
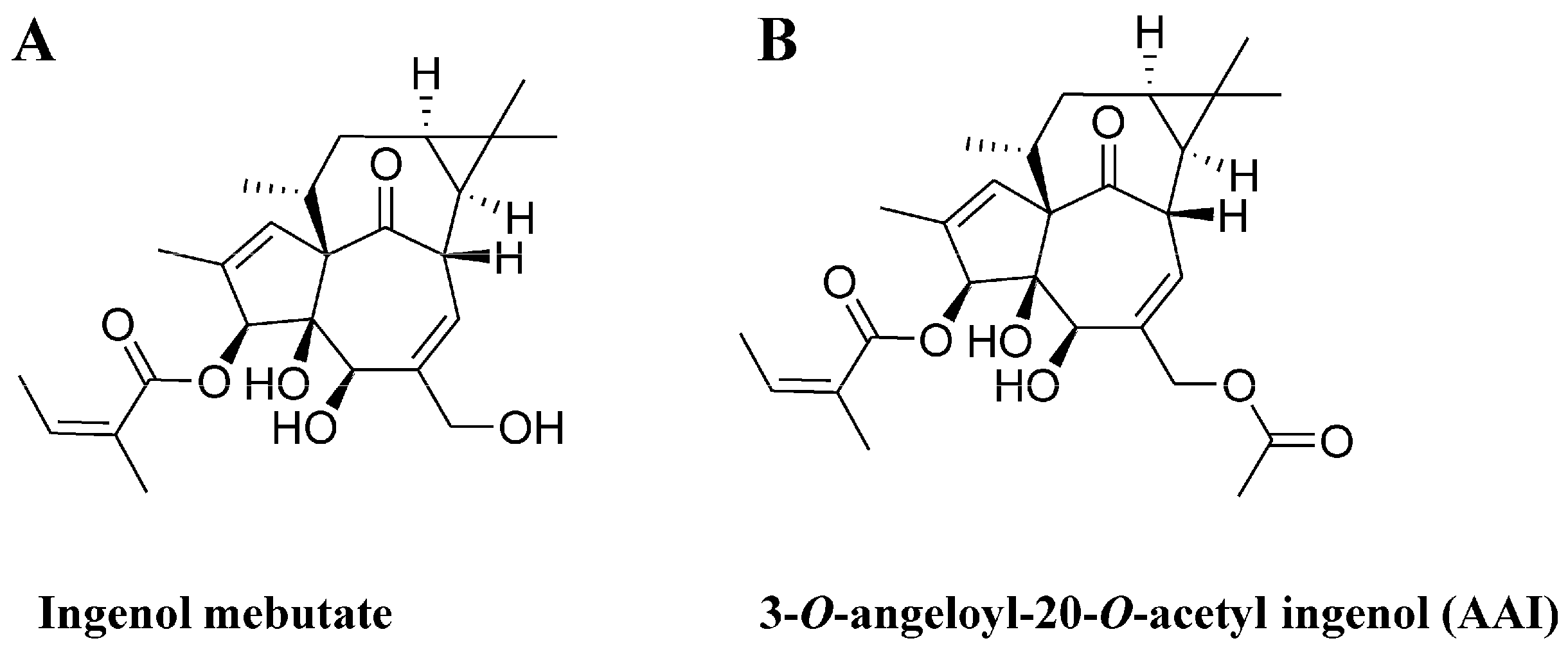
Synthesis and Cytotoxicity against K562 Cells of 3-O-Angeloyl-20-O-acetyl Ingenol, a Derivative of Ingenol Mebutate

 ,
,
Abstract
:
1. Introduction
2. Results
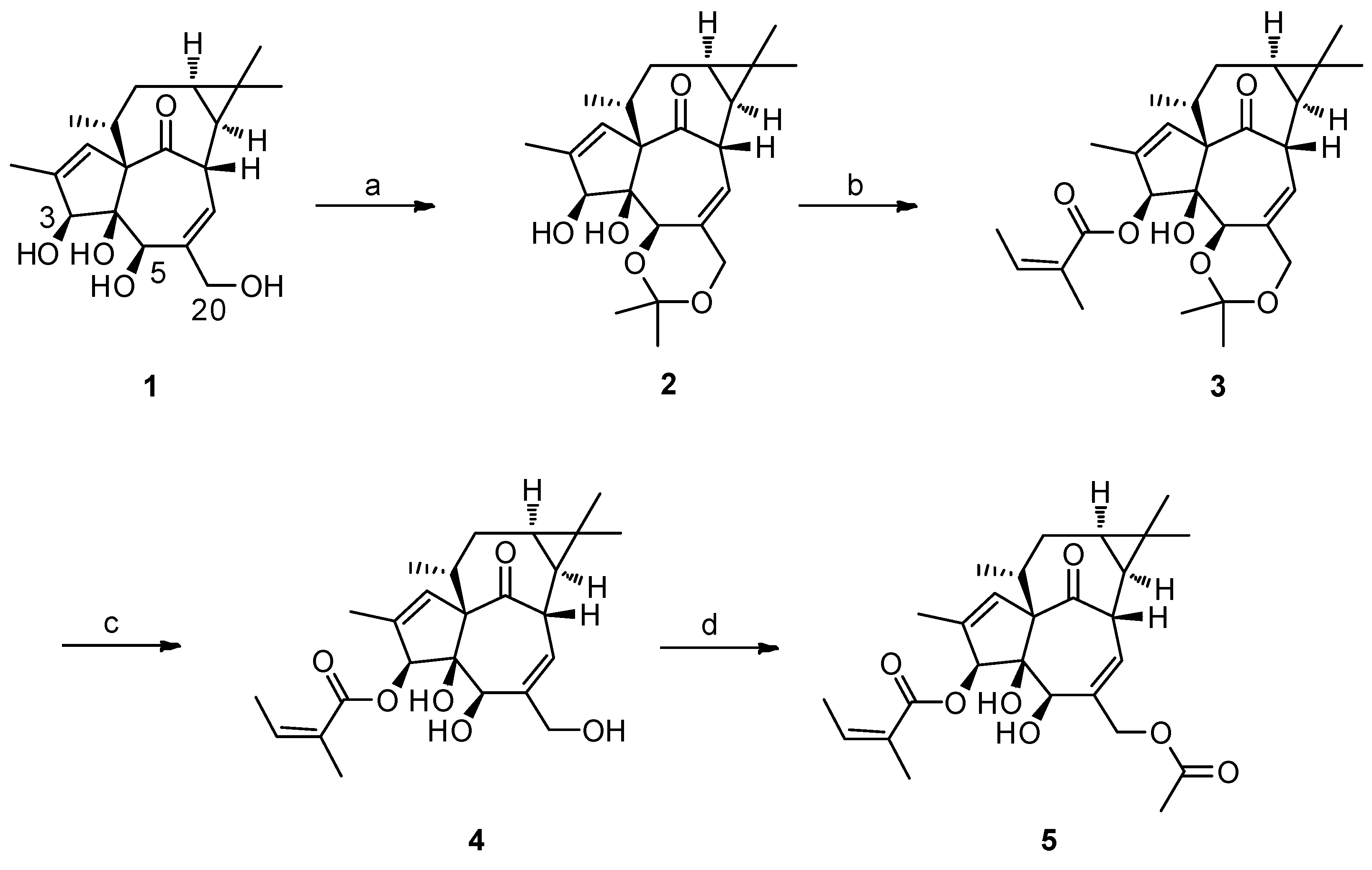
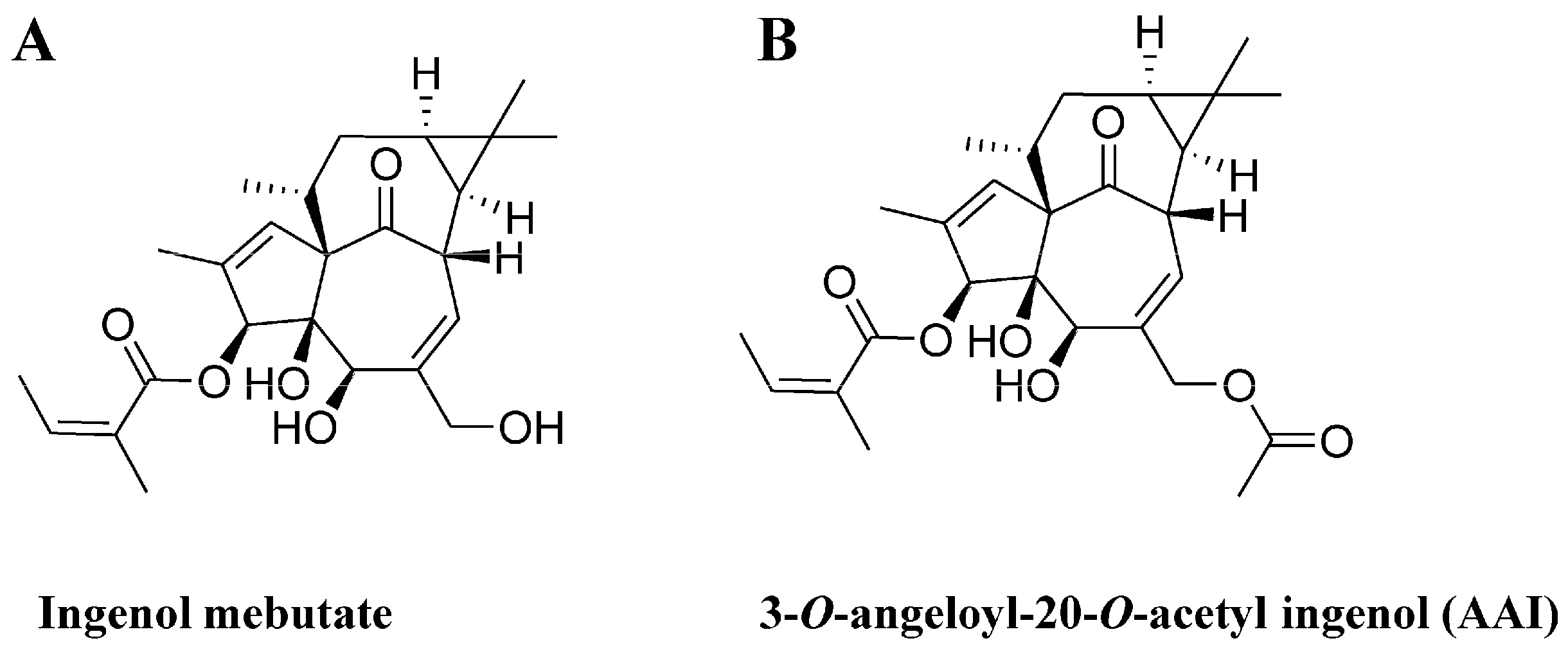
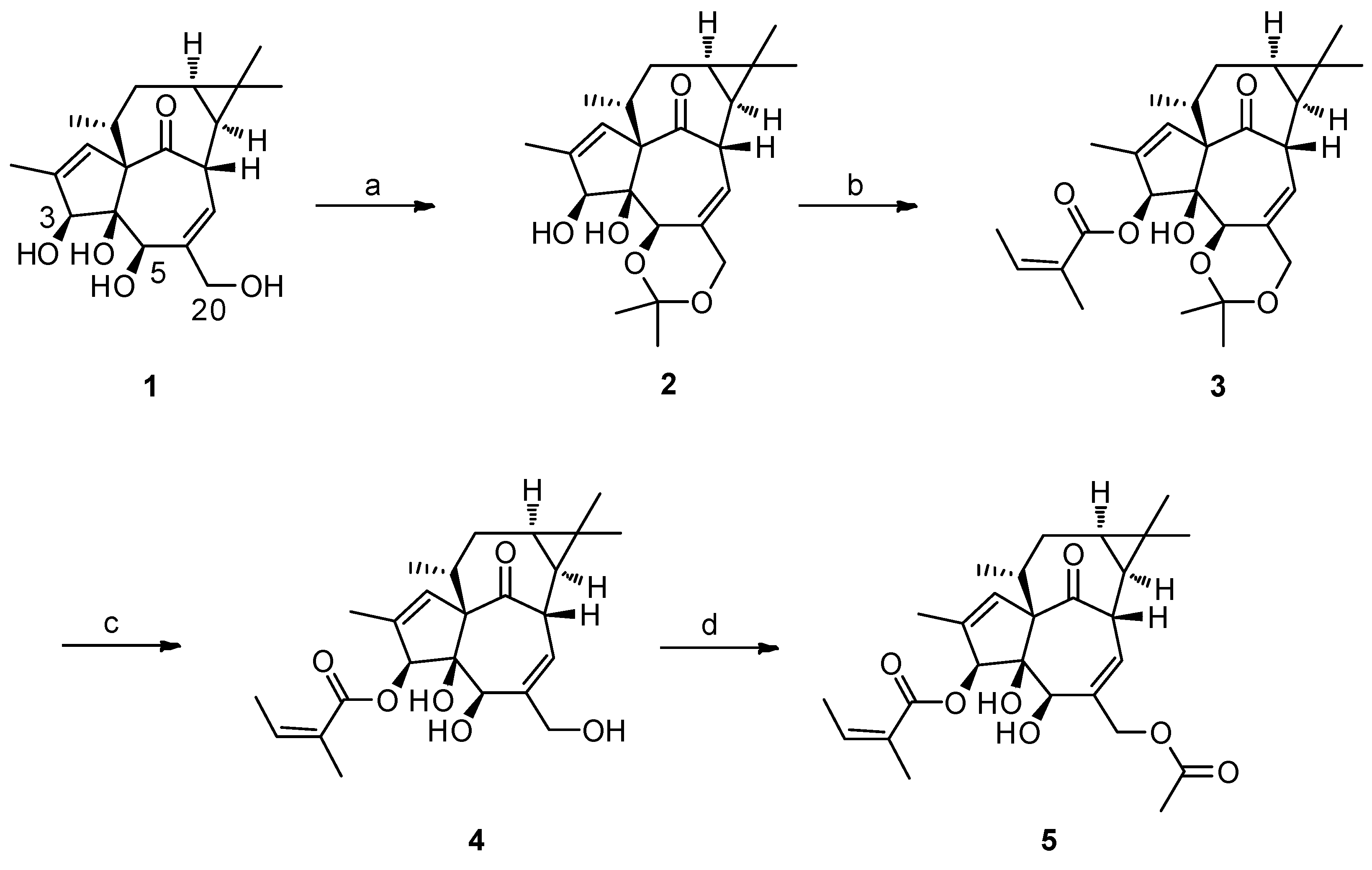
2.1. The Synthesis of 3-O-Angeloyl-20-O-acetyl ingenol (AAI)
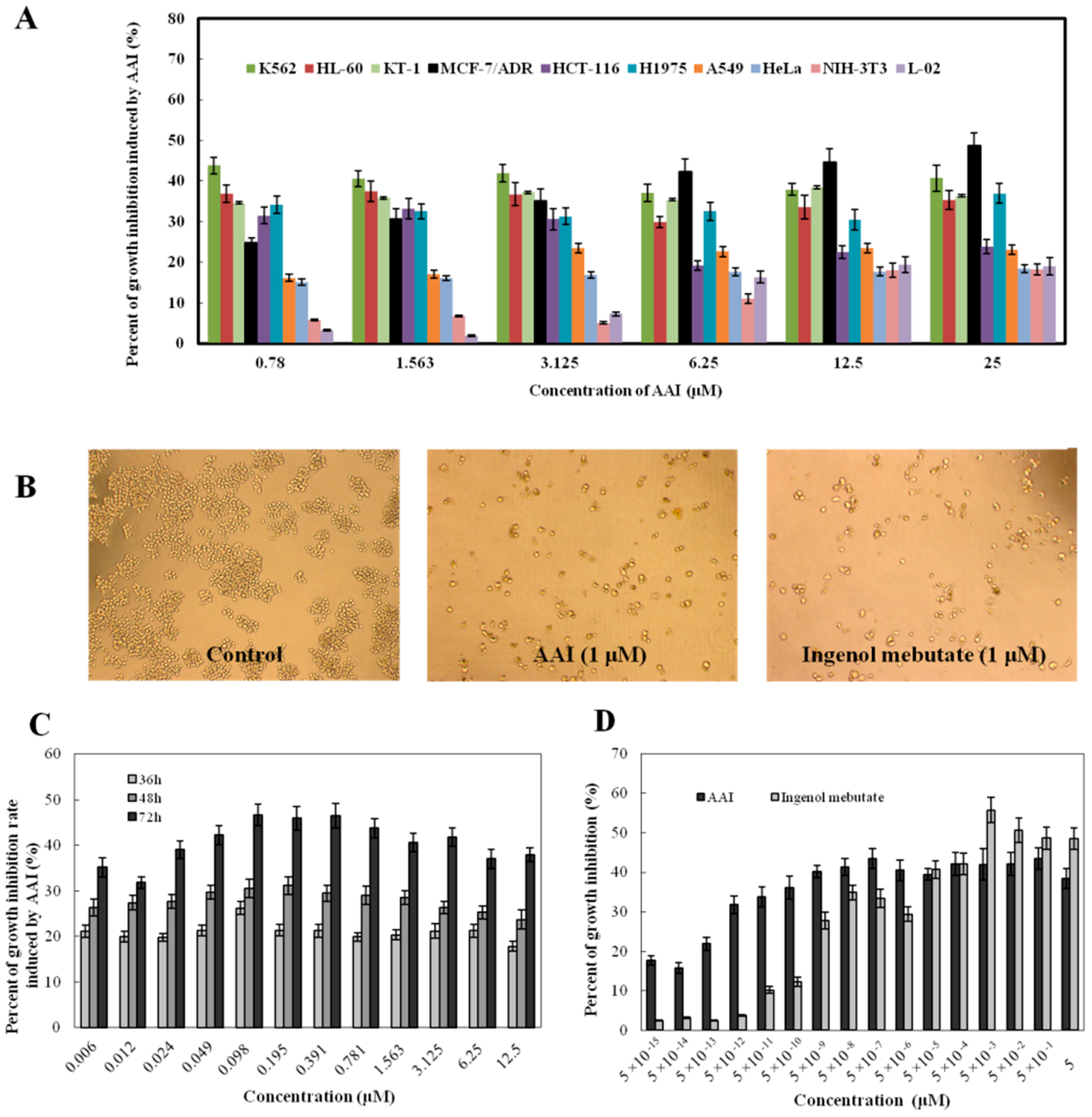
2.2. AAI Inhibits Cell Proliferation
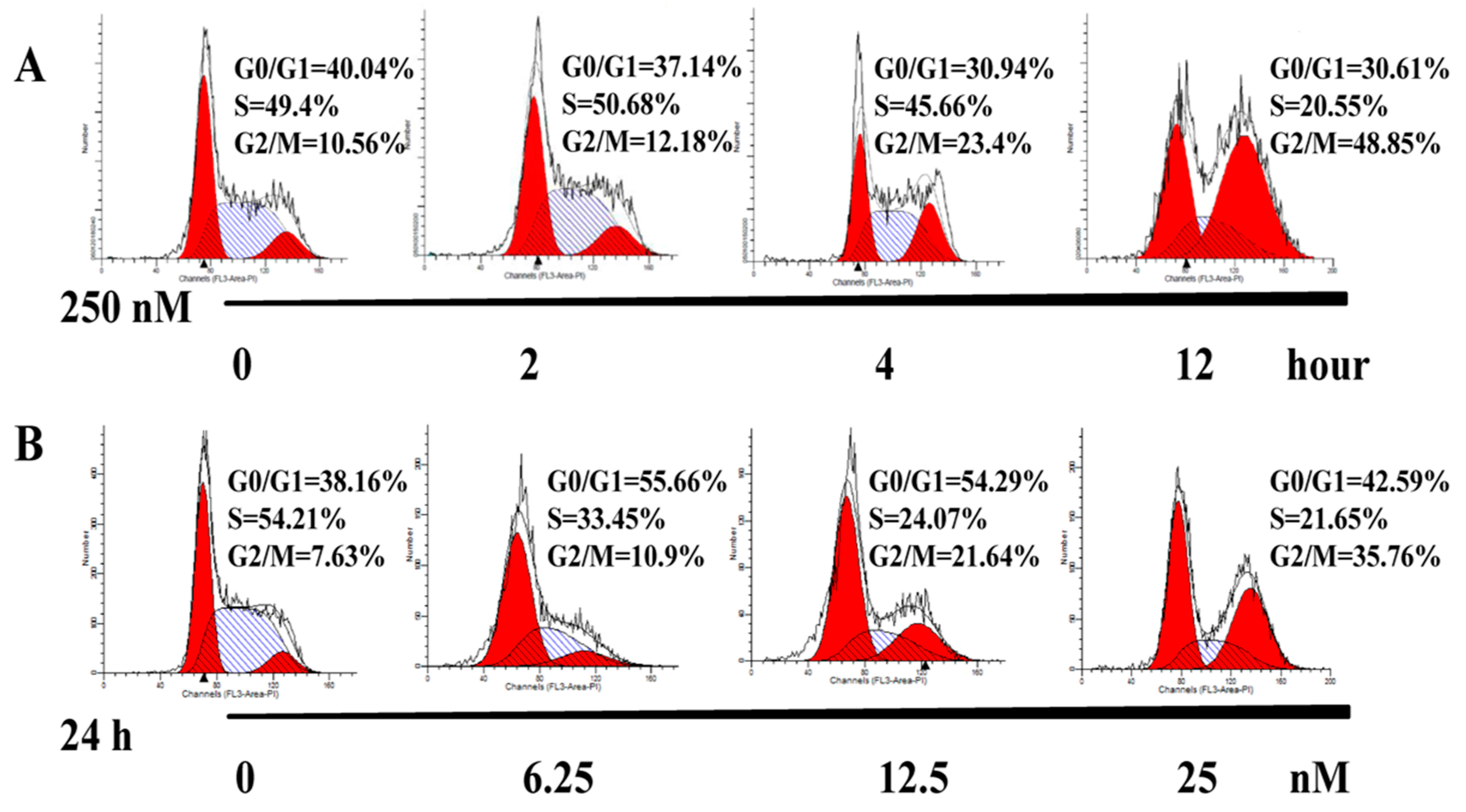
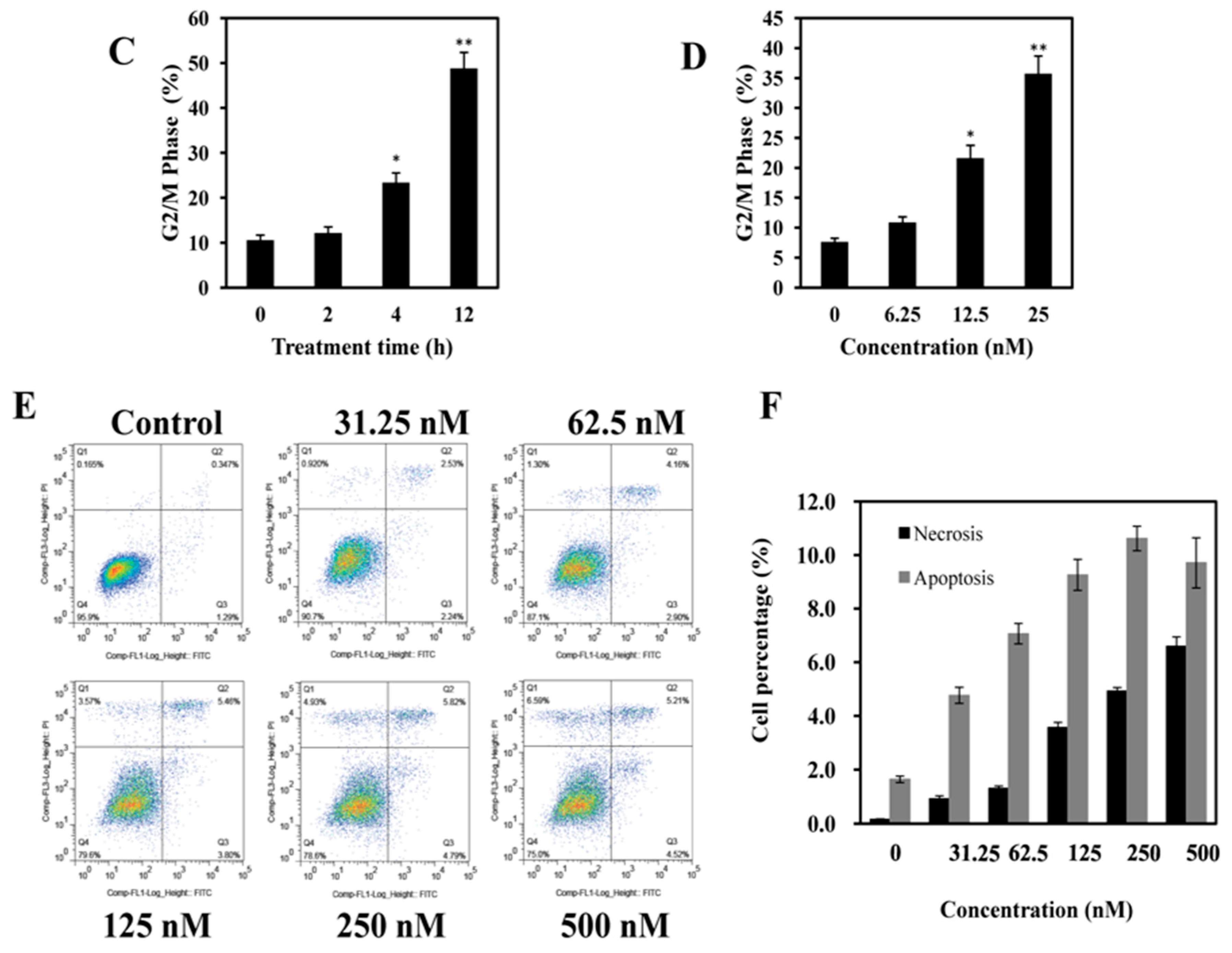
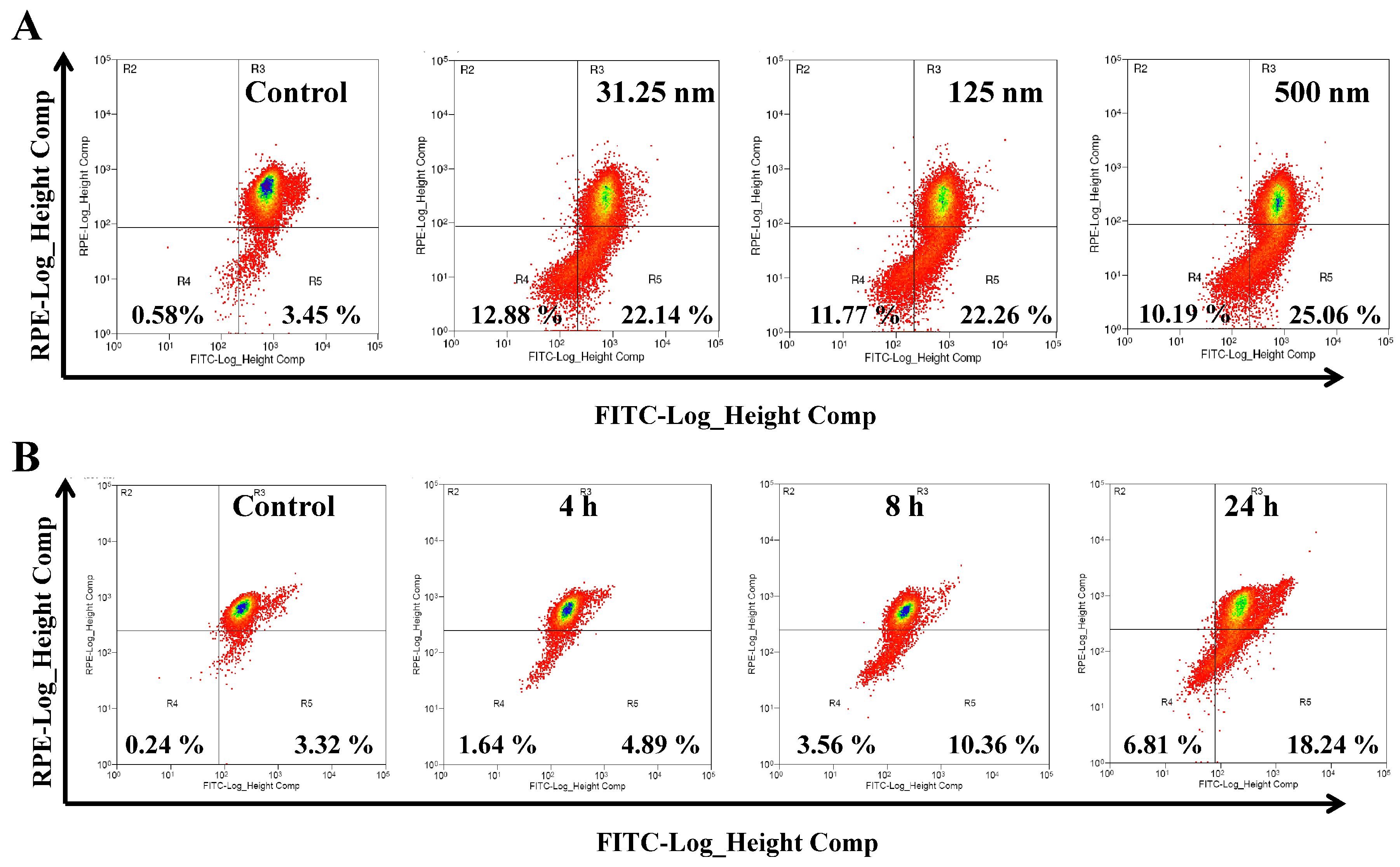
