
A Novel Pretreatment-Free Duplex Chamber Digital PCR Detection System for the Absolute Quantitation of GMO Samples

Abstract
:
1. Introduction
2. Results
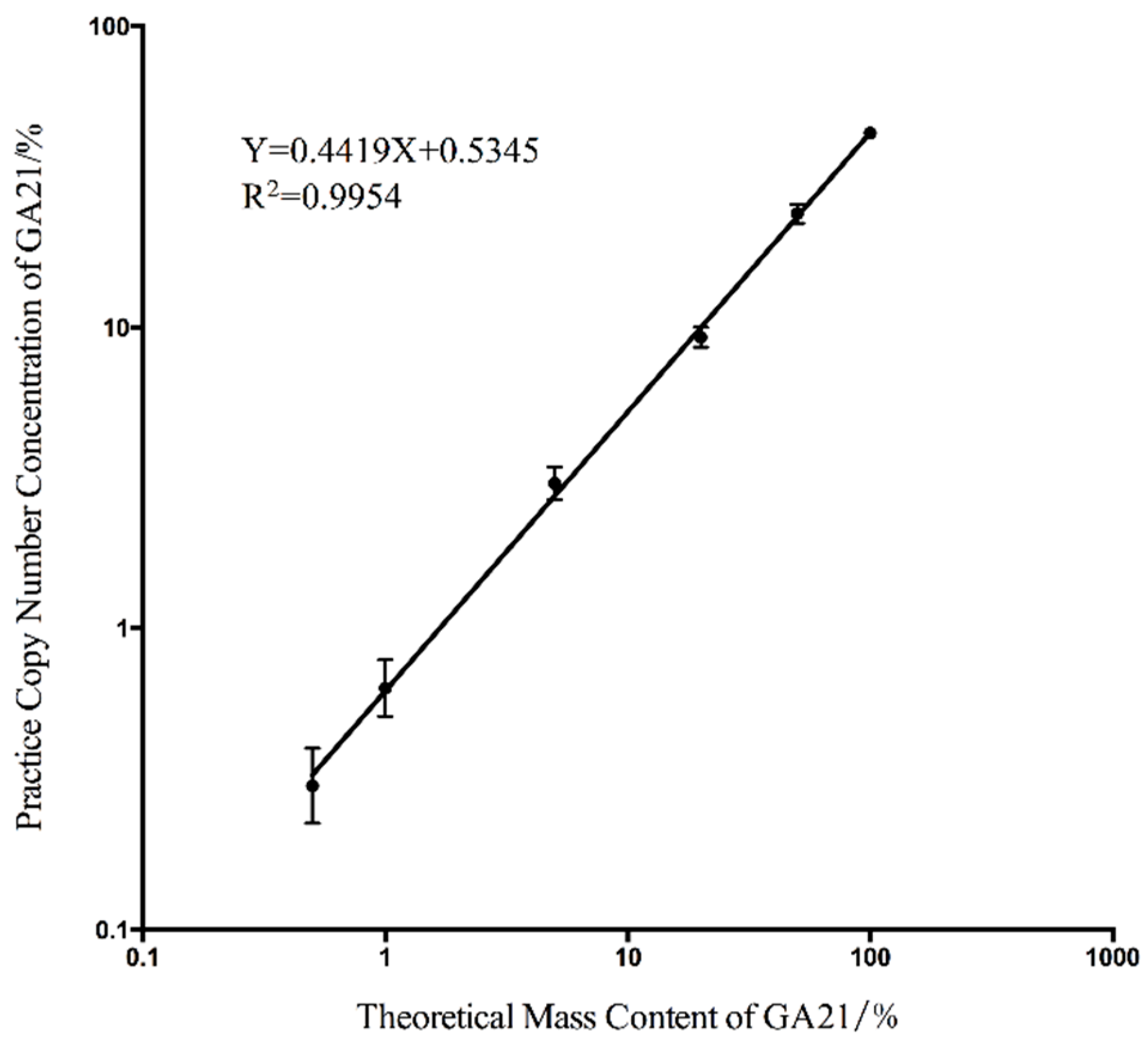
2.1. Determination of Dynamic Range and Limit of Quantitation
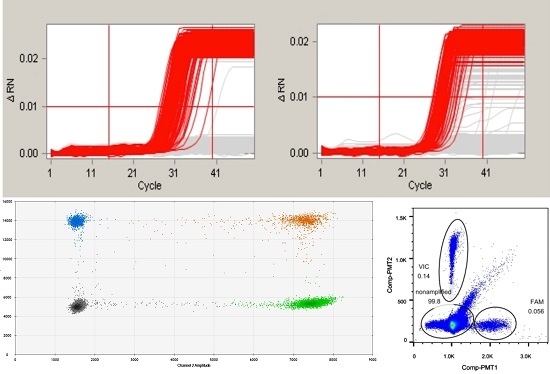
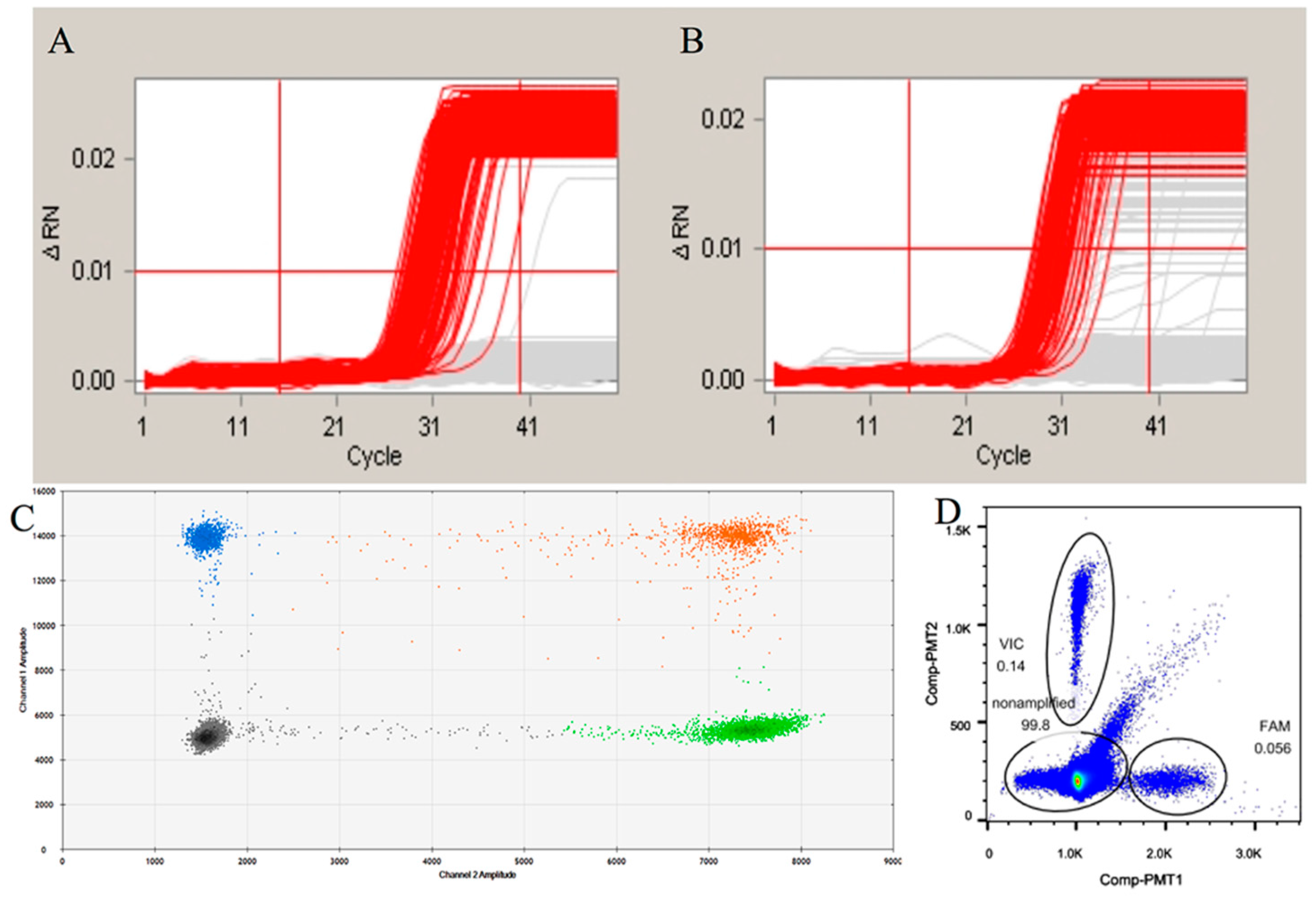
2.2. Evaluating the Sensitivity of the Duplex Digital Polymerase Chain Reaction (ddPCR)
2.3. Evaluation of the Specificity of the ddPCR
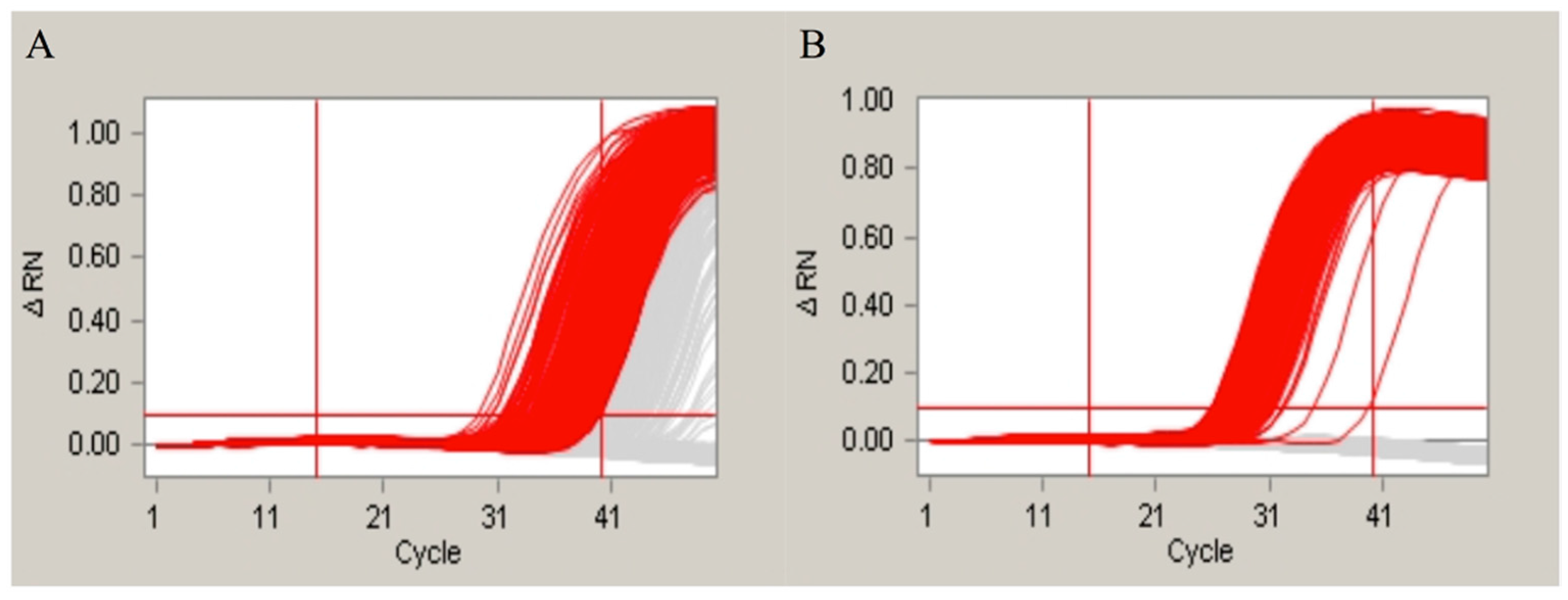
2.4. Evaluation of the Influence of the Pretreatment
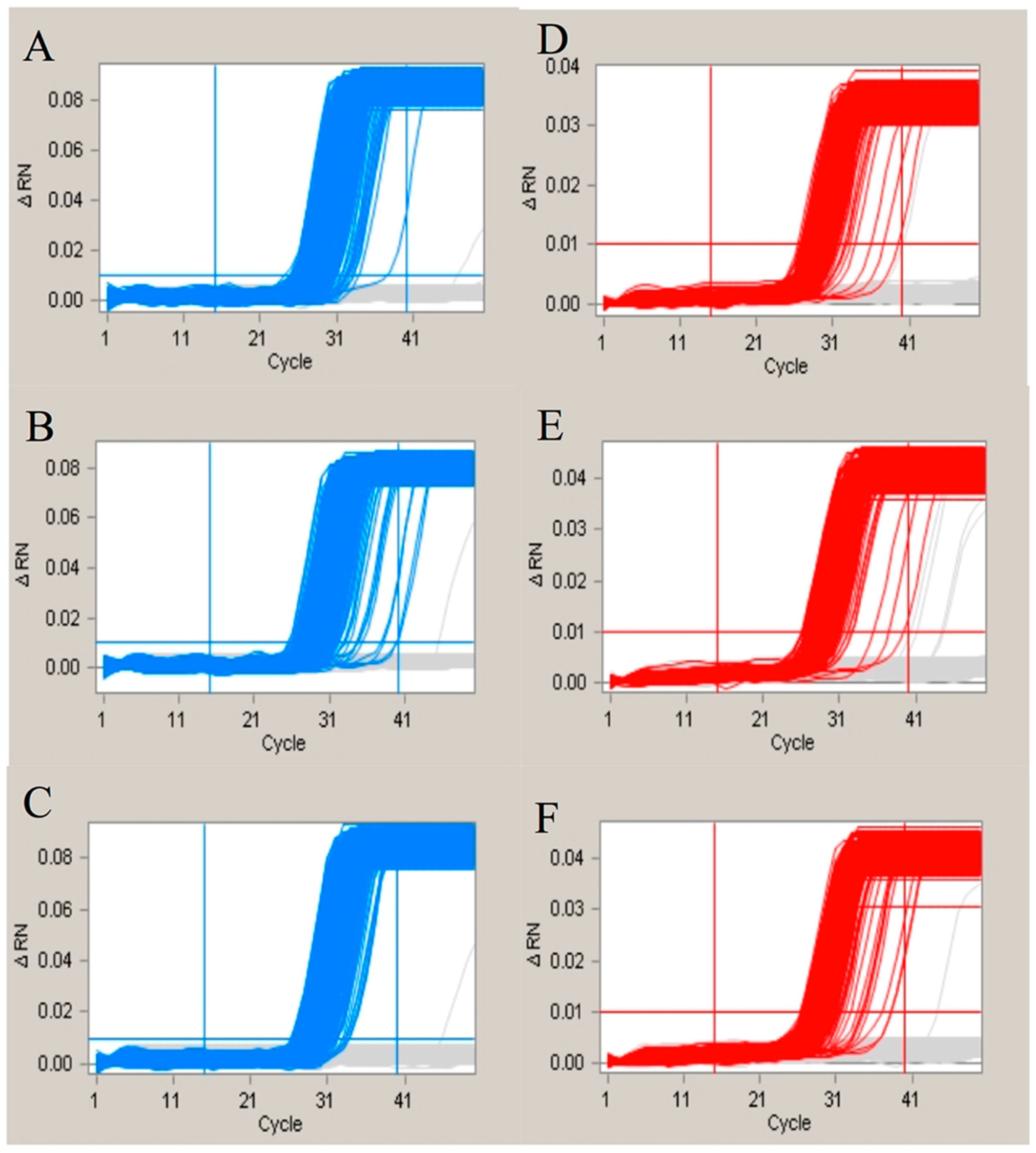
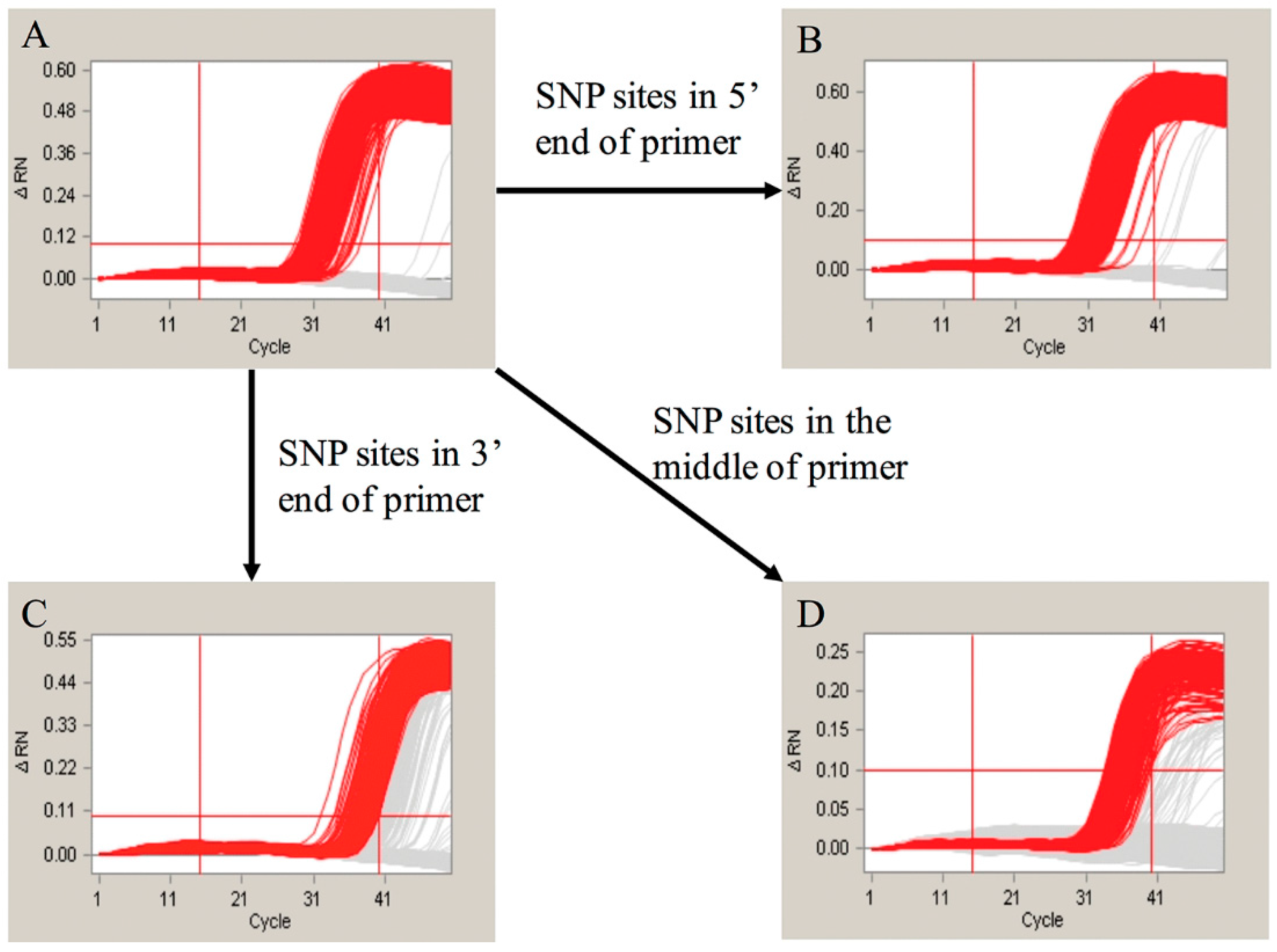
2.5. The Effect of the Nonspecific Hybridization Bases of the Probes
2.6. The Influence of the Single Nucleotide Polymorphism (SNP) Sites
3. Discussion
4. Materials and Methods
4.1. Transgenic Materials
4.2. Preparation of the Mixed Samples
4.3. Extraction of the Genome
4.4. Pretreatment of the DNA Samples
4.5. Primers and Probes
4.6. The Digital PCR
4.6.1. The Chamber-Based Digital PCR
4.6.2. The Droplet Digital PCR
