
Development of Therapeutic Chimeric Uricase by Exon Replacement/Restoration and Site-Directed Mutagenesis

Abstract
:
1. Introduction
2. Results
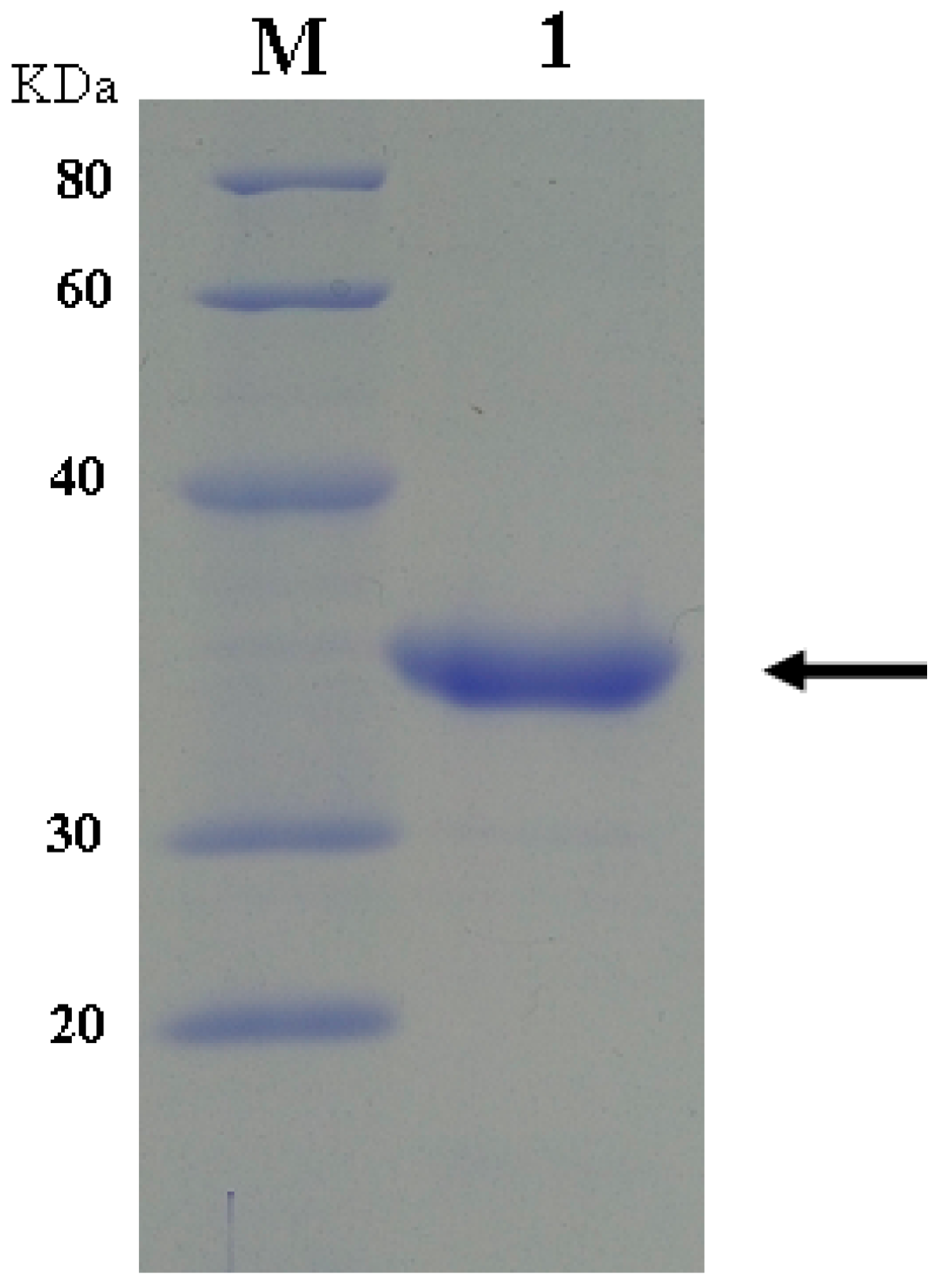
2.1. Expression and Purification of dHU and wPU
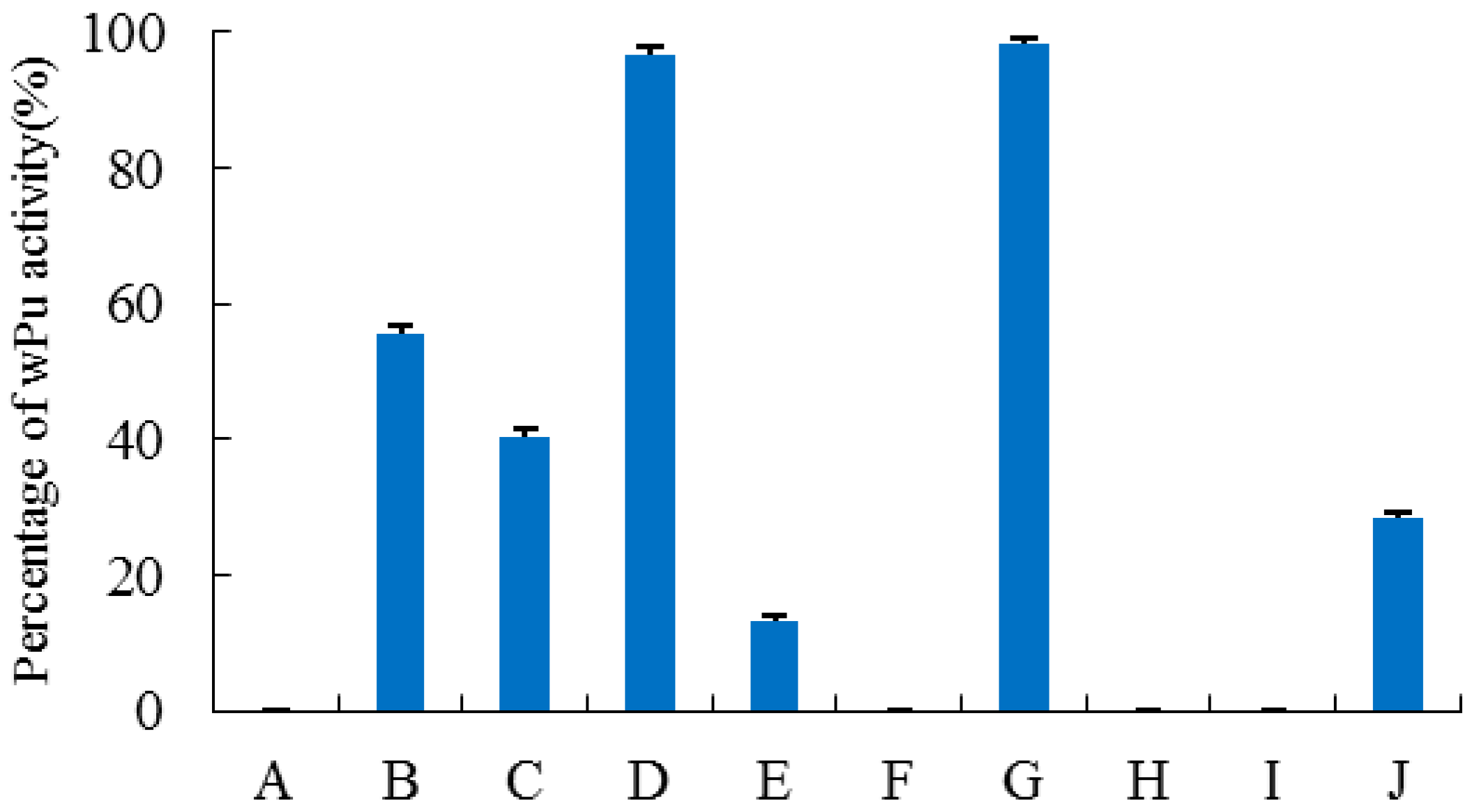
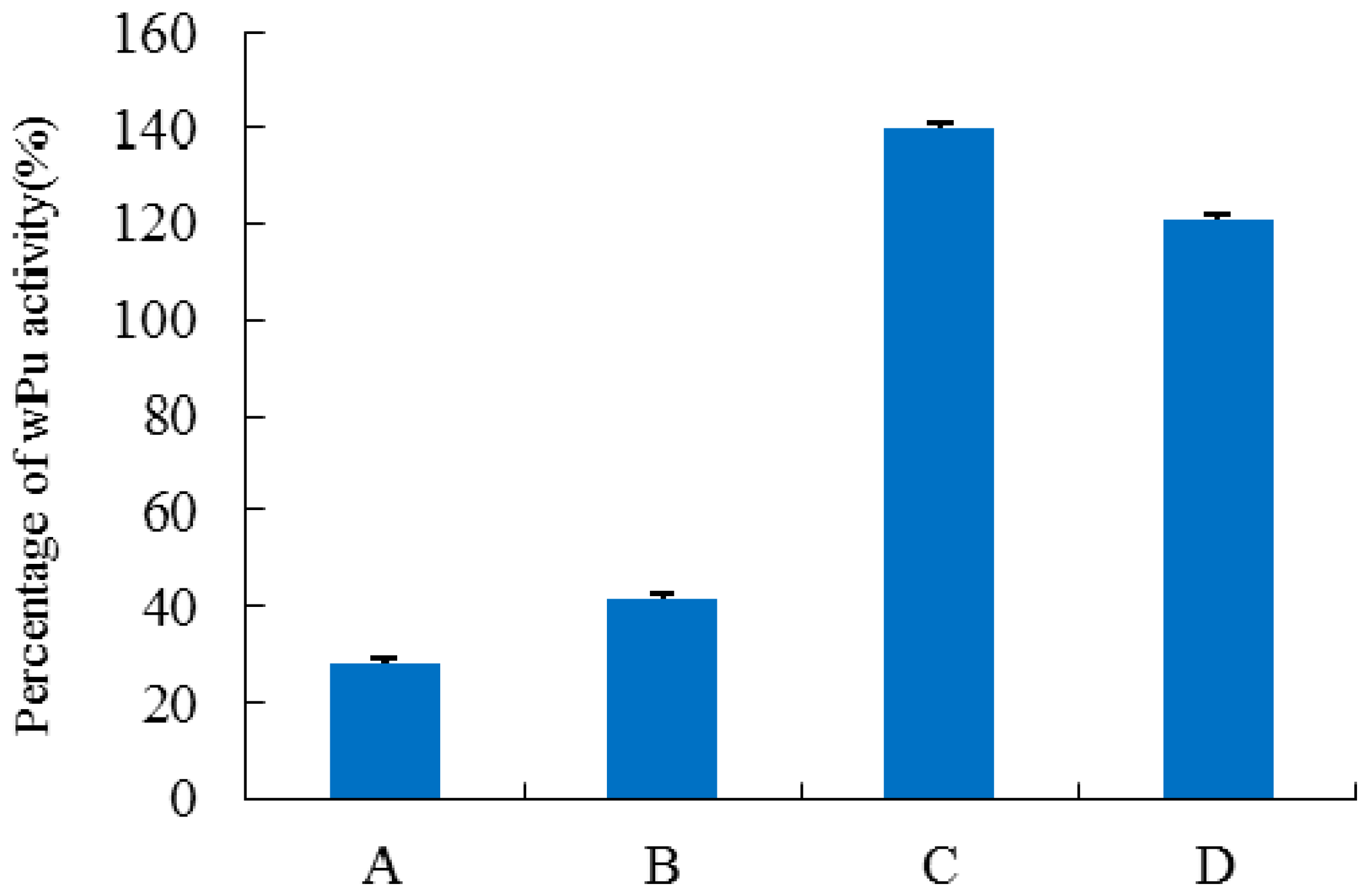
2.2. Construction, Purification, and Enzymatic Assay of Porcine–Human Uricases
2.3. Multiple Sequence Alignment and 3D Structure Modeling
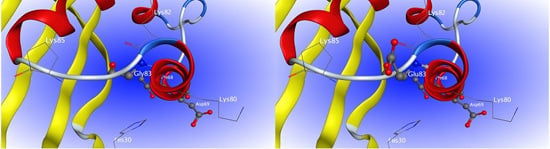
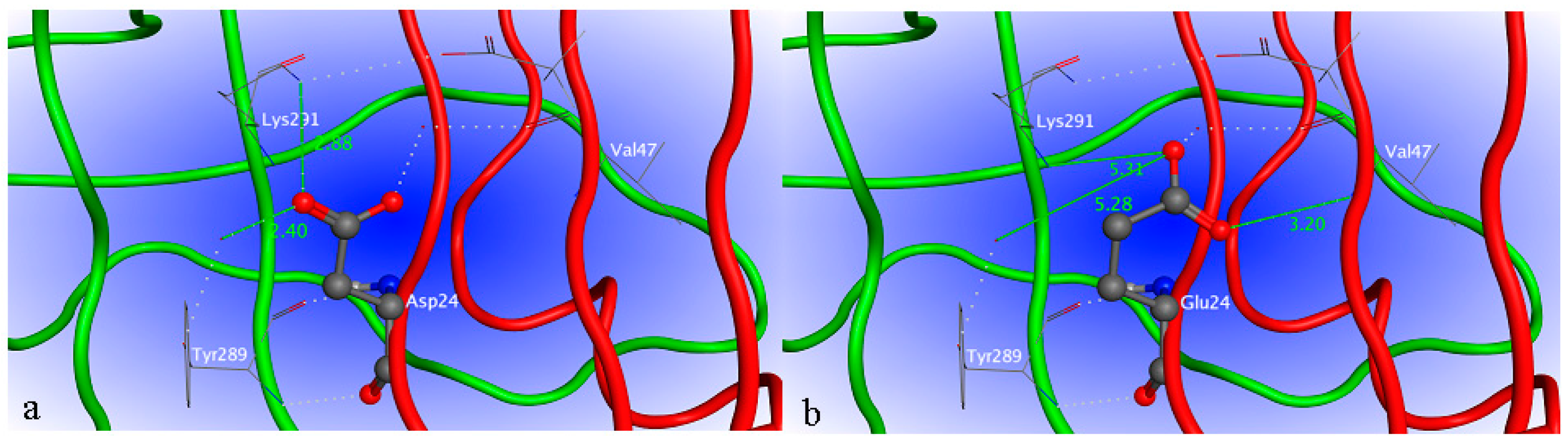
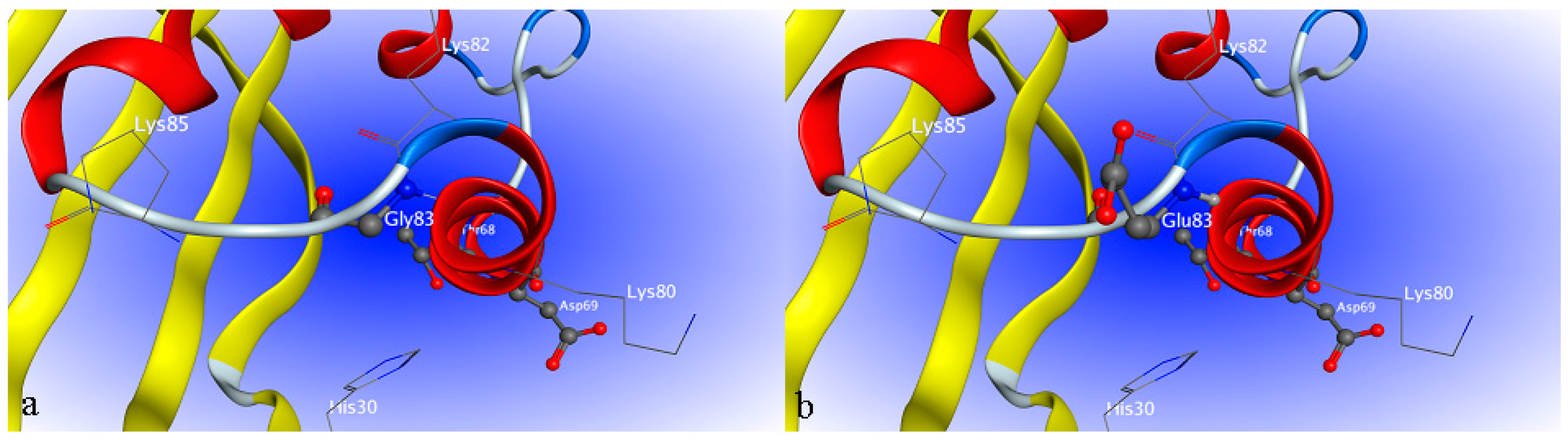
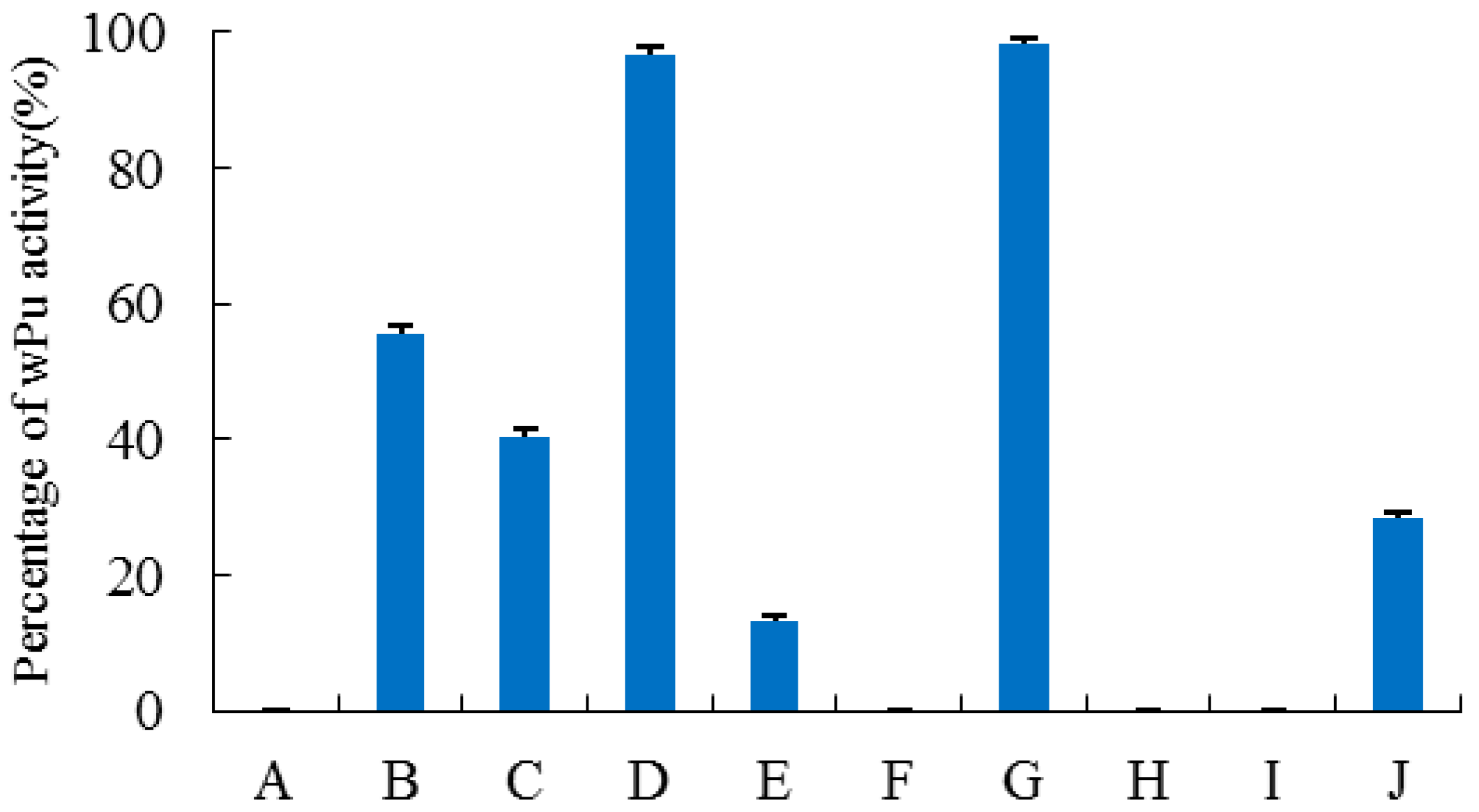
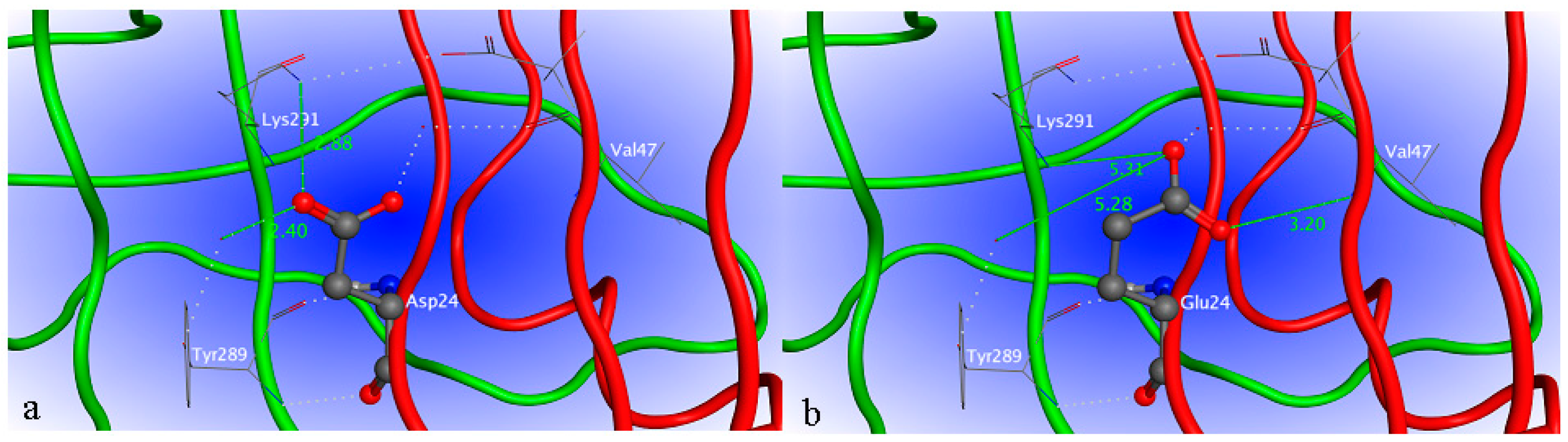
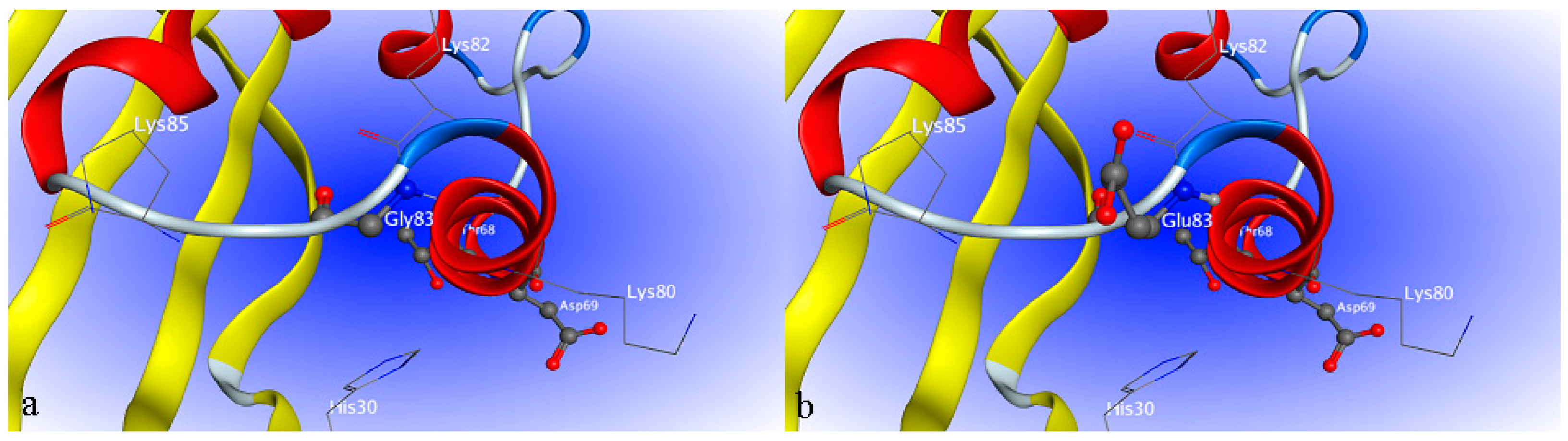
2.4. Site-Directed Mutagenesis
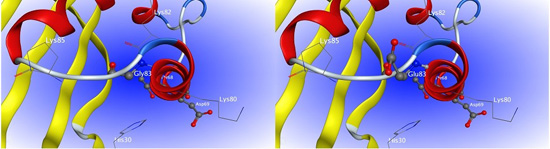
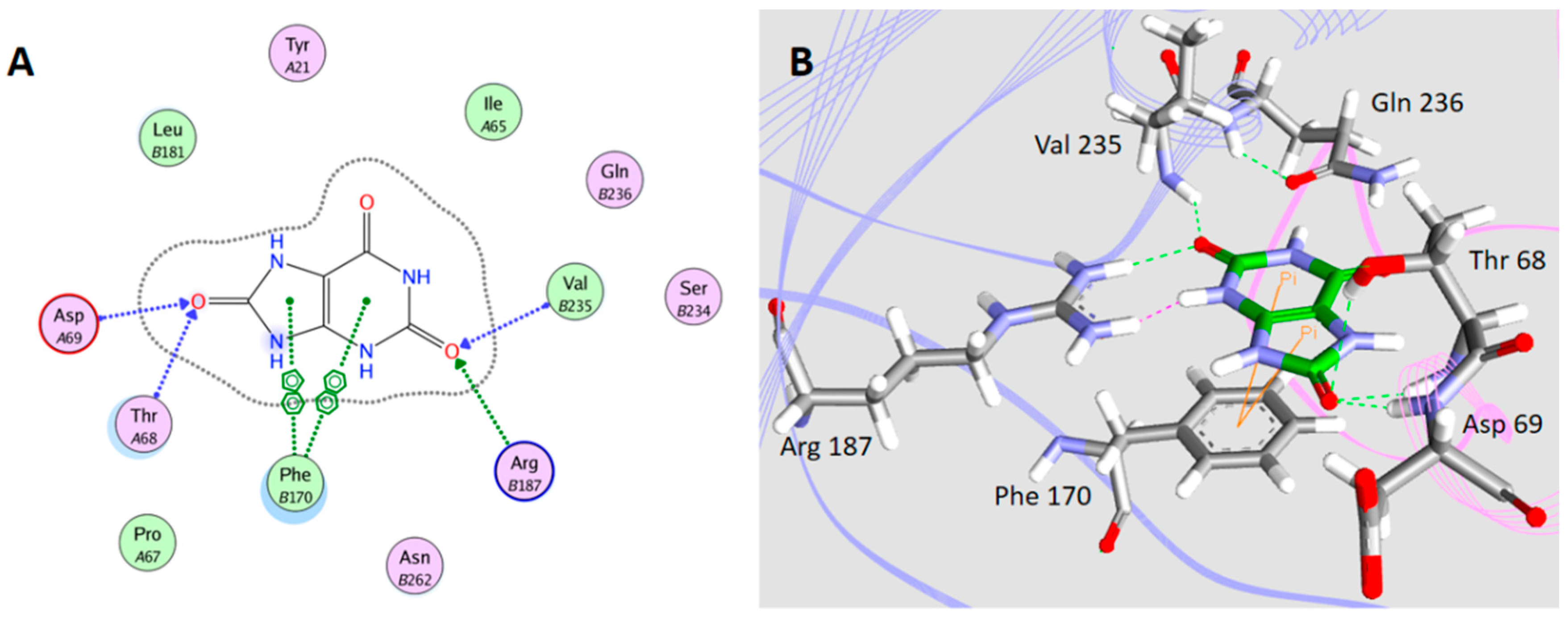
2.5. Interaction between H1-2P3H4P5-6H7-8 (E24D & E83G) and Uric Acid
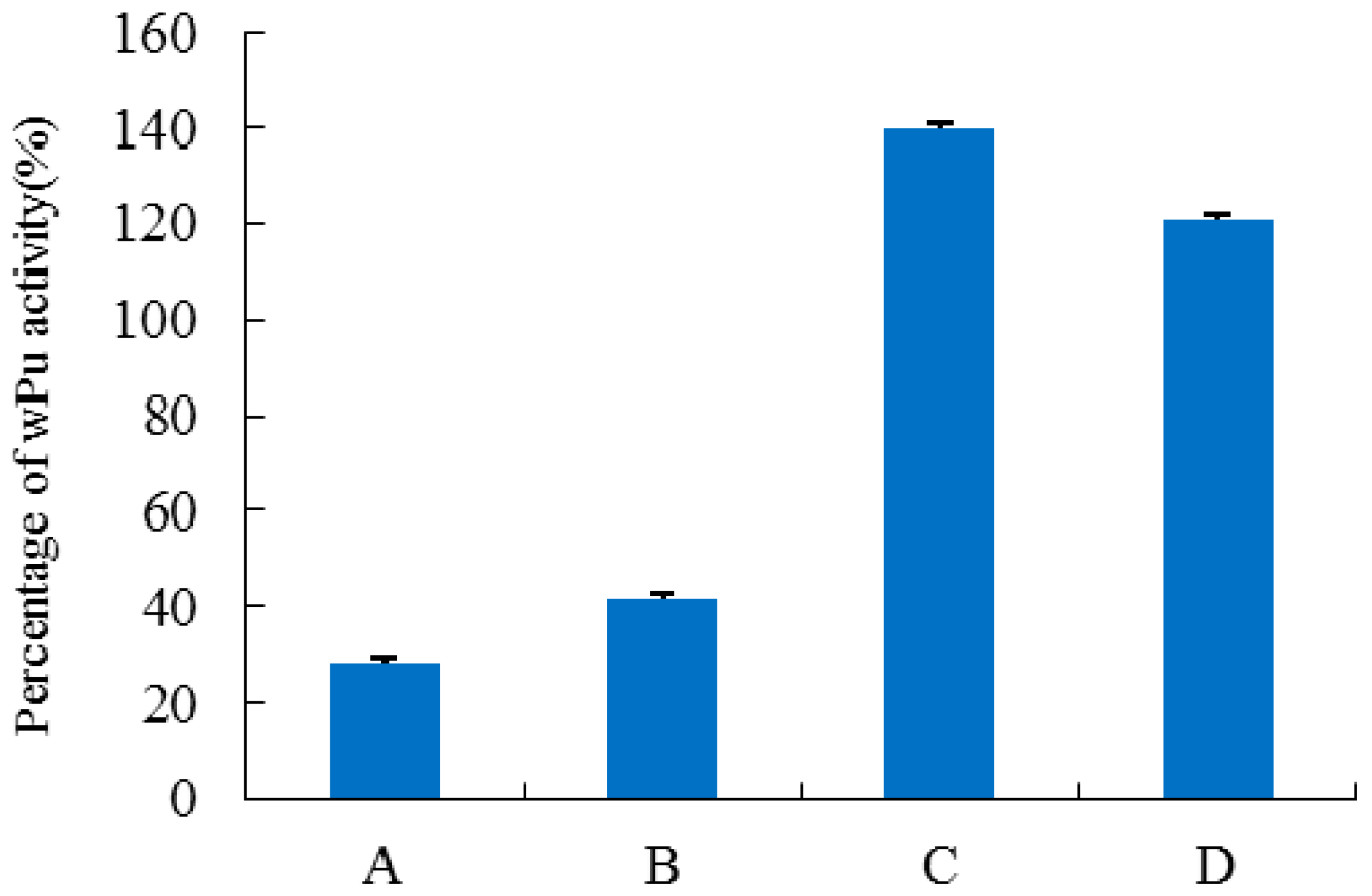
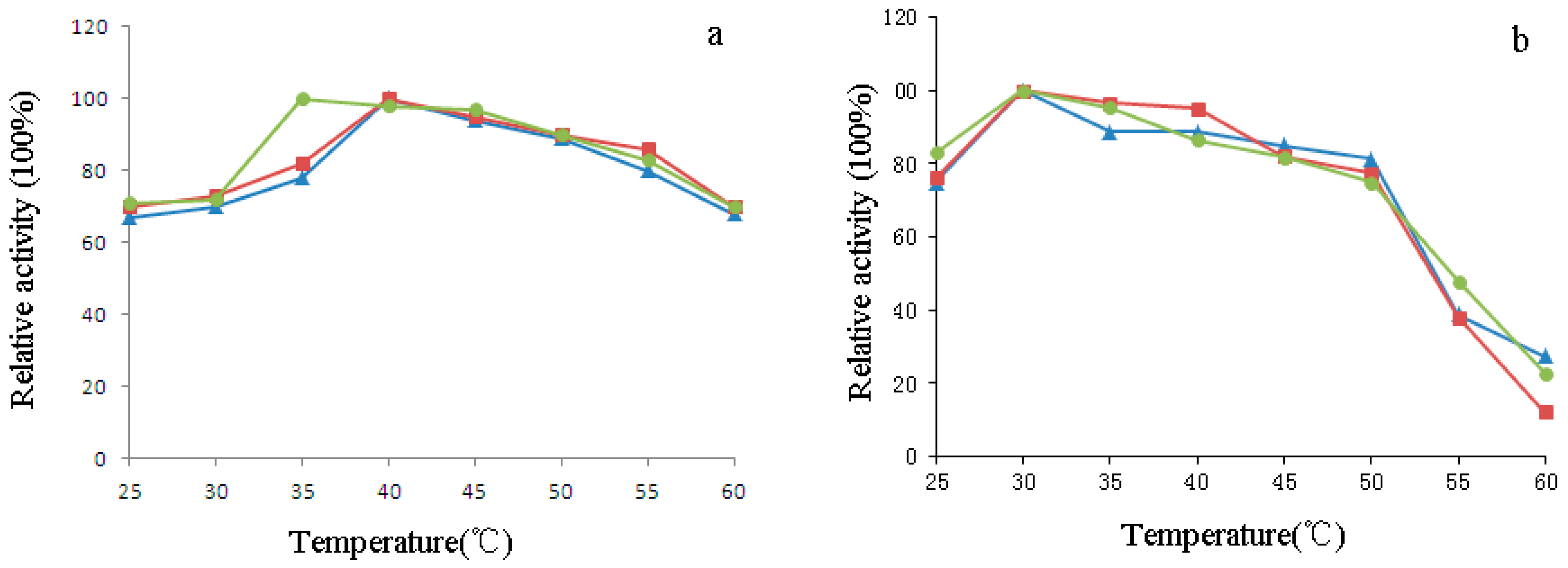
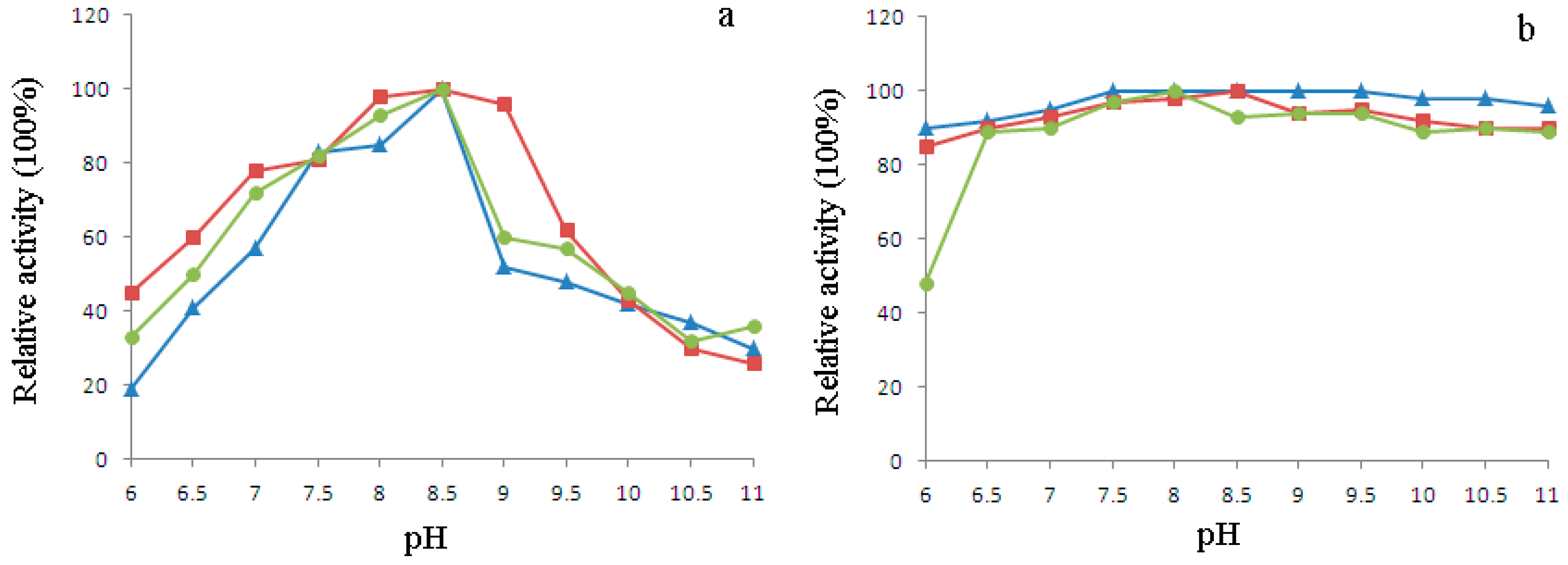
2.6. Characterization of H1-2P3H4P5-6H7-8 (E24D & E83G)
3. Discussion
4. Materials and Methods
4.1. Microorganisms, Vectors, and Materials
4.2. Construction of dHU, wPU, and Porcine-Human Chimeras
4.3. Expression and Purification of Porcine–Human Chimeras (PHC), wPU, and PBC
4.4. Protein Analysis and Enzymatic Assay
4.5. Multiple Protein Sequence Alignment and Homology Modeling