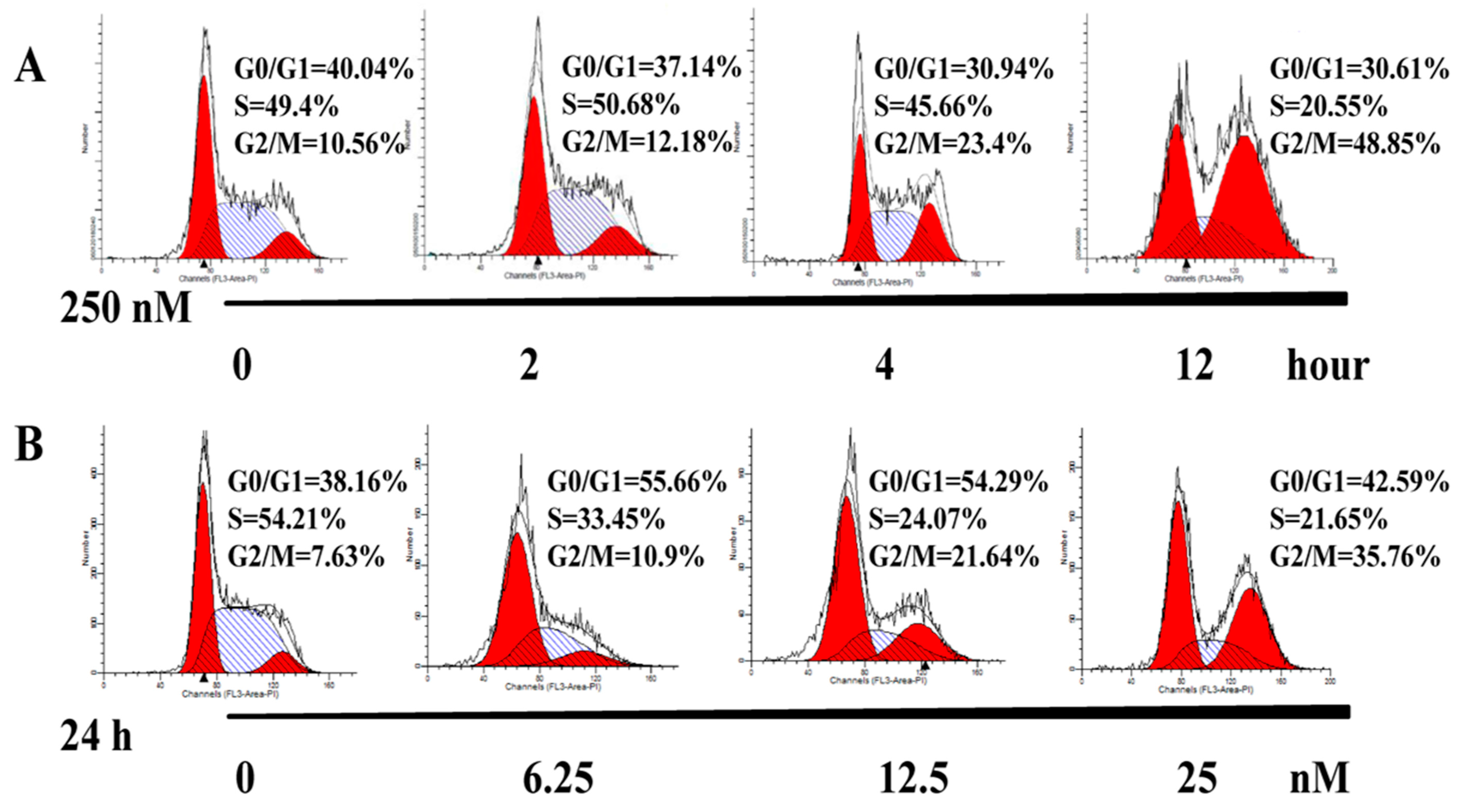
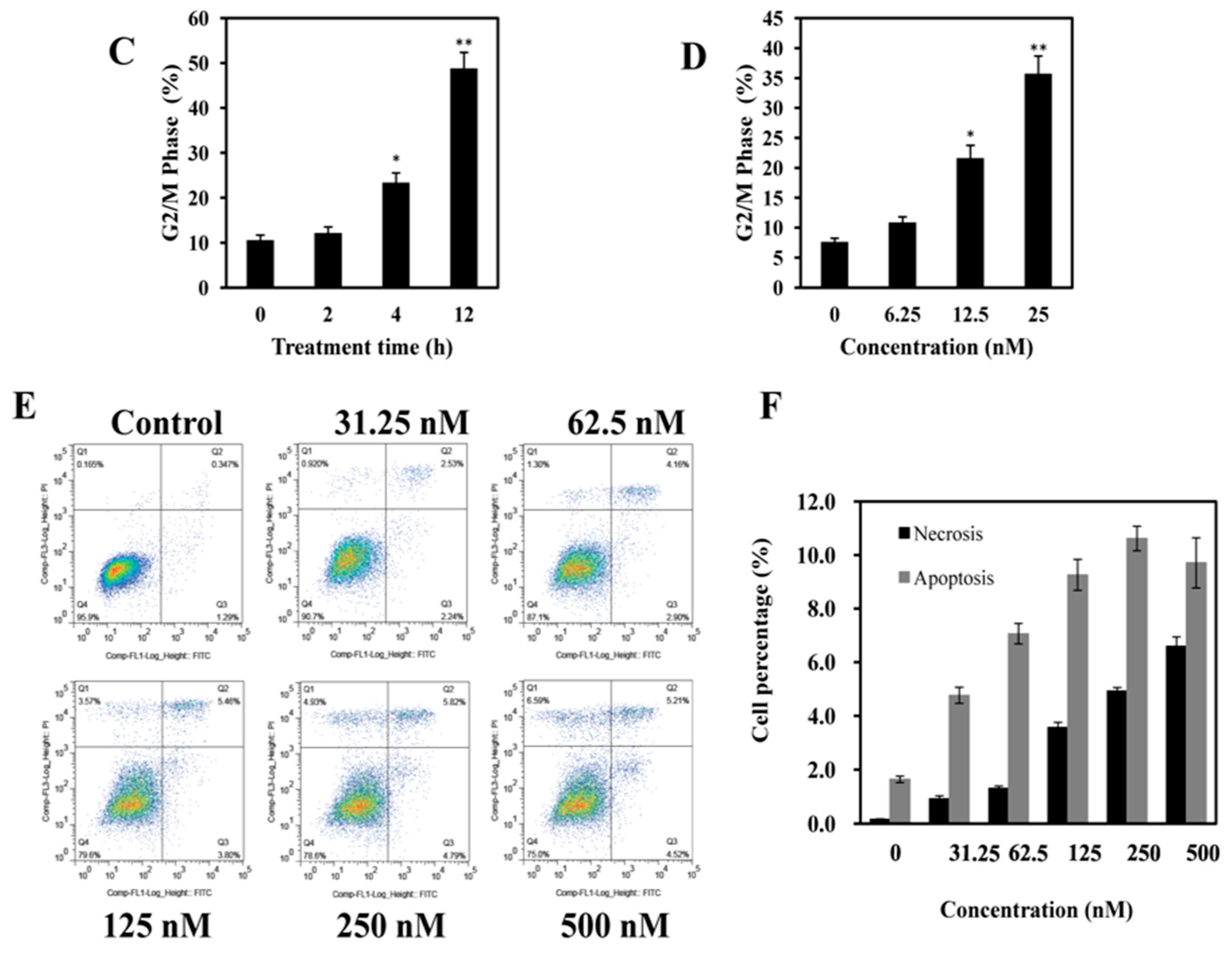
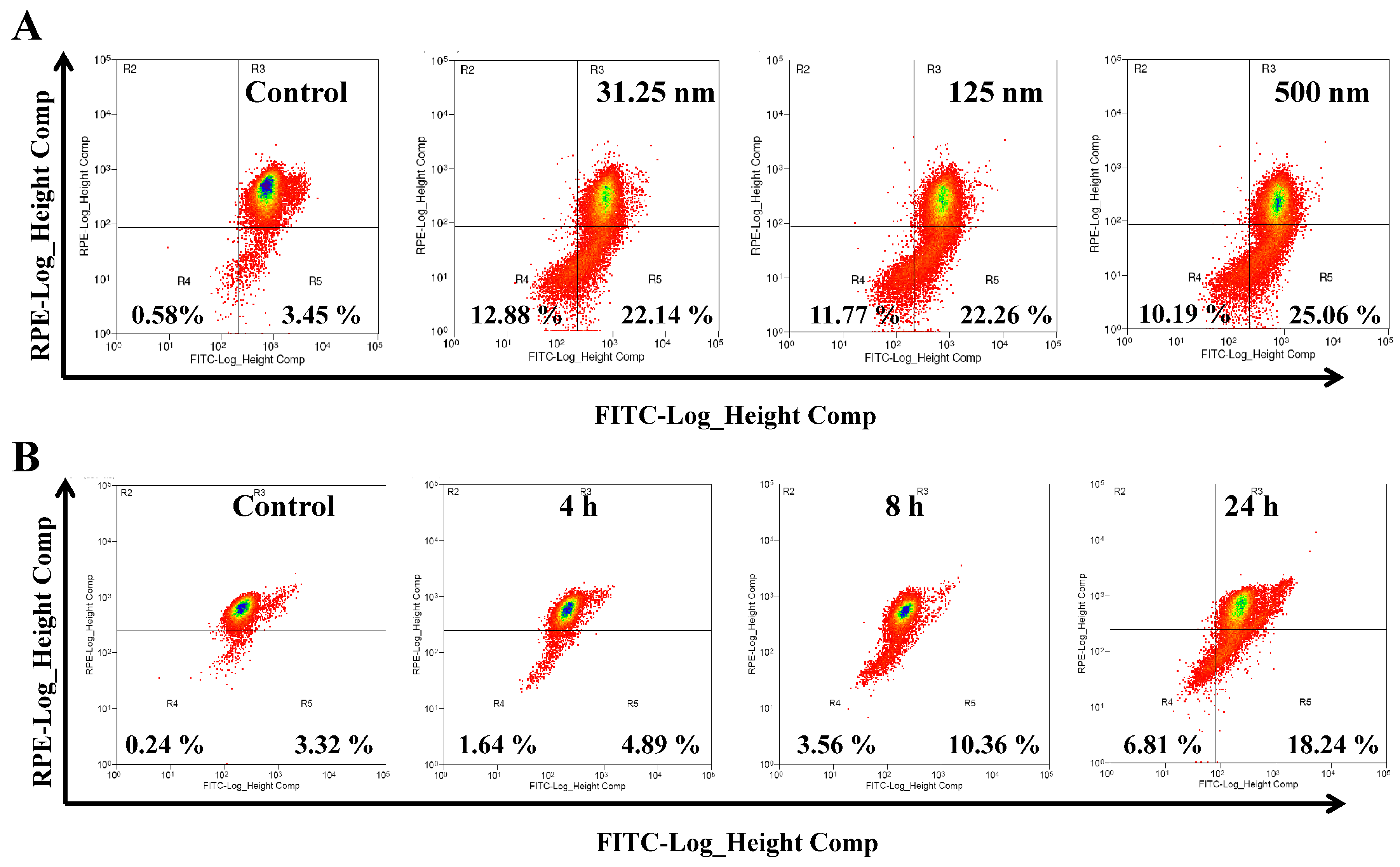
2.3. AAI Induces G2/M Phase Arrest, Apoptosis, and Necrosis in K562 Cells
2.4. AAI Disturbs Mitochondrial Membrane Potential (MMP) in K562 Cells
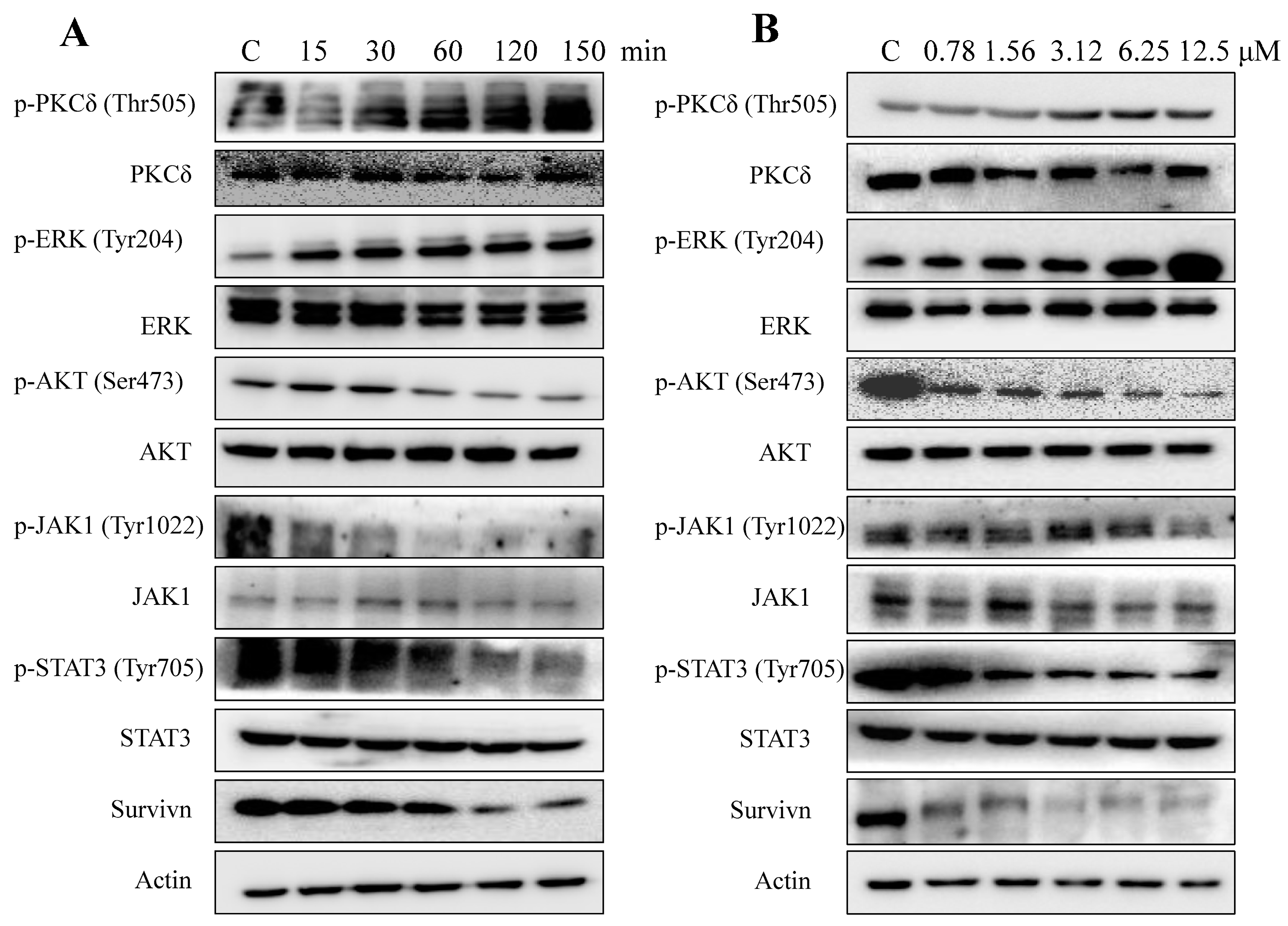
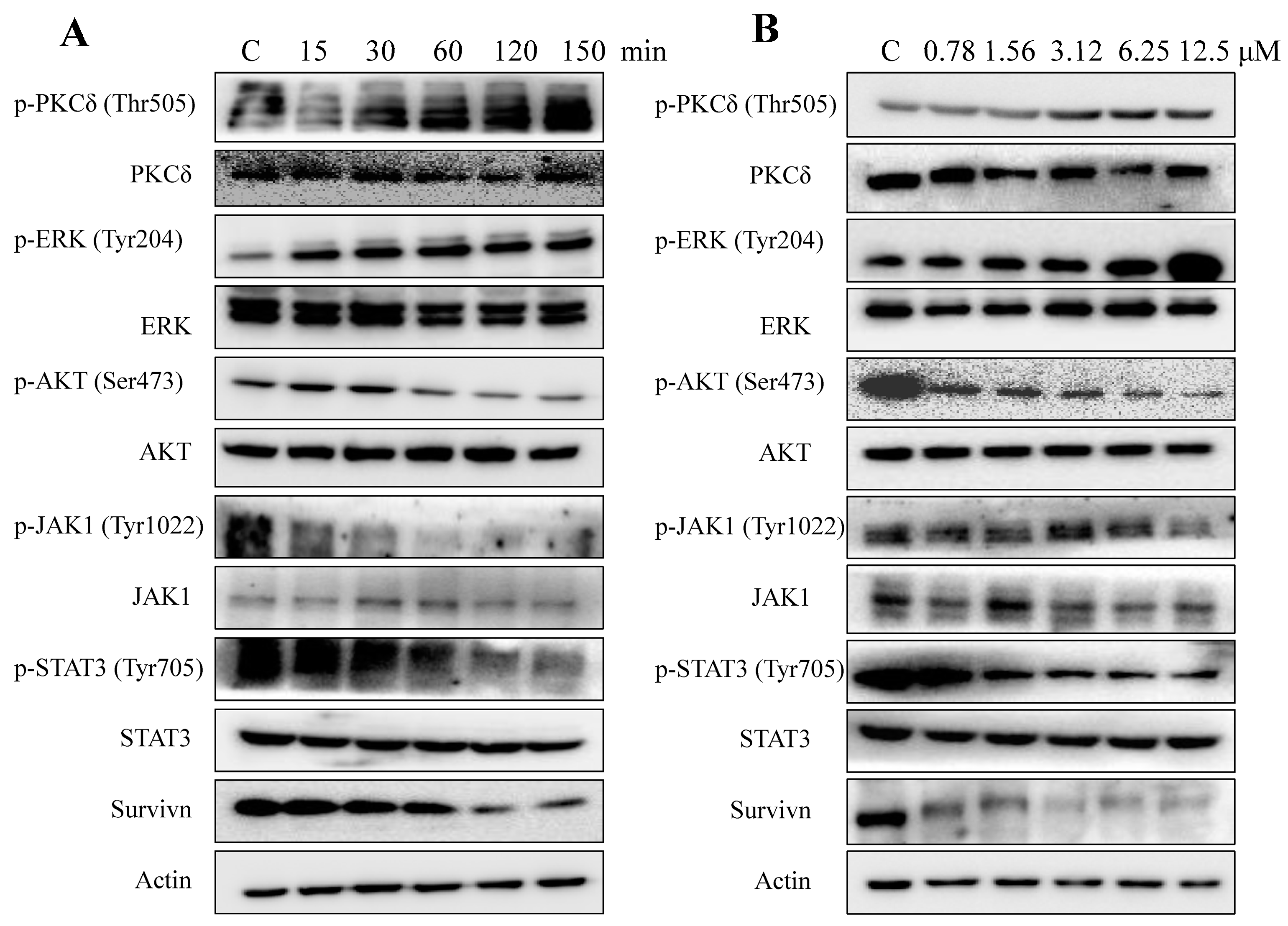
2.5. AAI Modulates Multiple Key Signaling Pathways in K562 Cells
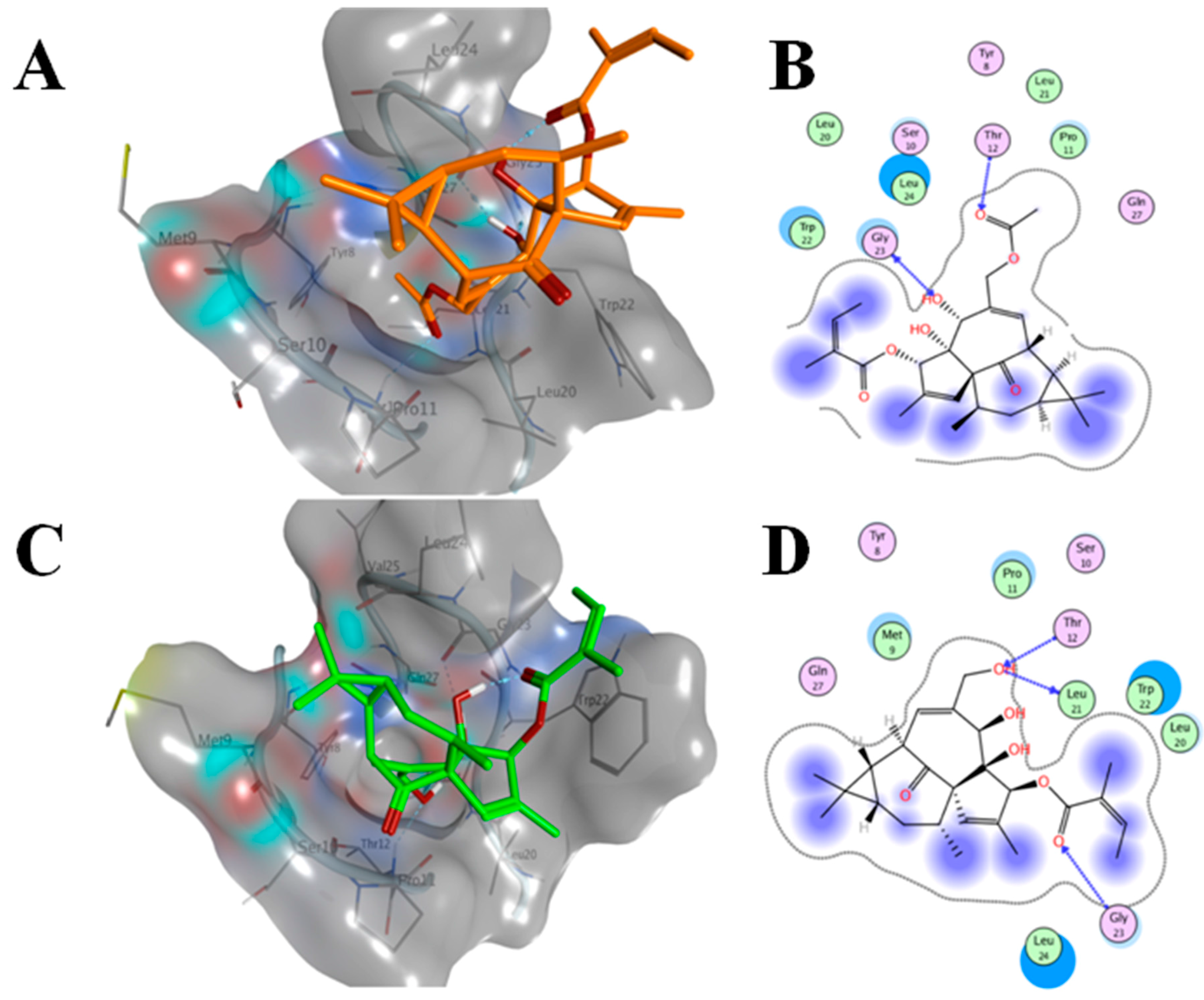
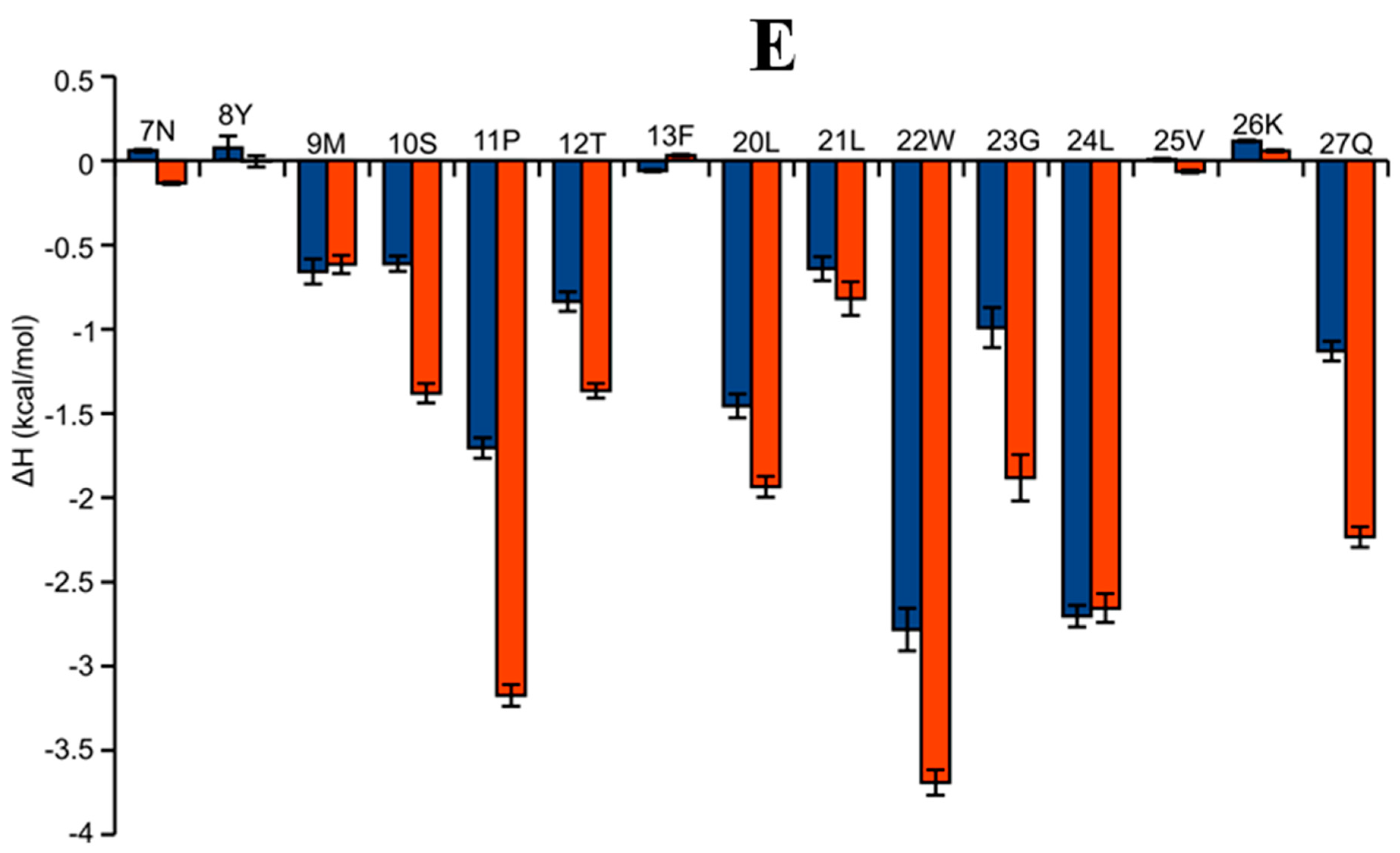
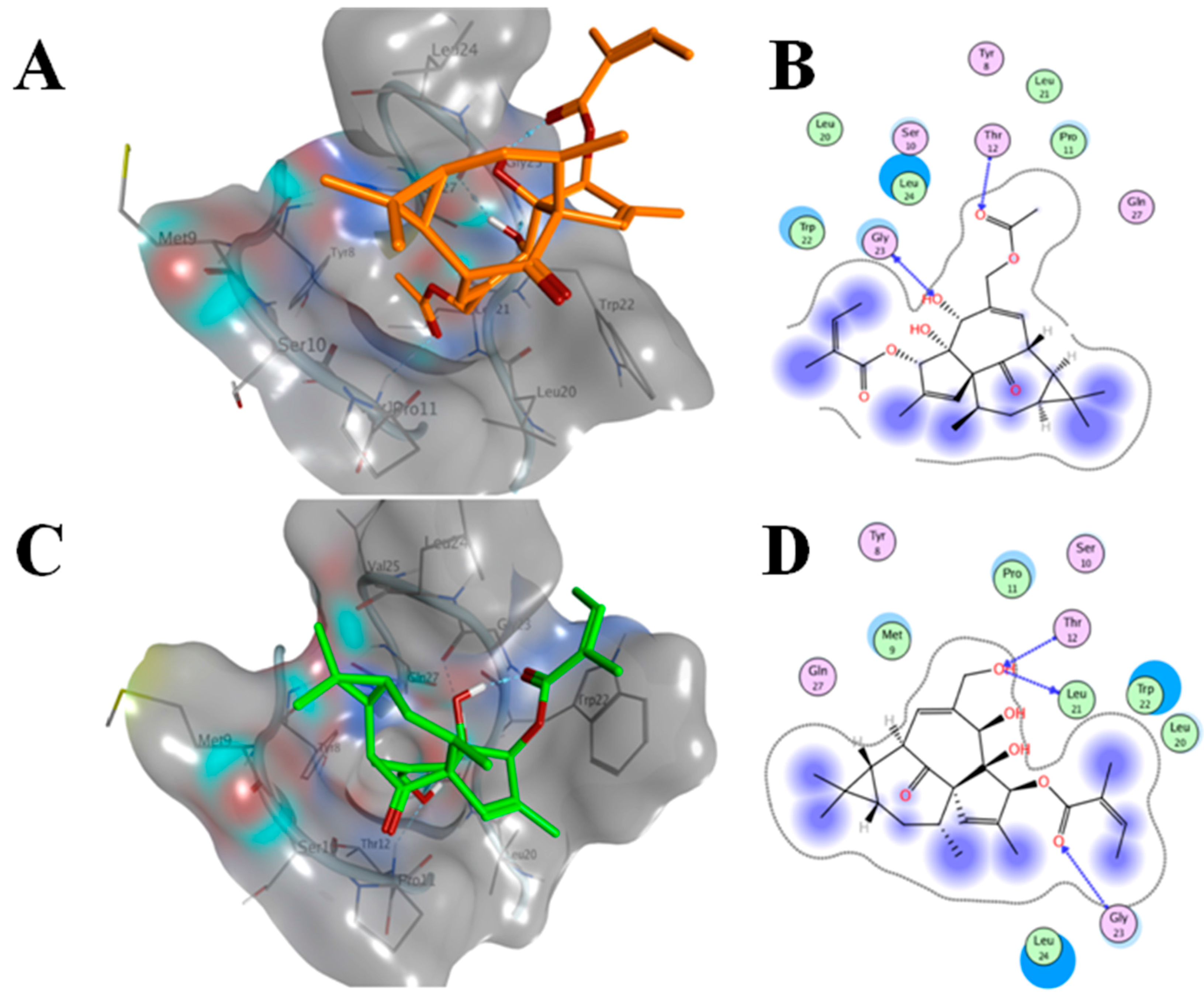
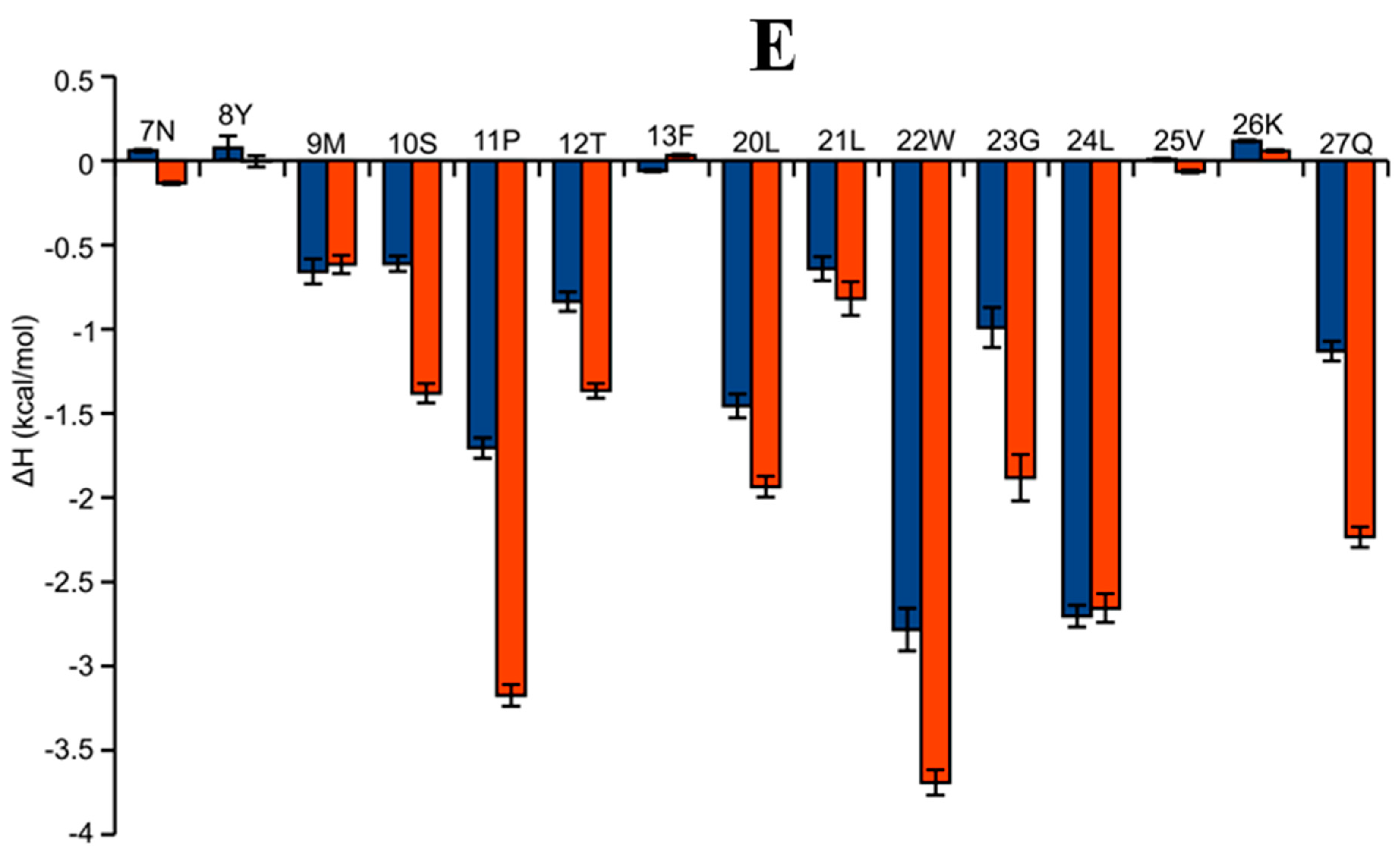