4.7. Data Analysis
4.8. Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PCR | polymerase chain reaction |
| GMO | genetically modified organism |
| SNP | single nucleotide polymorphisms |
| LOD | limit of detection |
| LOQ | limit of quantitation |
References
- Hamels, S.; Glouden, T.; Gillard, K.; Mazzara, M.; Debode, F.; Foti, N.; Sneyers, M.; Esteve-Nuez, T.; Pla, M.; Berben, G.; et al. A PCR-microarray method for the screening of genetically modified organisms. Eur. Food Res. Technol. 2009, 228, 531–541. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, W.; Zhai, Z.; Luo, Y.; Yan, X.; Zhang, N.; Huang, K. Universal primer-multiplex-polymerase chain reaction (UP-M-PCR) and capillary electrophoresis-laser-induced fluorescence analysis for the simultaneous detection of six genetically modified maize lines. J. Agric. Food Chem. 2011, 59, 5188–5194. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhai, Z.; Huang, K.; Zhang, N.; Yuan, Y.; Shang, Y.; Luo, Y. A novel universal primer-multiplex-PCR method with sequencing gel electrophoresis analysis. PLoS ONE 2012, 7, e22900. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Xu, W.; Zhai, Z.; Shi, H.; Luo, Y.; Chen, Z.; Huang, K. Universal primer-multiplex PCR approach for simultaneous detection of Escherichia coli, Listeria monocytogenes, and Salmonella spp. in food samples. J. Food Sci. 2009, 74, M446–M452. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Zhu, P.; Xu, W.; Guo, T.; Tian, W.; Luo, Y.; Huang, K. Single universal primer multiplex ligation-dependent probe amplification with sequencing gel electrophoresis analysis. Anal. Biochem. 2013, 443, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yuan, Y.; Luo, Y.; Bai, W.; Zhang, C.; Huang, K. Event-specific detection of stacked genetically modified maize Bt11 x GA21 by UP-M-PCR and real-time PCR. J. Agric. Food Chem. 2009, 57, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Xu, W.; Bai, W.; Zhai, Z.; Luo, Y.; Yan, X.; He, J.; Huang, K. Event-specific qualitative and quantitative PCR detection of LY038 maize in mixed samples. Food Control 2011, 22, 1287–1295. [Google Scholar] [CrossRef]