4.6. Site-Directed Mutagenesis of E24D and E83G
4.7. Simulation of Interaction between Uricase H1-2P3H4P5-6H7-8 (E24D & E83G) and Uric Acid
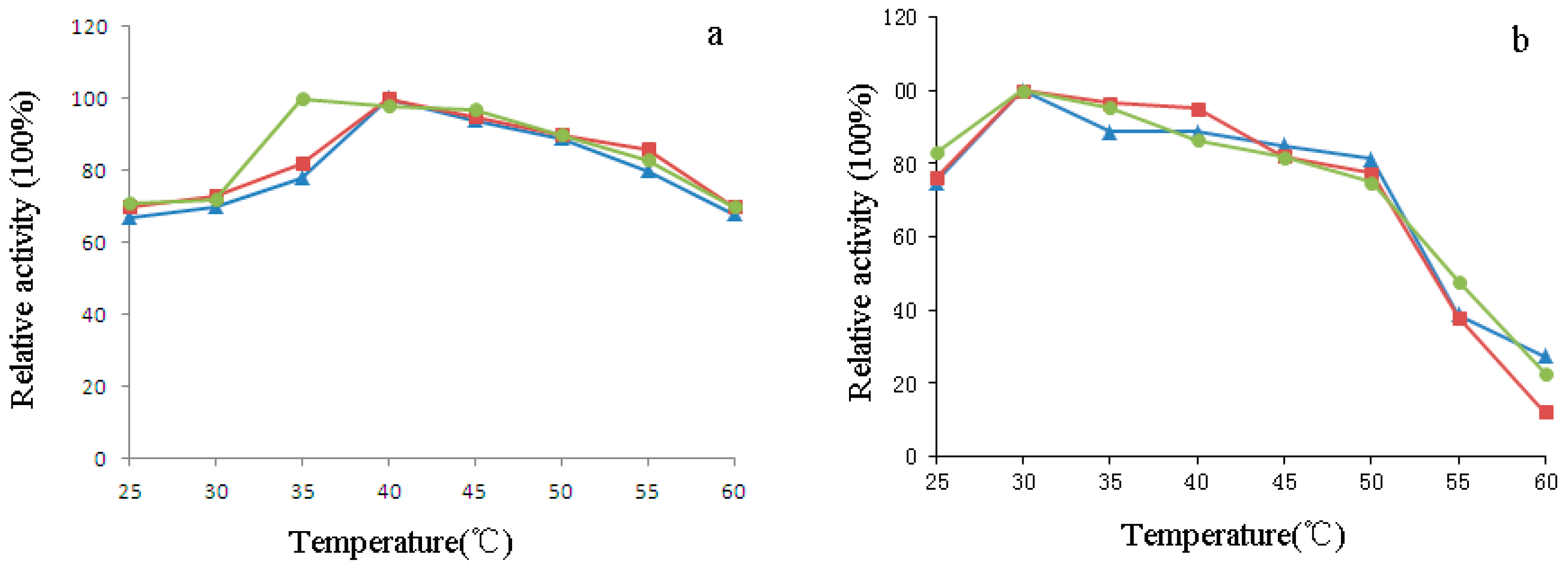
4.8. Effect of pH and Temperature on the Activity of H1-2P3H4P5-6H7-8 (E24D & E83G)
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kahn, K.; Serfozo, P.; Tipton, P.A. Identification of the true product of the urate oxidase reaction. J. Am. Chem. Soc. 1997, 119, 5435–5442. [Google Scholar] [CrossRef]
- Hayashi, S.; Fujiwara, S.; Noguchi, T. Evolution of urate-degrading enzymes in animal peroxisomes. Cell. Biochem. Biophys. 2000, 32, 123–129. [Google Scholar] [CrossRef]
- Ramazzina, I.; Folli, C.; Secchi, A.; Berni, R.; Percudani, R. Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat. Chem. Biol. 2006, 2, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Roch-Ramel, F.; Peters, G. Urinary excretion of uric acid in nonhuman mammalian species. Handb. Exp. Pharmacol. 1978, 51, 211–255. [Google Scholar]
- Wu, X.W.; Muzny, D.M.; Lee, C.C.; Caskey, C.T. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J. Mol. Evol. 1992, 34, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Segal, M.S.; Srinivas, T.; Ejaz, A.; Mu, W.; Roncal, C.; Sánchez-Lozada, L.G.; Gersch, M.; Rodriguez-Iturbe, B.; Kang, D.H.; et al. Essential hypertension, progressive renal disease, and uric acid: A pathogenetic link? J. Am. Soc. Nephrol. 