
Oncostatic-Cytoprotective Effect of Melatonin and Other Bioactive Molecules: A Common Target in Mitochondrial Respiration

Abstract
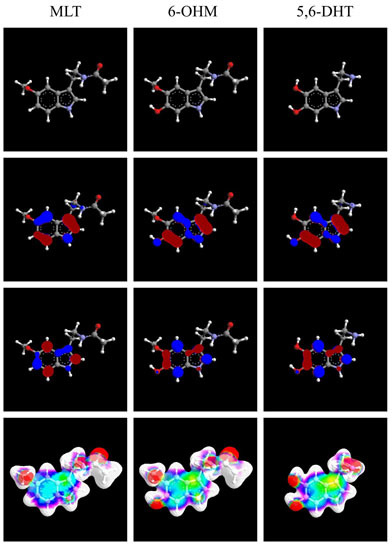
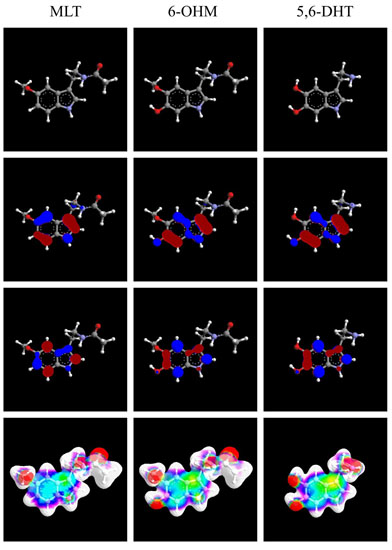
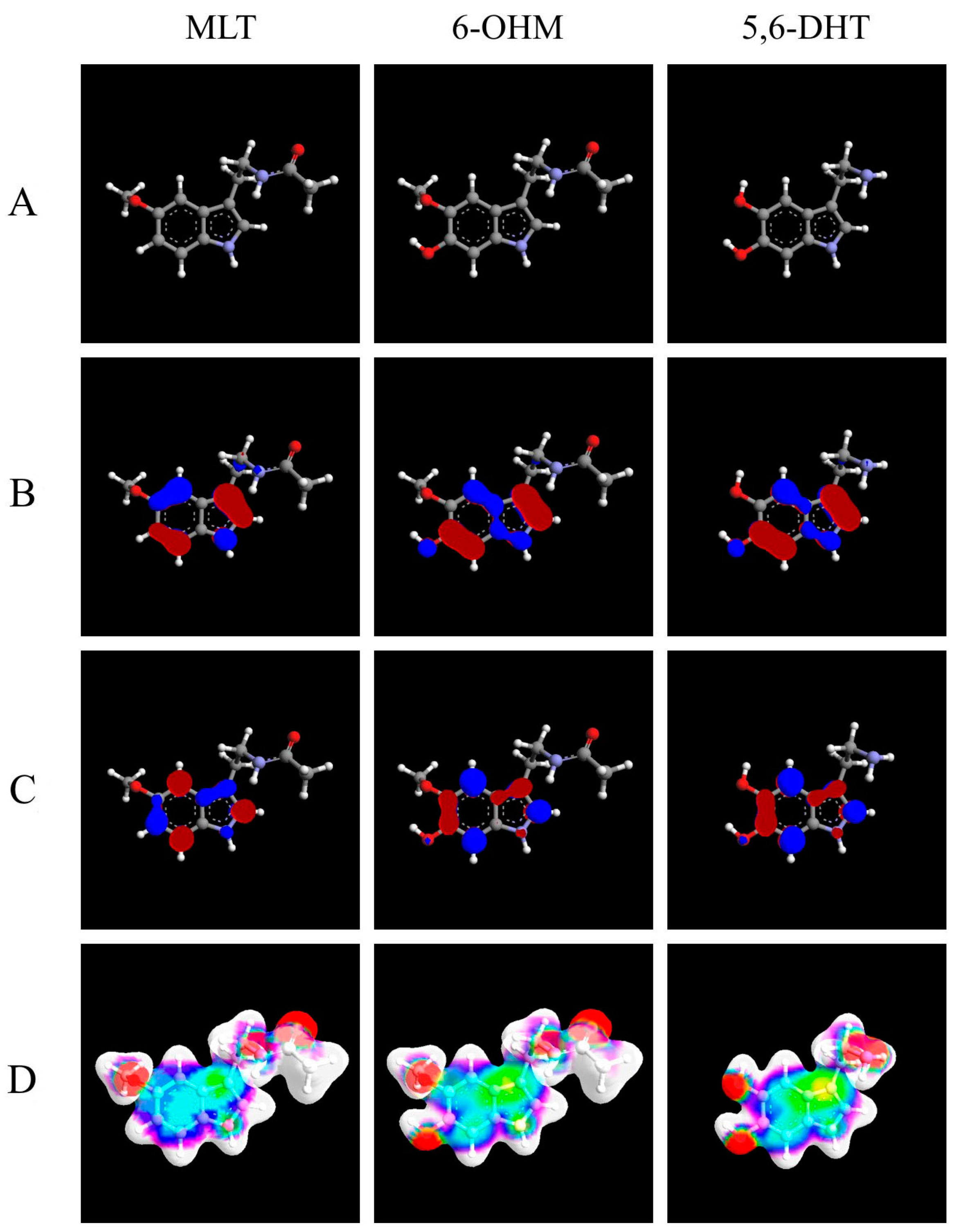
:
1. Introduction
2. Discussion
2.1. What Dosage of MLT?
2.2. Neoplasms, Metabolome and Cancer Stem Cells
2.3. Neoplasms and Mitochondria
2.4. Apoptosis, Aponecrosis and Necrosis: A Common Denominator
2.5. Melatonin and Mitochondria: A Developmental Liaison?
2.6. MLT and HIF-1α, Hypoxia-Reoxygenation
2.7. A Common Set of Mechanisms for Different Molecules?
- (a)
- the activity of respiratory complexes is diminished in many models of neurodegeneration;
- (b)
- likewise, the activity of respiratory complexes is strongly upregulated in neoplastic cells, which show UCP-mediated uncoupling and, at the same time, high respiratory consumption;
- (c)
- in response to a rise in the activity of complex IV and III, many neoplastic cells respond through the reduction of the enzymatic activity of these complexes in order to avoid catastrophic free radical events.
