol. Chem. 2011, 286, 8493–8506. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Sharma, L.K.; Vanegas, D.; Callaway, D.A.; Bai, Y.; Lechleiter, J.D.; Herman, B. A nonapoptotic role for CASP2/caspase 2: Modulation of autophagy. Autophagy 2014, 10, 1054–1070. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lee, P.Y.; Son, W.C.; Chi, S.W.; Park, B.C.; Kim, J.H.; Park, S.G. Identification of the novel substrates for caspase-6 in apoptosis using proteomic approaches. BMB Rep. 2013, 46, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Estornes, Y.; Aguileta, M.A.; Dubuisson, C.; de-Keyser, J.; Goossens, V.; Kersse, K.; Samali, A.; Vandenabeele, P.; Bertrand, M.J. RIPK1 promotes death receptor-independent caspase-8-mediated apoptosis under unresolved ER stress conditions. Cell Death Dis. 2015, 6, e1798. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Choi, H.Y.; Lee, E.R.; Kim, J.H.; Jeon, K.; Lee, H.J.; Cho, S.G. Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim. Biophys. Acta 2011, 1813, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, M.A.; Wang, W.; Rosales, K.R.; Welliver, M.X.; Pan, M.; Kong, M. The B55α subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell 2013, 50, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M.; Yellon, D.M. p53 down-regulation: A new molecular mechanism involved in ischaemic preconditioning. FEBS Lett. 2003, 555, 302–306. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld-Granot, G.; Krishnamurthy, J.; Kannan, K.; Toren, A.; Amariglio, N.; Givol, D.; Rechavi, G. A positive feedback mechanism in the transcriptional activation of Apaf-1 by p53 and the coactivator Zac-1. Oncogene 2002, 21, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Jangamreddy, J.R.; Los, M.J. Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat. Mon. 2012, 12, e6159. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Nepryakhina, O.K.; Saprunova, V.B.; Bakeeva, L.E.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta 2008, 1777, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Mattenberger, Y.; James, D.I.; Martinou, J.C. Fusion of mitochondria in mammalian cells is dependent on the mitochondrial inner membrane potential and independent of microtubules or actin. FEBS Lett. 2003, 538, 53–59. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.G.; Hollville, E.; Martin, S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014, 9, 1538–1553. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.R.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbé, S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Biala, A.K.; Kirshenbaum, L.A. The interplay between cell death signaling pathways in the heart. Trends Cardiovasc. Med. 2014, 24, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Fu, M. Cardiomyocyte apoptosis in heart development: Methods and protocols. Methods Mol. Biol. 2012, 843, 191–197. [Google Scholar] [PubMed]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Matsui, Y.; Hirotani, S.; Sakoda, H.; Asano, T.; Sadoshima, J. AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy 2007, 3, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Han, Z.; Ye, B.; Dai, Z.; Shan, P.; Lu, Z.; Dai, K.; Wang, C.; Huang, W. Berberine alleviates cardiac ischemia/reperfusion injury by inhibiting excessive autophagy in cardiomyocytes. Eur. J. Pharmacol. 2015, 762, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Ying, X.; Zhao, Y.; Yuan, A.; He, Q.; Tong, H.; Ding, S.; Liu, J.; Peng, X.; Gao, E.; et al. Vitamin D receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid. Redox Signal. 2015, 22, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.W.; Liu, J.; Liu, P.P.; Li, W.J.; Chang, F.; Miao, J.Y.; Zhao, J. Sphingosylphosphorylcholine protects cardiomyocytes against ischemic apoptosis via lipid raft/PTEN/Akt1/mTOR mediated autophagy. Biochim. Biophys. Acta 2015, 1851, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zhang, Y.J.; Yu, Y.H.; Yang, S.H.; Iqbal, J.; Mi, Q.Y.; Li, B.; Wang, Z.M.; Mao, W.X.; Xie, H.G.; et al. Berberine improves pressure overload-induced cardiac hypertrophy and dysfunction through enhanced autophagy. Eur. J. Pharmacol. 2014, 728, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gibson, M.E.; Li, Z.L.; Zhu, X.Y.; Jordan, K.L.; Lerman, A.; Lerman, L.O. Autophagy Portends the Level of Cardiac Hypertrophy in Experimental Hypertensive Swine Model. Am. J. Hypertens. 2016, 29, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, H.F.; Chen, H.H.; Lai, P.F.; Juan, S.H.; Chen, J.J.; Cheng, C.F. Activating transcription factor 3 protects against pressure-overload heart failure via the autophagy molecule Beclin-1 pathway. Mol. Pharmacol. 2014, 85, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Choi, A.M. Cross talk between autophagy and apoptosis in pulmonary hypertension. Pulm. Circ. 2012, 2, 407–414. [Google Scholar] [PubMed]