2005, 16, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Del, G.T.; Morris, E.; Cairo, M.S. Tumor lysis syndrome: Pathophysiology, definition, and alternative treatment approaches. Clin. Adv. Hematol. Oncol. 2005, 3, 54–61. [Google Scholar]
- Liebman, S.E.; Taylor, J.G.; Bushinsky, D.A. Uric acid nephrolithiasis. Curr. Rheumatol. Rep. 2007, 9, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Alderman, M.H.; Cohen, H.; Madhavan, S.; Kivlighn, S. Serum uric acid and cardiovascular events in successfully treated hypertensive patients. Hypertension 1999, 34, 144–150. [Google Scholar] [PubMed]
- Verdecchia, P.; Schillaci, G.; Reboldi, G.; Santeusanio, F.; Porcellati, C.; Brunetti, P. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension. The PIUMA study. Hypertension 2000, 36, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Sarawate, C.A.; Brewer, K.K.; Wenya, Y.; Patel, P.A.; Schumacher, H.R.; Saag, K.G.; Bakst, A.W. Gout medication treatment patterns and adherence to standards of care from a managed care perspective. Mayo Clin. Proc. 2006, 81, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Annemans, L.; Spaepen, E.; Gaskin, M.; Bonnemaire, M.; Malier, V.; Gilbert, T.; Nuki, G. Gout in the UK and Germany: Prevalence, comorbidities and management in general practice 2000–2005. Ann. Rheum. Dis. 2008, 67, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.Z.; Wallace, S.L. The allopurinol hypersensitivity syndrome. Unnecessary morbidity and mortality. Arthritis Rheum. 