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pacini, N.; Borziani, F. Action of melatonin on bone marrow depression induced by cyclophosphamide in acute toxicity phase. Neuro Endocrinol. Lett. 2009, 30, 582–591. [Google Scholar] [PubMed]
- Bubenik, G.A. Thirty four years since the discovery of gastrointestinal melatonin. J. Physiol. Pharmacol. 2008, 59 (Suppl. S2), 33–51. [Google Scholar] [PubMed]
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell. Endocrinol. 2012, 351, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Campos Costa, I.; Nogueira Carvalho, H.; Fernandes, L. Aging, circadian rhythms and depressive disorders: A review. Am. J. Neurodegener. Dis. 2013, 2, 228–246. [Google Scholar] [PubMed]
- Hirata, F.; Hayaishi, O.; Tokuyama, T.; Seno, S. In vitro and in vivo formation of two new metabolites of melatonin. J. Biol. Chem. 1974, 249, 1311–1313. [Google Scholar] [PubMed]
- Reiter, R.J.; Tan, D.X.; Cabrera, J.; D’Arpa, D. Melatonin and tryptophan derivatives as free radical scavengers and antioxidants. Adv. Exp. Med. Biol. 1999, 467, 379–387. [Google Scholar] [PubMed]
- Rich, A.; Farrugia, G.; Rae, J.L. Effects of melatonin on ionic currents in cultured ocular tissues. Am. J. Physiol. 1999, 276, C923–C929. [Google Scholar] [PubMed]
- Van den Top, M.; Buijs, R.M.; Ruijter, J.M.; Delagrange, P.; Spanswick, D.; Hermes, M.L. Melatonin generates an outward potassium current in rat suprachiasmatic nucleus neurones in vitro independent of their circadian rhythm. Neuroscience 2001, 107, 99–108. [Google Scholar] [CrossRef]
- Di Bella, L.; Bruschi, C.; Gualano, L. Melatonin effects on megakaryocyte membrane patch-clamp outward K+ current. Med. Sci. Monit. 2002, 8, BR527–BR531. [Google Scholar] [PubMed]
- Margheri, M.; Pacini, N.; Tani, A.; Nosi, D.; Squecco, R.; Dama, A.; Masala, E.; Francini, F.; Zecchi-Orlandini, S.; Formigli, L. Combined effects of melatonin and all-trans retinoic acid and somatostatin on breast cancer cell proliferation and death: Molecular basis for the anticancer effect of these molecules. Eur. J. Pharmacol. 2012, 681, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Korf, H.W. The pineal organ as a component of the biological clock. Phylogenetic and ontogenetic considerations. Ann. N. Y. Acad. Sci. 1994, 719, 13–42. [Google Scholar] [CrossRef] [PubMed]
- Shedpure, M.; Pati, A.K. The pineal gland: Structural and functional diversity. Indian J. Exp. Biol. 1995, 33, 625–640. [Google Scholar] [PubMed]
- Hardeland, R. Melatonin and 5-methoxytryptamine in non-metazoans. Reprod. Nutr. Dev. 1999, 39, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Burkhardt, S.; Manchester, L.C. Melatonin in plants. Nutr. Rev. 2001, 59, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Falcón, J.; Besseau, L.; Fuentès, M.; Sauzet, S.; Magnanou, E.; Boeuf, G. Structural and functional evolution of the pineal melatonin system in vertebrates. Ann. N. Y. Acad. Sci. 2009, 1163, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.B.; Case, J.D.; Takahashi, Y. Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands. J. Biol. Chem. 1960, 235, 1992–1997. [Google Scholar] [PubMed]
- Katagiri, E. Studies on the pineal gland; tumor proliferation and the pineal gland. Osaka Igakkai Zasshi 1944, 43, 315–320. [Google Scholar]
- Nakatani, M.; Ohara, Y.; Katagiri, E.; Nakano, K. Studien uber die zirbellosen weiblichen weissen ratten. Nippon Byori Gakkai Kaishi 1940, 30, 232–236. [Google Scholar]
- Sander, G.; Schmid, S. Pineal body and malignant growth. Krebsarzt 1952, 7, 300. [Google Scholar] [PubMed]
- Sander, G.; Schmid, S. Effects of implantations of pineal tissue and pineal extracts of malignancies in man. Wien. Klin. Wochenschr. 1952, 64, 505–508. [Google Scholar] [PubMed]
- Kitay, J.I.; Altschule, M.D. The Pineal Gland: A Review of the Physiologic Literature; Commonwealth Fund: Cambridge, MA, USA, 1954. [Google Scholar]
- Engel, P.; Fischl, S. Effect of pineal extracts on benzopyrene tumors. Z. Vitam. Horm. Fermentforsch. 1954, 6, 259–268. [Google Scholar] [PubMed]
- Drexler, J.; Meaney, T.F.; McCormack, L.J. The calcified pineal body and carcinoma. Clevel. Clin. Q. 1957, 24, 242–247. [Google Scholar] [CrossRef]
- Rodin, A.E. The growth and spread of walker 256 carcinoma in pinealectomized rats. Cancer Res. 1963, 23, 1545–1548. [Google Scholar] [PubMed]
- El-Domeiri, A.A.; Das Gupta, T.K. Reversal by melatonin of the effect of pinealectomy on tumor growth. Cancer Res. 1973, 33, 2830–2833. [Google Scholar] [PubMed]
- Starr, K.W. Hormonal imbalance and the sarcomata. Aust. N. Z. J. Surg. 1969, 39, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Starr, K.W. Growth and new growth: Environmental carcinogens in the process of human ontogeny. Prog. Clin. Cancer 1970, 4, 1–29. [Google Scholar] [PubMed]
- Smythe, G.A.; Lazarus, L. Growth hormone regulation by melatonin and serotonin. Nature 1973, 244, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Lenti, G.; Molinatti, G.M.; Pizzini, A. Antisomatotropic effect of lyophilized epiphyseal extract. Boll. Soc. Ital. Biol. Sper. 1953, 29, 192–194. [Google Scholar] [PubMed]
- Lenti, G.; Molinatti, G.M.; Pizzini, A. Experimental study on the antisomatotropic action of a lyophilized pineal gland extract. Folia Endocrinol. Mens. Incretologia Incretoterapia. 1955, 8, 923–931. [Google Scholar] [PubMed]
- Burns, J.K. Administration of melatonin to non-human primates and to women with breast carcinoma. J. Physiol. 1973, 229, 38P–39P. [Google Scholar] [PubMed]
- Tapp, E.; Blumfield, M. The weight of the pineal gland in malignancy. Br. J. Cancer 1970, 24, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, L.; Rossi, M.T.; Scalera, G. Perspectives in pineal functions. Prog. Brain Res. 1979, 52, 475–478. [Google Scholar] [PubMed]
- Antón-Tay, F.; Díaz, J.L.; Fernández-Guardiola, A. On the effect of melatonin upon human brain. Its possible therapeutic implications. Life Sci I 1971, 10, 841–850. [Google Scholar] [CrossRef]
- Papavasiliou, P.S.; Cotzias, G.C.; Düby, S.E.; Steck, A.J.; Bell, M.; Lawrence, W.H. Melatonin and parkinsonism. JAMA 1972, 221, 88–89. [Google Scholar] [CrossRef] [PubMed]