- Buh Gašparič, M.; Tengs, T.; La Paz, J.L.; Holst-Jensen, A.; Pla, M.; Esteve, T.; Žel, J.; Gruden, K. Comparison of nine different real-time PCR chemistries for qualitative and quantitative applications in GMO detection. Anal. Bioanal. Chem. 2010, 396, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Querci, M.; Foti, N.; Bogni, A.; Kluga, L.; Broll, H.; van den Eede, G. Real-time PCR-based ready-to-use multi-target analytical system for GMO detection. Food Anal. Methods 2009, 2, 325–336. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucl. Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Dingle, T.C.; Sedlak, R.H.; Linda, C.; Jerome, K.R. Tolerance of droplet-digital PCR vs. real-time quantitative PCR to inhibitory substances. Clin. Chem. 2013, 59, 1668–1669. [Google Scholar] [CrossRef] [PubMed]
- Clive, J. Global status of commercialized Biotech/GM crops: 2014. China Biotechnol. 2015, 35, 1–14. [Google Scholar]
- Bustin, S.; Benes, V.; Garson, J. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.; Huggett, J.F.; Bushell, C.A.; Cowen, S.; Scott, D.J.; Foy, C.A. Evaluation of digital PCR for absolute DNA quantification. Anal. Chem. 2011, 83, 6474–6484. [Google Scholar] [CrossRef] [PubMed]
- Miotto, E.; Saccenti, E.; Lupini, L.; Callegari, E.; Negrini, M.; Ferracin, M. Quantification of circulating miRNAs by droplet digital PCR: Comparison of EvaGreen- and TaqMan-based chemistries. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2638–2642. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, K.S.; Nizard, P.; Perez Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical relevance of KRAS-mutated sub-clones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Colston, B.W. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.J.; Burrell, A.M.; Foy, C.A. The applicability of digital PCR for the assessment of detection limits in GMO analysis. Eur. Food Res. Technol. 2010, 231, 353–362. [Google Scholar] [CrossRef]
- Demeke, T.; Gräfenhan, T.; Holigroski, M.; Fernando, U.; Bamforth, J.; Lee, S.J. Assessment of droplet digital PCR for absolute quantification of genetically engineered OXY235 canola and DP305423 soybean samples. Food Control 2014, 46, 470–474. [Google Scholar] [CrossRef]
- Morisset, D.; Štebih, D.; Milavec, M.; Gruden, K.; Žel, J. Quantitative Analysis of Food and Feed Samples with Droplet Digital PCR. PLoS ONE 2013, 8, e62583. [Google Scholar] [CrossRef] [PubMed]
- Corbisier, P.; Bhat, S.; Partis, L.; Rui Dan Xie, V.; Emslie, K.R. Absolute quantification of genetically modified MON810 maize (Zea mays L.) by digital polymerase chain reaction. Anal. Bioanal. Chem. 2010, 396, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Corlet, A.; Fouilloux, S.; Aure, D.Z.Æ. Impact of genetic structures on haploid genome-based quantification of genetically modified DNA: Theoretical considerations, experimental data in MON 810 maize kernels (Zea mays L.) and some practical applications. Transgenic Res. 2008, 17, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Larcher, S. Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing. Available online: http://gmo-crl.jrc.ec.europa.eu/doc/Min_Perf_Requirements_Analytical_methods.pdf (accessed on 13 April 2009).