1986, 29, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Arellano, F.; Sacristán, J.A. Alloputinol hypersensitivity syndrome: A review. Ann. Pharmacother. 1993, 27, 337–343. [Google Scholar] [PubMed]
- Goldman, S.C.; Holcenberg, J.S.; Finkelestein, J.Z.; Hutchinson, B.; Kreissman, S.; Johnson, F.L.; Tou, C.; Harvey, E.; Morris, E.; Cairo, M.S. A randomized comparison between rasburicase and allopurinol in children with lymphoma or leukemia at high risk for tumor lysis. Blood 2001, 97, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Jeha, S.C. Recombinant urate oxidase (rasburicase) in the prophylaxis and treatment of tumor lysis syndrome. Contrib. Nephrol. 2005, 147, 69–79. [Google Scholar] [PubMed]
- Cammalleri, L.; Malaguarnera, M. Rasburicase represents a new tool for hyperuricemia in tumor lysis syndrome and in gout. Int. J. Med. Sci. 2007, 4, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, Y.; Mechinaud, F.; Brethon, B.; Mialou, V.; Auvrignon, A.; Nelken, B.; Notz-Carrère, A.; Plantaz, D.; Patte, C.; Urbieta, M.; et al. SFCE recommendations for the management of tumor lysis syndrome (TLS) with rasburicase: An observational survey. J. Pediatr. Hematol. Oncol. 2008, 30, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.M.; Wortmann, R.L. Gout therapeutics: New drugs for an old disease. Lancet 2010, 377, 165–177. [Google Scholar] [CrossRef]