- García-Santos, G.; Antolín, I.; Herrera, F.; Martín, V.; Rodriguez-Blanco, J.; del Pilar Carrera, M.; Rodriguez, C. Melatonin induces apoptosis in human neuroblastoma cancer cells. J. Pineal Res. 2006, 41, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.; Martin, V.; Carrera, P.; García-Santos, G.; Rodriguez-Blanco, J.; Rodriguez, C.; Antolín, I. Tryptamine induces cell death with ultrastructural features of autophagy in neurons and glia: Possible relevance for neurodegenerative disorders. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Rizak, J.D.; Li, X.; Li, J.; Ma, Y. Melatonin treatment increases the transcription of cell proliferation-related genes prior to inducing cell death in C6 glioma cells in vitro. Oncol. Lett. 2013, 6, 347–352. [Google Scholar] [PubMed]
- Liu, L.; Xu, Y.; Reiter, R.J. Melatonin inhibits the proliferation of human osteosarcoma cell line MG-63. Bone 2013, 55, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Jardim-Perassi, B.V.; Arbab, A.S.; Ferreira, L.C.; Borin, T.F.; Varma, N.R.; Iskander, A.S.; Shankar, A.; Ali, M.M.; de Campos Zuccari, D.A.P. Effect of melatonin on tumor growth and angiogenesis in xenograft model of breast cancer. PLoS ONE 2014, 9, e85311. [Google Scholar]
- Grant, S.G.; Melan, M.A.; Latimer, J.J.; Witt-Enderby, P.A. Melatonin and breast cancer: Cellular mechanisms, clinical studies and future perspectives. Expert Rev. Mol. Med. 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Seely, D.; Wu, P.; Fritz, H.; Kennedy, D.A.; Tsui, T.; Seely, A.J.; Mills, E. Melatonin as adjuvant cancer care with and without chemotherapy: A systematic review and meta-analysis of randomized trials. Integr. Cancer Ther. 2012, 11, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pandi-Perumal, S.R.; Brzezinski, A.; Bhatnagar, K.P.; Cardinali, D.P. Melatonin, immune function and cancer. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Pagano, E.S.; Scacchi Bernasconi, P.A.; Reynoso, R.; Scacchi, P. Melatonin and mitochondrial dysfunction in the central nervous system. Horm. Behav. 2013, 63, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiasl, N.; Banaei, S.; Alihemmati, A.; Baradaran, B.; Azimian, E. The anti-inflammatory effect of erythropoietin and melatonin on renal ischemia reperfusion injury in male rats. Adv. Pharm. Bull. 2014, 4, 49–54. [Google Scholar] [PubMed]
- Kang, J.W.; Cho, H.I.; Lee, S.M. Melatonin inhibits mTOR-dependent autophagy during liver ischemia/reperfusion. Cell. Physiol. Biochem. 2014, 33, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Martín, V.; Herrera, F.; García-Santos, G.; Rodriguez-Blanco, J.; Casado-Zapico, S.; Sánchez-Sánchez, A.M.; Suárez, S.; Puente-Moncada, N.; Anítua, M.J.; Antolín, I. Mechanisms involved in the pro-apoptotic effect of melatonin in cancer cells. Int. J. Mol. Sci. 2013, 14, 6597–6613. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.P.; da Silva, T.G.; Teixeira, Á.A.; de Medeiros, P.L.; Teixeira, V.W.; Alves, L.C.; dos Santos, F.A.B. Melatonin effect on the ultrastructure of Ehrlich ascites tumor cells, lifetime and histopathology in Swiss mice. Life Sci. 2013, 93, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.P.; da Silva, T.G.; Teixeira, A.A.; de Medeiros, P.L.; Teixeira, V.W.; Alves, L.C.; dos Santos, F.A.B.; Silva, E.C. Ultrastructural aspects of melatonin cytotoxicity on Caco-2 cells in vitro. Micron 2014, 59, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.; Sanchez, A.; Ferguson, J.A.; Balmer, C.; Daniel, C.; Cohn, A.; Robinson, W.A. Melatonin therapy of advanced human malignant melanoma. Melanoma Res. 1991, 1, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. On the stem cell origin of Cancer. Am. J. Pathol. 2010, 176, 2584–2494. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 2013, 108, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.S.; Lee, H.Y.; Nam, J.S. Cancer stem cells: The “Achilles Heel” of chemo-resistant tumors. Recent Pat. Anticancer Drug Discov. 2015, 10, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.C.; Lorenzi, M.V. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget 2010, 1, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Jiang, H.; Li, C.; Tian, M.; Zhang, H. Molecular imaging in tracking tumor stem-like cells. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, E.; Bassi, D.E. The mesenchymal tumor microenvironment: A drug-resistant niche. Cell Adhes. Migr. 2012, 6, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pacini, N.; Borziani, F. Cancer stem cell theory and the Warburg effect, two sides of the same coin? Int. J. Mol. Sci. 2014, 15, 8893–8930. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Olivotto, M.; Arcangeli, A.; Caldini, R.; Chevanne, M.; Cipolleschi, M.G.; Dello Sbarba, P. Metabolic aspects of cell cycle regulation in normal and cancer cells. Toxicol. Pathol. 1984, 12, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nichols, K.; Nathan, C.A.; Zhao, Y. Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark. 2013, 13, 377–383. [Google Scholar] [PubMed]
- Kuai, X.Y.; Ji, Z.Y.; Zhang, H.J. Mitochondrial uncoupling protein 2 expression in colon cancer and its clinical significance. World J. Gastroenterol. 2010, 16, 5773–5778. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Domann, F.E. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic. Biol. Med. 2009, 47, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Gambini, J.; Gomez-Cabrera, M.C.; Borras, C.; Valles, S.L.; Lopez-Grueso, R.; Martinez-Bello, V.E.; Herranz, D.; Pallardo, F.V.; Tresguerres, J.A.F.; Serrano, M.; et al. Free [NADH]/[NAD+] regulates sirtuin expression. Arch. Biochem. Biophys. 2011, 512, 24–29. [Google Scholar] [CrossRef]
- Legname, A.H.; Salomón de Legname, H. Changes in the oxidative metabolism during maturation of amphibian oocytes. J. Embryol. Exp. Morphol. 1980, 59, 175–186. [Google Scholar] [PubMed]