- Broothaerts, W.; Corbisier, P.; Schimmel, H.; Trapmann, S.; Vincent, S.; Emons, H. A single nucleotide polymorphism (SNP839) in the adh1 reference gene affects the quantitation of genetically modified maize (Zea mays L.). J. Agric. Food Chem. 2008, 56, 8825–8831. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Zhu, P.; Wang, C.; Huang, K.; Du, Z.; Tian, W.; Wang, Q.; Wang, H.; Xu, W.; Zhu, S. A highly sensitive and specific method for the screening detection of genetically modified organisms based on digital PCR without pretreatment. Sci. Rep. 2015, 5, 12715. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, E.; McCabe, E.; Burgess, C.; McGuinness, S.; Barry, T.; Duffy, G.; Whyte, P.; Fanning, S. Development of a real-time multiplex PCR assay for the detection of multiple Salmonella serotypes in chicken samples. BMC Microbiol. 2008, 8, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgrader, P.; Young, S.; Yuan, B.; Primeau, M.; Christel, L.A.; Pourahmadi, F.; Northrup, M.A. A battery-powered notebook thermal cycler for rapid multiplex real-time PCR analysis. Anal. Chem. 2001, 73, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Gryson, N.; Messens, K.; Dewettinck, K. Evaluation and optimisation of five different extraction methods for soy DNA in chocolate and biscuits. Extraction of DNA as a first step in GMO analysis. J. Sci. Food Agric. 2004, 84, 1357–1363. [Google Scholar] [CrossRef]
- Mazzara, M.; Cordeil, S.; van den Eede, G. Event-Specific Method for the Quantitation of Maize Line MON863 Using Real-Time PCR. Available online: http://gmo-crl.jrc.ec.europa.eu/gmomethods/docs/QT-EVE-ZM-009.pdf (accessed on 16 February 2005).
- Delobel, C.; Foti, N.; Mazzara, M.; van den Eede, G. Event-specific Method for the Quantification of Maize Event ga21 Using Real-time PCR Protocol. Available online: http://gmo-crl.jrc.ec.europa.eu/gmomethods/docs/QT-EVE-ZM-014.pdf (accessed on 30 March 2010).
- Arumuganathan, K.; Earle, E. Nuclear DNA content of some important plant species. Plant Mol. Biol. Report. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Lun, F.M.F.; Chiu, R.W.K.; Chan, K.C.A.; Tak, Y.L.; Tze, K.L.; Lo, Y.M.D. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin. Chem. 2008, 54, 1664–1672. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass Concentration a | GA21 Event-Specific Gene b | Endogenous Gene | Copy Number Concentration d | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 c | 2 | 3 | RSD | 1 | 2 | 3 | RSD | 1 | 2 | 3 | RSD | |
| 100% | 261 | 282 | 282 | 3.60% | 617 | 635 | 617 | 1.36% | 42.3% | 44.4% | 45.7% | 3.17% |
| 50% | 140 | 122 | 150 | 8.44% | 565 | 563 | 601 | 3.03% | 24.8% | 21.7% | 25% | 6.34% |
| 20% | 45 | 50 | 44 | 5.66% | 505 | 497 | 501 | 0.65% | 8.9% | 10.1% | 8.8% | 6.37% |
| 5% | 16 | 17 | 14 | 7.96% | 511 | 491 | 509 | 1.79% | 3.1% | 3.4% | 2.7% | 9.35% |
| 1% | 3 | 4 | 3 | 14.14% | 505 | 480 | 514 | 2.88% | 0.6% | 0.8% | 0.5% | 19.69% |
| 0.5% | 2 | 2 | 1 | 24.28% | 597 | 524 | 532 | 5.93% | 0.3% | 0.4% | 0.3% | 24.22% |
| 0.1% | 1 | 0 | 1 | – | 588 | 556 | 571 | 2.80% | 0.3% | 0 | 0.3% | – |
| Negative | 0 | 0 | 0 | – | 600 | 589 | 591 | 0.99% | 0 | 0 | 0 | – |