- Sundy, J.S.; Becker, M.A.; Baraf, H.S.; Barkhuizen, A.; Moreland, L.W.; Huang, W.; Waltrip, R.W.; Maroli, A.N.; Horowitz, Z.; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol‑conjugated uricase) in patients with treatment‑failure gout: Results of a phase II randomized study. Arthritis Rheum. 2008, 58, 2882–2891. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, N.; Yasothan, U.; Kirkpatrick, P. Pegloticase. Nat. Rev. Drug. Discov. 2011, 10, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Zhang, C.; Ma, X.; Mei, X.; Hu, C. Humanized Recombinant Uricase and Mutants Thereof. U.S. Patent US8586535 B2, 19 November 2013. [Google Scholar]
- Rong, J.; Kuang, H.; Sun, Z. Preparation and Application of PEG Recombinant Pig-Human Urate Oxidase Fusion Protein. CN Patent CN102260653 B, 3 April 2013. [Google Scholar]
- Yang, X.; Yuan, Y.; Zhan, C.G.; Liao, F. Uricase as therapeutic agent to treat refractory gout: Current states and future directions. Drug Dev. Res. 2012, 73, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Yeldandi, A.V.; Patel, Y.D.; Liao, M.; Kao, F.T.; Rao, M.S.; Reddy, J.K.; Le Beau, M.M. Localization of the human urate oxidase gene (UOX) to 1p22. Cytogenet. Cell Genet. 