- Wales, R.G. Measurement of metabolic turnover in single mouse embryos. J. Reprod. Fertil. 1986, 76, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by phasor fluorescence lifetime microscopyof NADH. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.K.; Andrews, L.M.; Markham, J.; Jones, M.R.; Stringari, C.; Digman, M.A.; Gratton, E. Phasor-FLIM analysis of NADH distribution and localization in the nucleus of live progenitor myoblast cells. Microsc. Res. Tech. 2012, 75, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Damm, V.T.; Cox, R.T. Drosophila clueless is highly expressed in larval neuroblasts, affects mitochondrial localization and suppresses mitochondrial oxidative damage. PLoS ONE 2013, 8, e54283. [Google Scholar] [CrossRef] [PubMed]
- Magnani, M.; Stocchi, V.; Dachà, M.; Fornaini, G. Hexokinase in developing rabbit erythroid cells. Biochim. Biophys. Acta 1984, 802, 346–351. [Google Scholar] [CrossRef]
- Smith, D.G.; Sturmey, R.G. Parallels between embryo and cancer cell metabolism. Biochem. Soc. Trans. 2013, 41, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prigione, A.; Adjaye, J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int. J. Dev. Biol. 2010, 54, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Terzic, A. Induced pluripotent stem cells: An emerging theranostics platform. Clin. Pharmacol. Ther. 2011, 89, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65, 5996–6000. [Google Scholar] [CrossRef] [PubMed]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro, O.M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2015, 34, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Teslaa, T.; Teitell, M.A. Pluripotent stem cell energy metabolism: An update. EMBO J. 2015, 34, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Phipps, C.; Molavian, H.; Kohandel, M. A microscale mathematical model for metabolic symbiosis: Investigating the effects of metabolic inhibition on ATP turnover in tumors. J. Theor. Biol. 2015, 366, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Jarmusch, A.K.; Pirro, V.; Alfaro, C.M.; González-Serrano, A.F.; Niemann, H.; Wheeler, M.B.; Rabel, R.A.C.; Hallett, J.E.; Houser, R.; et al. Ambient ionisation mass spectrometry for lipid profiling and structural analysis of mammalian oocytes, preimplantation embryos and stem cells. Reprod. Fertil. Dev. 2015, 27, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Liu, X.; Bu, P.; Lin, C.S.; Rakhilin, N.; Locasale, J.W.; Shen, X. A metabolic signature of colon cancer initiating cells. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2014, 2014, 4759–4762. [Google Scholar] [PubMed]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox regulation in cancer stem cells. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Huqi, A.; Jaswal, J.S.; Mori, J.; Paulin, R.; Haromy, A.; Onay-Besikci, A.; Ionescu, L.; Thébaud, B.; Michelakis, E.; et al. Effect of fatty acids on human bone marrow mesenchymal stem cell energy metabolism and survival. PLoS ONE 2015, e0120257. [Google Scholar]
- Li, Y.; Luo, S.; Ma, R.; Liu, J.; Xu, P.; Zhang, H.; Tang, K.; Ma, J.; Zhang, Y.; Liang, X.; et al. Upregulation of cytosolic phosphoenolpyruvate carboxykinase is a critical metabolic event in melanoma cells that repopulate tumors. Cancer Res. 2015, 75, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Marsboom, G.; Glick, D.; Zhang, Y.; Toth, P.T.; Jones, N.; Malik, A.B.; Rehman, J. Bioenergetic shifts during transitions between stem cell states (2013 Grover Conference series). Pulm. Circ. 2014, 4, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.; Joy, S.; McKay, T. Cell signalling pathways underlying induced pluripotent stem cell reprogramming. World J. Stem Cells 2014, 6, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Kruspig, B.; Zhivotovsky, B.; Gogvadze, V. Mitochondrial substrates in cancer: Drivers or passengers? Mitochondrion 2014, 19 Pt A, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect. Mitochondrion 2014, 19, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Alarcòn, T. Metabostemness: A new cancer hallmark. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Montagna, M.K.; Sumi, N.J.; Joo, W.D.; Graham, E.; Mor, G. Multiple blocks in the engagement of oxidative phosphorylation in putative ovarian cancer stem cells: Implication for maintenance therapy with glycolysis inhibitors. Oncotarget 2014, 5, 8703–8715. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.W.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Vander Heiden, M.G.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.; Deli, A.; Karlsson, A.; Martins, L.M. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zheng, H.; Yu, W.M.; Cooper, T.M.; Bunting, K.D.; Qu, C.K. Maintenance of mouse hematopoietic stem cells ex vivo by reprogramming cellular metabolism. Blood 2015, 125, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Doki, Y.; Mori, M.; Ishii, H. Metabolism enzyme controls cancer stemness. Nihon Rinsho 2015, 73, 745–750. [Google Scholar] [PubMed]
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P., Jr. Differentiation of human neural stem cells into motor neurons stimulates mitochondrial biogenesis and decreases glycolytic flux. Stem Cells Dev. 2015, 24, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Béroud, C.; Demont, J.; Bouvier, R.; Schägger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Putignani, L.; Raffa, S.; Pescosolido, R.; Rizza, T.; Del Chierico, F.; Leone, L.; Aimati, L.; Signore, F.; Carrozzo, R.; Callea, F.; et al. Preliminary evidences on mitochondrial injury and impaired oxidative metabolism in breast cancer. Mitochondrion 2012, 12, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Vélez, J.; Hail, N., Jr.; Konopleva, M.; Zeng, Z.; Kojima, K.; Samudio, I.; Andreeff, M. Mitochondrial uncoupling and the reprograming of intermediary metabolism in leukemia cells. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Kjellin, H.; Johansson, H.; Höög, A.; Lehtiö, J.; Jakobsson, P.J.; Kjellman, M. Differentially expressed proteins in malignant and benign adrenocortical tumors. PLoS ONE 2014, e87951. [Google Scholar] [CrossRef] [PubMed]