2.6. AAI and Ingenol Mebutate Bind PKCδ in a Similar Manner
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Gerneral Procedure for the Synthesis of 3-O-Angeloyl-20-O-acetyl ingenol
4.3. Cell Lines and Cell Culture
4.4. Cell Proliferation Inhibition Assay
4.5. Cell Cycle Distribution Assay
4.6. Apoptosis and Necrosis Assay by Annexin V-FITC/PI Double Staining
4.7. MMP Assay
4.8. Western Blotting Assay
4.9. Computational Docking
4.10. MD Simulations
4.11. Binding Energy Calculations
4.12. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aditya, S.; Gupta, S. Ingenol mebutate: A novel topical drug for actinic keratosis. Indian Dermatol. Online J. 2013, 4, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Tzogani, K.; Nagercoil, N.; Hemmings, R.J.; Samir, B.; Gardette, J.; Demolis, P.; Salmonson, T.; Pignatti, F. The European Medicines Agency approval of ingenol mebutate (Picato) for the cutaneous treatment of non-hyperkeratotic, non-hypertrophic actinic keratosis in adults: Summary of the scientific assessment of the Committee for Medicinal Products for Human Use (CHMP). Eur. J. Dermatol. 2014, 24, 457–463. [Google Scholar] [PubMed]