1992, 61, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yuan, C.; Hwang, H.T.; Zhao, X.; Ramkrishna, D.; Liu, D.; Varma, A. Engineering surface hydrophobicity improves activity of Bacillus thermocatenulatuslipase 2 enzyme. Biotechnol. J. 2015, 10, 1762–1769. [Google Scholar]
- Stephens, D.E.; Khan, F.I.; Singh, P.; Bisetty, K.; Singh, S.; Permaul, K. Creation of thermostable and alkaline stable xylanase variants by DNA shuffling. J. Biotechnol. 2014, 187, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Heng, C.; Braker, J.D.; Chan, V.J.; Lee, C.C.; Jordan, D.B.; Yuan, L.; Wagschal, K. Directed evolution of GH43 β-xylosidase XylBH43 thermal stability and L186 saturation mutagenesis. J. Ind. Microbiol. Biotechnol. 2014, 41, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, N.; Wang, T.; Xie, G.; Zhang, Z.; Li, H.; Yuan, J.; Sun, Z.; Chen, J. DNA shuffling of uricase gene leads to a more “human like” chimeric uricase with increased uricolytic activity. Int. J. Biol. Macromol. 2016, 82, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Satta, Y.; Takenaka, O.; Takahata, N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol. Biol. Evol. 2002, 19, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S. Ancient insights into uric acid metabolism in primates. Proc. Natl. Acad. Sci. USA 2014, 1111, 3657–3658. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J. Purifying Proteins for Proteomics: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2004. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Conley, T.G.; Priest, D.G. Thermodynamics and stoicheiometry of the binding of substrate analogues to uricase. Biochem. J. 1980, 187, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Kalckar, H.M. Differential spectrophotometry of purine compounds by means of specific enzymes; determination of hydroxypurine compounds. J. Biol. Chem. 1947, 167, 429–443. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uricase | Km (Μm) | kcat/(s) | kcat/Km/(s/μM) | Specific Activity | Homology with dHU (%) |
|---|---|---|---|---|---|
| dHU | ND | ND | ND | ND | 100 |
| wPU | 10.12 ± 0.10 | 23.54 ± 0.03 | 2.33 | 4.32 ± 0.07 | 87.5 |
| H1-2P3H4P5-6H7-8 (E24D & E83G) | 5.37 ± 0.09 | 30.95 ± 0.01 | 5.76 | 6.03 ± 0.06 | 91.45 |
| PBC | 7.51 ± 0.04 | 27.24 ± 0.03 | 3.62 | 5.19 ± 0.08 | 89.5 |
| Strain/Plasmid | Description | Source/Reference |
|---|---|---|
| Strain | ||
| E.coli BL 21 StarTM (DE3) | Cloning host | Invitrogen |
| Plasmid | ||
| pET-22b(+) | T7 promoter, AmpR | EMD Biosciences |
| pET-22b(+)_H1-2 | coding for H1-2P3-8 | This study |
| pET-22b(+)_H3 | coding for P1-2H3P4-8 | This study |
| pET-22b(+)_H4 | coding for P1-3H4P5-8 | This study |
| pET-22b(+)_H5 | coding for P1-4H5P6-8 | This study |
| pET-22b(+)_H6 | coding for P1-5H6P7-8 | This study |