- Lynen, F. Die rolle der phosphorsaeure bei dehydrierungsvorgaengen und ihre biologische bedeutung. Naturwissenschaften 1951, 30, 398–406. [Google Scholar] [CrossRef]
- Weinhouse, S. Glycolysis, respiration, and anomalous gene expression in experimental hepatomas: G.H.A. Clowes memorial lecture. Cancer Res. 1972, 32, 2007–2016. [Google Scholar] [PubMed]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008, 22, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Jaña, F.; Faini, F.; Lapier, M.; Pavani, M.; Kemmerling, U.; Morello, A.; Maya, J.D.; Jara, J.; Parra, E.; Ferreira, J. Tumor cell death induced by the inhibition of mitochondrial electron transport: The effect of 3-hydroxybakuchiol. Toxicol. Appl. Pharmacol. 2013, 272, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondrial regulation of oxygen sensing. Adv. Exp. Med. Biol. 2010, 661, 339–354. [Google Scholar] [PubMed]
- Chong, S.J.; Low, I.C.; Pervaiz, S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion 2014, 19 Pt A, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Li, X.; Wang, M.Q.; Qian, J.; Zheng, K.; Bian, H.W.; Han, N.; Wang, J.H.; Pan, J.W.; Zhu, M.Y. Protective effects of ETC complex III and cytochrome c against hydrogen peroxide-induced apoptosis in yeast. Free Radic. Res. 2014, 48, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Sennoune, S.R.; Bakunts, K.; Martínez, G.M.; Chua-Tuan, J.L.; Kebir, Y.; Attaya, M.N.; Martínez-Zaguilán, R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: Distribution and functional activity. Am. J. Physiol. Cell Physiol. 2004, 286, C1443–C1452. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS complex III in breast cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53 (Suppl. S1), S130–S135. [Google Scholar] [CrossRef] [PubMed]
- Low, I.C.; Chen, Z.X.; Pervaiz, S. Bcl-2 modulates resveratrol-induced ROS production by regulating mitochondrial respiration in tumor cells. Antioxid. Redox Signal. 2010, 13, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Kim, A.; Yoshihara, T.; Tobita, S.; Takeuchi, T.; Higuchi, M. Mitochondrial respiratory function induces endogenous hypoxia. PLoS ONE 2014, 9, e88911. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, B.; Farzadi, S.; Ghuman, M.; Hughes, F.J. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells 2014, 32, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997, 17, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Miriyala, S.; Fang, F.; Bakthavatchalu, V.; Noel, T.; Schell, D.M.; Wang, C.; st. Clair, W.H.; St. Clair, D.K. Manganese superoxide dismutase deficiency triggers mitochondrial uncoupling and the Warburg effect. Oncogene 2015, 34, 4229–4237. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Ball, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.L.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Y.; Xie, F.Z.; Zhai, C.; Stern, J.S.; Liu, Y.; Liu, S.L. The role of cellular oxidative stress in regulating glycolysis energy metabolism in hepatoma cells. Mol. Cancer 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Fliedner, S.M.; Kaludercic, N.; Jiang, X.S.; Hansikova, H.; Hajkova, Z.; Sladkova, J.; Limpuangthip, A.; Backlund, P.S.; Wesley, R.; Martiniova, L.; et al. Warburg effect’s manifestation in aggressive pheochromocytomas and paragangliomas: Insights from a mouse cell model applied to human tumor tissue. PLoS ONE 2012, 7, e40949. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Kalen, A.L.; Xiao, Z.; Veenstra, T.D.; Chaudhuri, L.; Venkataraman, S.; Reigan, P.; Buettner, G.R.; Goswami, P.C. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Res. 2012, 72, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. The protonmotive Q cycle: A general formulation. FEBS Lett. 1975, 59, 137–139. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [PubMed]
- Kerr, J.F. History of the events leading to the formulation of the apoptosis concept. Toxicology 2002, 181–182, 471–474. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Brinkmann, E.; Gallo, M.; Pastan, I. Cloning and characterization of a cellular apoptosis susceptibility gene, the human homologue to the yeast chromosome segregation gene CSE1. Proc. Natl. Acad. Sci. USA 1995, 92, 10427–10431. [Google Scholar] [CrossRef] [PubMed]
- Ruckenstuhl, C.; Büttner, S.; Carmona-Gutierrez, D.; Eisenberg, T.; Kroemer, G.; Sigrist, S.J.; Fröhlich, K.-U.; Madeo, F. The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for Cancer. PLoS ONE 2009, 4, e4592. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhong, D.; Wu, X.; Sha, M.; Kang, L.; Ding, Z. Voltage-gated potassium channel Kv1.3 is highly expressed in human osteosarcoma and promotes osteosarcoma growth. Int. J. Mol. Sci. 2013, 14, 19245–19256. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Biasutto, L.; Managò, A.; Gulbins, E.; Zoratti, M.; Szabò, I. Intracellular ion channels and cancer. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Vucic, D.; Stennicke, H.R.; Pisabarro, M.T.; Salvesen, G.S.; Dixit, V.M. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 2000, 10, 1359–1366. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Silke, J.; Ekert, P.G.; Pakusch, M.; Kaufmann, H.; Connolly, L.M.; Day, C.L.; Tikoo, A.; Burke, R.; Wrobel, C.; et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 2002, 277, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pandi-Perumal, S.R.; Maestroni, G.J.; Esquifino, A.I.; Hardeland, R.; Cardinali, D.P. Role of melatonin in neurodegenerative diseases. Neurotox. Res. 2005, 7, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.X.; Zhang, S.H.; Wang, X.M.; Wu, J.B. Inhibition of mitochondria responsible for the anti-apoptotic effects of melatonin during ischemia-reperfusion. J. Zhejiang Univ. Sci. B 2006, 7, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.E.; Cosma, G.N.; Frank, A.A.; Wells, R.L.; Gardner, H.S., Jr. Disruption of mitochondrial respiration by melatonin in MCF-7 cells. Toxicol. Appl. Pharmacol. 