- Hampson, P.; Wang, K.; Milverton, L.; Ersvaer, E.; Bruserud, O.; Lord, J.M. Kinetics of ERK1/2 activation determine sensitivity of acute myeloid leukaemia cells to the induction of apoptosis by the novel small molecule ingenol 3-angelate (PEP005). Apoptosis 2010, 15, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Akar, U.; Tekedereli, I.; Alpay, S.N.; Barria, M.; Gezgen, B.; zhang, N.; Coombes, K.; Kornblau, S.; Lopez-Berestein, G. PKCδ regulates translation initiation through PKR and eIF2α in response to retinoic acid in acute myeloid leukemia cells. Leuk. Res. Treat. 2012, 2012, 482905. [Google Scholar]
- Clamp, A.; Jayson, G.C. The clinical development of the bryostatins. Anticancer Drugs 2002, 13, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Mans, D.R.; Regner, A.; Schwartsmann, G. Targeting protein kinase C: New therapeutic opportunities against high-grade malignant gliomas? Oncologist 2002, 7, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Grue-Sorensen, G.; Mansson, K.; Vedso, P.; Soor, A.; Stahlhut, M.; Bertelsen, M.; Engell, K.M.; Högberg, T. Syntheses, biological evaluation and SAR of ingenol mebutate