| pET-22b(+)_H7-8 | coding for P1-6H7-8 | This study |
| pET-22b(+)_P6 | coding for H1-5P6H7-8 | This study |
| pET-22b(+)_P5-6 | coding for H1-4P5-6H7-8 | This study |
| pET-22b(+)_P3-5-6 | coding for H1-2P3H4P5-6H7-8 | This study |
| pET-22b(+)_24 | coding for H1-2P3H4P5-6H7-8(E24D) | This study |
| pET-22b(+)_24–83 | coding for H1-2P3H4P5-6H7-8(E24D & E83G) | This study |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, G.; Yang, W.; Chen, J.; Li, M.; Jiang, N.; Zhao, B.; Chen, S.; Wang, M.; Chen, J. Development of Therapeutic Chimeric Uricase by Exon Replacement/Restoration and Site-Directed Mutagenesis. Int. J. Mol. Sci. 2016, 17, 764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050764
Xie G, Yang W, Chen J, Li M, Jiang N, Zhao B, Chen S, Wang M, Chen J. Development of Therapeutic Chimeric Uricase by Exon Replacement/Restoration and Site-Directed Mutagenesis. International Journal of Molecular Sciences. 2016; 17(5):764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050764
Chicago/Turabian StyleXie, Guangrong, Weizhen Yang, Jing Chen, Miaomiao Li, Nan Jiang, Baixue Zhao, Si Chen, Min Wang, and Jianhua Chen. 2016. "Development of Therapeutic Chimeric Uricase by Exon Replacement/Restoration and Site-Directed Mutagenesis" International Journal of Molecular Sciences 17, no. 5: 764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050764