2001, 171, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, L.; Radogna, F.; Accorsi, A.; Cristina Albertini, M.; Gualandi, G.; Ghibelli, L. Melatonin as a modulator of apoptosis in B-lymphoma cells. Ann. N. Y. Acad. Sci. 2009, 1171, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, I.; Espino, J.; Marchena, A.M.; Barriga, C.; Paredes, S.D.; Rodríguez, A.B.; Pariente, J.A. Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol. Cell. Biochem. 2011, 353, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; del Castillo-Vaquero, A.; Miro-Moran, A.; Tapia, J.A.; Salido, G.M. Melatonin reduces pancreatic tumor cell viability by altering mitochondrial physiology. J. Pineal Res. 2011, 50, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sánchez, A.M.; Martín, V.; García-Santos, G.; Rodríguez-Blanco, J.; Casado-Zapico, S.; Suarez-Garnacho, S.; Antolín, I.; Rodriguez, C. Intracellular redox state as determinant for melatonin antiproliferative vs cytotoxic effects in cancer cells. Free Radic. Res. 2011, 45, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, J.; Cabrera, J.; Estévez, F.; Loro, J.; Reiter, R.J.; Quintana, J. Melatonin induces apoptosis through a caspase-dependent but reactive oxygen species-independent mechanism in human leukemia Molt-3 cells. J. Pineal Res. 2013, 55, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuna-Castroviejo, D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357. [Google Scholar] [CrossRef]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J.; Miyahara, Y. Hepatic mitochondrial dysfunction in senescence-accelerated mice: Correction by long-term, orally administered physiological levels of melatonin. J. Pineal Res. 2002, 33, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Toso, C.F.; Ricci, C.R.; de Mignone, I.R.; Reyes, P.; Linares, L.M.; Albornoz, L.E.; Cardinali, D.P.; Zaninovich, A. In vitro effect of melatonin on oxygen consumption in liver mitochondria of rats. Neuro Endocrinol. Lett. 2003, 24, 341–344. [Google Scholar] [PubMed]
- Sousa, S.C.; Castilho, R.F. Protective effect of melatonin on rotenone plus Ca2+-induced mitochondrial oxidative stress and PC12 cell death. Antioxid. Redox Signal. 2005, 7, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Toso, C.F.; Rebagliati, I.R.; Ricci, C.R.; Linares, L.M.; Albornoz, L.E.; Cardinali, D.P.; Zaninovich, A. Effect of melatonin treatment on oxygen consumption by rat liver mitochondria. Amino Acids 2006, 31, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Cheshchevik, V.T.; Dremza, I.K.; Lapshina, E.A.; Zabrodskaya, S.V.; Kujawa, J.; Zavodnik, I.B. Corrections by melatonin of liver mitochondrial disorders under diabetes and acute intoxication in rats. Cell. Biochem. Funct. 2011, 29, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Zavodnik, I.B.; Lapshina, E.A.; Cheshchevik, V.T.; Dremza, I.K.; Kujawa, J.; Zabrodskaya, S.V.; Reiter, R.J. Melatonin and succinate reduce rat liver mitochondrial dysfunction in diabetes. J. Physiol. Pharmacol. 2011, 62, 421–427. [Google Scholar] [PubMed]
- Girish, K.S.; Paul, M.; Thushara, R.M.; Hemshekhar, M.; Shanmuga Sundaram, M.; Rangappa, K.S.; Kemparaju, K. Melatonin elevates apoptosis in human platelets via ROS mediated mitochondrial damage. Biochem. Biophys. Res. Commun. 2013, 438, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Ashraf, M. Melatonin protection against lethal myocyte injury induced by doxorubicin as reflected by effects on mitochondrial membrane potential. J. Mol. Cell. Cardiol. 2002, 34, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B.; Sambamurti, K.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Pappolla, M.A. A novel endogenous indole protects rodent mitochondria and extends rotifer lifespan. PLoS ONE 2010, 5, e10206. [Google Scholar] [CrossRef] [PubMed]
- López, A.; García, J.A.; Escames, G.; Venegas, C.; Ortiz, F.; López, L.C.; Castroviejo, D.A. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J. Pineal Res. 2009, 46, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Sainz, R.M.; Mayo, J.C.; Leon, J.; Hardeland, R.; Poeggeler, B.; Reiter, R.J. Interactions between melatonin and nicotinamide nucleotide: NADH preservation in cells and in cell-free systems by melatonin. J. Pineal Res. 2005, 39, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Prunet-Marcassus, B.; Ambid, L.; Viguerie-Bascands, N.; Pénicaud, L.; Casteilla, L. Evidence for a direct effect of melatonin on mitochondrial genome expression of Siberian hamster brown adipocytes. J. Pineal Res. 2001, 30, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Uguz, A.C.; Cig, B.; Espino, J.; Bejarano, I.; Naziroglu, M.; Rodríguez, A.B.; Pariente, J.A. Melatonin potentiates chemotherapy-induced cytotoxicity and apoptosis in rat pancreatic tumor cells. J. Pineal Res. 2012, 53, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Semak, I.; Naumova, M.; Korik, E.; Terekhovich, V.; Wortsman, J.; Slominski, A. A novel metabolic pathway of melatonin: Oxidation by cytochrome C. Biochemistry 2005, 44, 9300–9307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Zhang, Y.; Zhang, B.X. The role of mitochondrial complex III in melatonin-induced ROS production in cultured mesangial cells. J. Pineal Res. 2011, 50, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, H.M.; Wu, L.P.; Tan, D.X.; Kamat, A.; Li, Y.-Q.; Katz, M.S.; Abboud, H.E.; Reiter, R.J.; Zhang, B.-X. Impaired mitochondrial complex III and melatonin responsive reactive oxygen species generation in kidney mitochondria of db/db mice. J. Pineal Res. 2011, 51, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.L.; Zhang, H.M.; Zhang, H.; Kamat, A.; Yeh, C.K.; Zhang, B.X. A melatonin-based fluorescence method for the measurement of mitochondrial complex III function in intact cells. J. Pineal Res. 2013, 55, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Q.; Yang, Q.H.; Xu, R.K.; Xu, J.N. Elevated levels of mitochonrial respiratory complexes activities and ATP production in 17-β-estradiol-induced prolactin-secretory tumor cells in male rats are inhibited by melatonin in vivo and in vitro. Chin. Med. J. 2013, 126, 4724–4730. [Google Scholar] [PubMed]