| Entry | Total Droplets | Pretreatments Including | Pretreatment-Free | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Copy Number of Endogene 1 | Copy Number of Event-Specific Gene | The GMO Content by Copy Number | Copy Number of Endogene | Copy Number of Event-Specific Gene | The GMO Content by Copy Number | ||||||||
| RainDrop | 5,000,000 | 5825 | 5136 | 2281 | 2016 | 39.2% | 39.3% | 7939 | 7099 | 3493 | 3220 | 44.0% | 45.4% |
| Bio-Rad | 20,000 | 5937 | 5648 | 2333 | 2236 | 39.3% | 39.6% | 7815 | 7449 | 3430 | 3232 | 43.9% | 43.4% |
| Primer Name | Sequence of Duplex Probe | The Hybridization Part | The Hybridization ΔG 1 |
|---|---|---|---|
| GA21-P-4 | AATCAGGGCTCATTTTCTCGCTCCTCA | ….GAGA….-3′….CTCG….-5′ | −6.31 kcal/mol |
| GA21-P-5 | ….GAGCC….-3′….CTCGG….-5′ | −9.38 kcal/mol | |
| A135-P-4 | TTTCTCAACAGCAGGTGGGTCCGGGT | ….GCTG….-3′….CGAC….-5′ | −6.69 kcal/mol |
| A135-P-5 | ….TGCTG….-3′….ACGAC….-5′ | −8.65 kcal/mol |
| Target Genes | Primers/Probes Name | Primers/Probes Sequence | Reference |
|---|---|---|---|
| Adh1 reference gene | Adh1-70F | 5′-CCTTCTTGGCGGCTTATCTG-3′ | [33] |
| Adh1-70R | 5′-CCAGCCTCATGGCCAAAG-3′ | ||
| Adh1-70P | 5′-VIC-CTTAGGGGCAGACTCCCGTGTTCCCT-BHQ1-3′ | ||
| Adh1-135F | 5′- CGTCGTTTCCCATCTCTTCCTCC-3′ | [34] | |
| Adh1-135R | 5′-CCACTCCGAGACCCTCAGTC-3′ | ||
| Adh1-135P | 5′-VIC-AATCAGGGCTCATTTTCTCGCTCCTCA-BHQ1-3′ | ||
| GA21 event-specific gene | GA21-F | 5′-CGTTATGCTATTTGCAACTTTAGAACA-3′ | [34] |
| GA21-R | 5′-GCGATCCTCCTCGCGTT-3′ | ||
| GA21-P | 5′-FAM-TTTCTCAACAGCAGGTGGGTCCGGGT-BHQ1-3′ | ||
| Primers for unspecific hybridization | GA21-P-4 | 5′-TTTCTCAAGAGCAGGTGGGTCCGGGT-3′ | This research |
| GA21-P-5 | 5′-TTTCTCAAGAGCCGGTGGGTCCGGGT-3′ | ||
| A135-P-4 | 5′-AATCAGGGCTGATTTTCTCGCTCCTCA-3′ | ||
| A135-P-5 | 5′-AATCAGTGCTGATTTTCTCGCTCCTCA-3′ | ||
| Primers for SNP researches 2 | A70-R-M-A-G | 5′-CCAGCCTCGTGGCCAAAG-3′ | This research |
| A135-F-M-A-C | 5′-CGTCGTTTCCCCTCTCTTCCTCC-3′ | ||
| A135-F-M-A-G | 5′-CGTCGTTTCCCGTCTCTTCCTCC-3′ | ||
| A135-F-M-A-T | 5′-CGTCGTTTCCCTTCTCTTCCTCC-3′ | ||
| A135-F-3′-C-A | 5′-CGTCGTTTCCCATCTCTTCCTCA-3′ | ||
| A135-F-3′-C-T | 5′-CGTCGTTTCCCATCTCTTCCTCT-3′ | ||
| A135-F-3′-C-G | 5′-CGTCGTTTCCCATCTCTTCCTCG-3′ | ||
| A135-F-5′-C-G | 5′-GGTCGTTTCCCATCTCTTCCTCC-3′ | ||
| A135-F-5′-C-A | 5′-AGTCGTTTCCCATCTCTTCCTCC-3′ | ||
| A135-F-5′-C-T | 5′-TGTCGTTTCCCATCTCTTCCTCC-3′ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, P.; Wang, C.; Huang, K.; Luo, Y.; Xu, W. A Novel Pretreatment-Free Duplex Chamber Digital PCR Detection System for the Absolute Quantitation of GMO Samples. Int. J. Mol. Sci. 2016, 17, 402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030402
Zhu P, Wang C, Huang K, Luo Y, Xu W. A Novel Pretreatment-Free Duplex Chamber Digital PCR Detection System for the Absolute Quantitation of GMO Samples. International Journal of Molecular Sciences. 2016; 17(3):402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030402
Chicago/Turabian StyleZhu, Pengyu, Chenguang Wang, Kunlun Huang, Yunbo Luo, and Wentao Xu. 2016. "A Novel Pretreatment-Free Duplex Chamber Digital PCR Detection System for the Absolute Quantitation of GMO Samples" International Journal of Molecular Sciences 17, no. 3: 402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030402