- Ouyang, C.; You, J.; Xie, Z. The interplay between autophagy and apoptosis in the diabetic heart. J. Mol. Cell. Cardiol. 2014, 71, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; He, C.; Zou, M.H. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011, 7, 1254–1255. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Xie, Z. Regulation of interplay between autophagy and apoptosis in the diabetic heart: New role of AMPK. Autophagy 2013, 9, 624–625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, L.; Qiao, Y.; Zhou, X.; Wu, G.; Wang, L.; Peng, Y.; Dong, X.; Huang, H.; Si, L.; et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE 2013, 8, e75927. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Condition | Autophagy | Apoptosis | Molecular Event | References |
|---|---|---|---|---|---|
| Bcl-2/Beclin-1 | Normal | ↓ | ↓ | Beclin-1 binding to Bcl-2 | [48,49,50,51,52,53,54,55,56] |
| Starvation | ↑ | ↓ | Bcl-2-Beclin-1 complex disruption; promoting autophagosome formation | [57,58,59,60,61] | |
| Long-term starvation | ↑ | ↓ | Phosphorylated Bcl-2 binding to Bax; preserving the mitochondrial membrane integrity; preventing pro-apoptotic proteins releasing into cytoplasm | [62] | |
| Extreme starvation | ↓ | ↑ | Hyper-phosphorylated Bcl-2 dissociation from Bax; caspase 3 cleavage | [62] | |
| ATG4 | Normal | ↑ | ↓ | Covalent attachment ATG8 to PE and delipidation of ATG8 at the lysosomal fusion stage | [63] |
| Drug intervention | ↓ | ↑ | The N-terminal fragment of ATG4D cleaving and delipidating GABARAP-L1, decreasing autophagosome formation; the C-terminal fragment recruiting to mitochondrial matrix, promoting mitochondria-mediated apoptosis | [64,65] | |
| ATG5 | Normal | ↑ | ↓ | Promoting autophagosome formation | [66] |
| Apoptotic stimuli | ↓ | ↑ | Calpains cleaving ATG5 and truncated ATG5 interacting with Bcl-XL and triggering cytochrome c release and caspase activation | [67] | |
| ATG12 | Normal | ↑ | ↓ | Promoting autophagosome formation | [66] |
| Apoptotic stimuli | ↓ | ↑ | Non-conjugated ATG12 binding to and inhibiting Mcl-1 and Bcl-2, promoting mitochondrial apoptosis; ATG12 could be directly ubiquitinated, promoting its proteasomal degradation and proteasome inhibitor-mediated apoptosis | [68,69] | |
| Caspase-2 | Lack of caspase-2 | ↑ | ↓ | Inhibiting caspase-2-dependent apoptosis | [70,71] |
| Caspase-3 | Staurosporine inducing | ↓ | ↑ | Caspase-3 cleaving Beclin-1 | [72] |
| IL-3 withdrawal from culture medium | ↓ | ↑ | C-terminal fragment of Beclin-1 localizing at mitochondria and sensitizing cells to apoptosis | [73] | |
| Caspase-6 | Apoptotic stimuli | ↓ | ↑ | Caspase-6 cleaving p62 and ATG3 | [74] |
| Arginine deprivation | ↓ | ↑ | Caspase-6 cleaving ATG5 and Beclin-1 | [75] | |
| Caspase-8 | Death receptor-triggered apoptosis | ↓ | ↑ | Caspase-8 cleaving ATG3 | [76] |
| Caspase-9 | Interaction with Atg7 | ↑ | ↓ | Caspase-9 interacting with ATG7 and promoting the ATG7-dependent formation of autophagosomal LC3-II; hindering the recruitment and processing of caspase-9 in apoptosome | [77] |
| Inhibition of caspase-9 | ↓ | ↑ | Blocking autophagic flux and inducing cell death | [78] | |
| p53 | Normal | ↓ | ↑ | In cytoplasm, p53 promoting the pro-apoptotic proteins and inhibiting Bcl-2, triggering the intrinsic apoptotic pathway; inactivating AMPK and mTOR signaling; in nucleus, p53 increasing TRAIL and Fas receptor, initiating the extrinsic apoptotic pathway; p53 activating Apaf-1 of the apoptosome | [79,80,81] |
| Genotoxic stress | ↑ | ↓ | Transcriptional activation of DRAM, promoting autolysosome formation | [82] | |
| Nutrient deprivation | ↓ | ↑ | p53 post-transcriptionally down-regulating LC3 and controlling autophagic flux | [83] | |
| FLIP | Virus infection | ↓ | ↑ | FLIP competing with LC3 for binding of ATG3 and inhibiting LC3 lipidation, suppressing autophagy | [84] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Gao, P.; Zhang, J. Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. Int. J. Mol. Sci. 2016, 17, 332. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030332
Li M, Gao P, Zhang J. Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. International Journal of Molecular Sciences. 2016; 17(3):332. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030332
Chicago/Turabian StyleLi, Meng, Ping Gao, and Junping Zhang. 2016. "Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases" International Journal of Molecular Sciences 17, no. 3: 332. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030332