- Acuña-Castroviejo, D.; Carretero, M.; Doerrier, C.; López, L.C.; García-Corzo, L.; Tresguerres, J.A.; Escames, G. Melatonin protects lung mitochondria from aging. Age 2012, 34, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Tutton, P.J.; Barkla, D.H. Cytotoxicity of 5,6-dihydroxytryptamine in dimethylhydrazine-induced carcinomas of rat colon. Cancer Res. 1977, 37, 1241–1244. [Google Scholar] [PubMed]
- Klemm, H.P.; Baumgarten, H.G.; Schlossberger, H.G. Polarographic measurements of spontaneous and mitochondria-promoted oxidation of 5,6- and 5,7-dihydroxytryptamine. J. Neurochem. 1980, 35, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, H.G.; Jenner, S.; Klemm, H.P. Serotonin neurotoxins: Recent advances in the mode of administration and molecular mechanism of action. J. Physiol. 1981, 77, 309–314. [Google Scholar]
- Singh, S.; Dryhurst, G. Further insights into the oxidation chemistry and biochemistry of the serotonergic neurotoxin 5,6-dihydroxytryptamine. J. Med. Chem. 1990, 33, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Shellard, S.A.; Whelan, R.D.; Hill, B.T. Growth inhibitory and cytotoxic effects of melatonin and its metabolites on human tumour cell lines in vitro. Br. J. Cancer 1989, 60, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Sakano, K.; Oikawa, S.; Hiraku, Y.; Kawanishi, S. Oxidative DNA damage induced by a melatonin metabolite, 6-hydroxymelatonin, via a unique non-O-quinone type of redox cycle. Biochem. Pharmacol. 2004, 68, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- AbuHammad, S.; Zihlif, M. Gene expression alterations in doxorubicin resistant MCF7 breast cancer cell line. Genomics 2013, 101, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Li, Y.L.; Zhao, L.; Zhang, C.L. Role of CYP1A2 1F polymorphism in cancer risk: Evidence from a meta-analysis of 46 case-control studies. Gene 2013, 524, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Bosin, T.R.; Jonsson, G.; Beck, O. On the occurrence of 5-methoxytryptamine in brain. Brain Res. 1979, 173, 79–88. [Google Scholar] [CrossRef]
- Uemura, T.; Yamamoto, A.; Miura, R.; Yamano, T. Microsomal methoxymelanin formation from 6-hydroxymelatonin. FEBS Lett. 1980, 122, 237–240. [Google Scholar] [CrossRef]
- Beck, O.; Jonsson, G. In vivo formation of 5-methoxytryptamine from melatonin in rat. J. Neurochem. 1981, 36, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Young, I.M.; Leone, R.M.; Francis, P.; Stovell, P.; Silman, R.E. Melatonin is metabolized to N-acetyl serotonin and 6-hydroxymelatonin in man. J. Clin. Endocrinol. Metab. 1985, 60, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Young, I.M.; Leone, R.M.; Silman, R.E. The mass spectrometric analysis of the urinary metabolites of melatonin and its deuterated analogues, confirming their identity as N-acetylserotonin and 6-hydroxymelatonin. Biomed. Mass Spectrom. 1985, 12, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Idle, J.R.; Byrd, L.G.; Krausz, K.W.; Küpfer, A.; Gonzalez, F.J. Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D6. Pharmacogenetics 2003, 13, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin metabolism in the central nervous system. Curr. Neuropharmacol. 2010, 8, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Erkoç, Ş.; Erkoç, F.; Keskinc, N. Theoretical investigation of melatonin and its hydroxy isomers. J. Mol. Struct. 2002, 587, 73–79. [Google Scholar] [CrossRef]
- Dai, M.; Cui, P.; Yu, M.; Han, J.; Li, H.; Xiu, R. Melatonin modulates the expression of VEGF and HIF-1 α induced by CoCl2 in cultured cancer cells. J. Pineal Res. 2008, 44, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hwang, M.S.; Suh, S.I.; Baek, W.K. Melatonin down-regulates HIF-1 α expression through inhibition of protein translation in prostate cancer cells. J. Pineal Res. 2009, 46, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, A.; Papazafiri, P. Attenuation of oxidative stress in HL-1 cardiomyocytes improves mitochondrial function and stabilizes Hif-1α. Free Radic. Res. 2005, 39, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Carbajo-Pescador, S.; Ordoñez, R.; Benet, M.; Jover, R.; García-Palomo, A.; Mauriz, J.L.; González-Gallego, J. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 2013, 109, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Choi, J.S.; Kang, I.; Kim, K.W.; Jeong, C.H.; Jeong, J.W. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J. Pineal Res. 2013, 54, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, D.; Lacasse, A.A.; Reiter, R.J.; Vaillancourt, C. Melatonin: The watchdog of villous trophoblast homeostasis against hypoxia/reoxygenation-induced oxidative stress and apoptosis. Mol. Cell. Endocrinol. 2013, 381, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Frangioni, G.; Borgioli, G.; Bianchi, S. Melatonin, melanogenesis, and hypoxic stress in the newt, Triturus carnifex. J. Exp. Zool. A Comp. Exp. Biol. 2003, 296, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Frangioni, G.; Santoni, M.; Bianchi, S.; Franchi, M.; Fuzzi, G.; Marcaccini, S.; Varlani, C.; Borgioli, G. Function of the hepatic melanogenesis in the newt, Triturus carnifex. J. Exp. Zool. A Comp. Exp. Biol. 2005, 303, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Hahn, D.W.; Srivastava, L.; Kumaresan, P.; Turner, C.W. Effect of pinealectomy and melatonin on feed consumption and thyroid hormone secretion rate. Proc. Soc. Exp. Biol. Med. 1966, 122, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar] [PubMed]
- Dong, L.F.; Jameson, V.J.; Tilly, D.; Cerny, J.; Mahdavian, E.; Marín-Hernández, A.; Hernández-Esquivel, L.; Rodríguez-Enríquez, S.; Stursa, J.; Witting, P.K.; et al. Mitochondrial targeting of vitamin E succinate enhances its pro-apoptotic and anti-cancer activity via mitochondrial complex II. J. Biol. Chem. 2011, 286, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Li, P.M.; Li, Y.L.; Liu, B.; Wang, W.J.; Wang, Y.Z.; Li, Z. Curcumin inhibits MHCC97H liver cancer cells by activating ROS/TLR-4/caspase signaling pathway. Asian Pac. J. Cancer Prev. 2014, 15, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Sassi, N.; Mattarei, A.; Azzolini, M.; Szabo’, I.; Paradisi, C.; Zoratti, M.; Biasutto, L. Cytotoxicity of mitochondria-targeted resveratrol derivatives: Interactions with respiratory chain complexes and ATP synthase. Biochim. Biophys. Acta 2014, 1837, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Santarelli, L.; Alleva, R.; Dong, L.F.; Neuzil, J. Redox-active and redox-silent compounds: Synergistic therapeutics in cancer. Curr. Med. Chem. 2015, 22, 552–568. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr. Cytochrome oxidase deficiency in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1991, 640, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; He, P.; Shen, Y.; Li, R. New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: Deficiency of estrogen receptor-β. J. Alzheimers Dis. 2012, 30, 545–558. [Google Scholar] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Amadoro, G.; La Piana, G.; Calissano, P.; Atlante, A. Glycolytic enzyme upregulation and numbness of mitochondrial activity characterize the early phase of apoptosis in cerebellar granule cells. Apoptosis 2015, 20, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Erion, D.; Beiler, R.; Liu, Z.W.; Abizaid, A.; Zigman, J.; Elsworth, J.D.; Savitt, J.M.; DiMarchi, R.; Tschöp, M.; et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 2009, 29, 14057–14065. [Google Scholar] [CrossRef] [PubMed]
- Besson, M.T.; Dupont, P.; Fridell, Y.W.; Liévens, J.C. Increased energy metabolism rescues glia-induced pathology in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3372–3382. [Google Scholar] [CrossRef] [PubMed]
- Krako, N.; Magnifico, M.C.; Arese, M.; Meli, G.; Forte, E.; Lecci, A.; Manca, A.; Giuffre, A.; Mastronicola, D.; Sarti, P.; et al. Characterization of mitochondrial dysfunction in the 7PA2 cell model of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 37, 747–758. [Google Scholar]
- Cruz-Bermúdez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernández-Moreno, M.Á.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, S.; Fichna, J.; Lewandowska, U. Polyphenols as mitochondria-targeted anticancer drugs. Cancer Lett. 2015, 366, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jang, H.; Kim, T.W.; Kang, B.H.; Lee, S.E.; Jeon, Y.K.; Chung, H.D.; Choi, J.; Shin, J.; Cho, E.-J.; et al. Core pluripotency factors directly regulate metabolism in embryonic stem cell to maintain pluripotency. Stem Cells 2015. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, R.; Magalhães-Novais, S.; Mesquita, K.A.; Baldeiras, I.; Sousa, I.S.; Tavares, L.C.; Barbosa, I.A.; Oliveira, P.J.; Vega-Naredo, I. Melatonin antiproliferative effects require active mitochondrial function in embryonal carcinoma cells. Oncotarget 2015, 6, 17081–17096. [Google Scholar] [CrossRef] [PubMed]
- Rimmelé, P.; Liang, R.; Bigarella, C.L.; Kocabas, F.; Xie, J.; Serasinghe, M.N.; Chipuk, J.; Sadek, H.; Zhang, C.C.; Ghaffari, S. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 2015, 16, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Saini, N.; Bakhshi, S.; Pushker, N.; Sen, S.; Sharma, A.; Kaur, J.; Kashyap, S. Prognostic significance of mitochondrial oxidative phosphorylation complexes: Therapeutic target in the treatment of retinoblastoma. Mitochondrion 2015, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Voloboueva, L.A.; Stary, C.M.; Giffard, R.G. Physiologically normal 5% O2 supports neuronal differentiation and resistance to inflammatory injury in neural stem cell cultures. J. Neurosci. Res. 2015, 93, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Lalrohlui, F.; Ghatak, S.; Zohmingthanga, J.; Lallawmzuali, D.; Pautu, J.L.; Kumar, N.S. Mitochondrial complex I and V gene polymorphisms associated with breast cancer in mizo-mongloid population. Breast Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Vatrinet, R.; Iommarini, L.; Kurelac, I.; de Luise, M.; Gasparre, G.; Porcelli, A.M. Targeting respiratory complex I to prevent the Warburg effect. Int. J. Biochem. Cell Biol. 2015, 63, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Biener-Ramanujan, E.; Yang, W.; Ramanujan, V.K. Targeting metabolic plasticity in breast cancer cells via mitochondrial complex I modulation. Breast Cancer Res. Treat. 2015, 150, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Sarti, P.; Magnifico, M.C.; Altieri, F.; Mastronicola, D.; Arese, M. New evidence for cross talk between melatonin and mitochondria mediated by a circadian-compatible interaction with nitric oxide. Int. J. Mol. Sci. 2013, 14, 11259–11276. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Origin of eukaryotic cells. In Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth; Yale University: New Haven, CT, USA, 1970. [Google Scholar]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Molecule | MLT | 6-OHM | 5,6-DHT |
|---|---|---|---|
| Heat of formation | −31.8208 kcal/mol | −77.0263 kcal/mol | −28.3233 kcal/mol |
| self-consistent field energy | −67,768.8155 kcal/mol | −75,162.8503 kcal/mol | −57,634.9772 kcal/mol |
| Molecule | Serotonin | N-Acetylserotonin | MLT | 6-OHM | 5,6-DHT |
|---|---|---|---|---|---|
| XLogP3 | 0.2 | 1 | 1.4 | 0.85 | −0.1 |
| Hydrogen bond donor count | 2 | 3 | 2 | 3 | 4 |
| Hydrogen bond acceptor count | 2 | 2 | 2 | 3 | 3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacini, N.; Borziani, F. Oncostatic-Cytoprotective Effect of Melatonin and Other Bioactive Molecules: A Common Target in Mitochondrial Respiration. Int. J. Mol. Sci. 2016, 17, 341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030341
Pacini N, Borziani F. Oncostatic-Cytoprotective Effect of Melatonin and Other Bioactive Molecules: A Common Target in Mitochondrial Respiration. International Journal of Molecular Sciences. 2016; 17(3):341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030341
Chicago/Turabian StylePacini, Nicola, and Fabio Borziani. 2016. "Oncostatic-Cytoprotective Effect of Melatonin and Other Bioactive Molecules: A Common Target in Mitochondrial Respiration" International Journal of Molecular Sciences 17, no. 3: 341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030341