analogues for treatment of actinic keratosis and non-melanoma skin cancer. Bioorg. Med. Chem. Lett. 2013, 23, 5624–5629. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.A.; Cozzi, S.-J.; Pierce, C.J.; Pavey, S.J.; Parsons, P.G.; Boyle, G.M. The induction of senescence-like growth arrest by protein kinase C-activating diterpene esters in solid tumor cells. Investig. New Drugs 2010, 28, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Kazanietz, M.G.; Blumberg, P.M.; Hurley, J.H. Crystal structure of the Cys2 activator-binding domain of protein kinase Cδ in complex with phorbol ester. Cell 1995, 81, 917–924. [Google Scholar] [CrossRef]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Ijichi, K.; Konno, K.; Yokota, T.; Tokuhisa, K.; Katsuura, K.; Uemura, D.; Shigeta, S.; Baba, M. Ingenol derivatives, ingredient of ‘Kansui’, are highly potent inhibitor of HIV. Antivir. Res. 1995, 26, A228. [Google Scholar] [CrossRef]
- Nambudiri, V. From home remedy to cancer treatment: A history of ingenol mebutate and Euphorbia peplus in dermatology. J. Am. Acad. Dermatol. 2013, 68, AB33. [Google Scholar]
- Ogbourne, S.M.; Suhrbier, A.; Jones, B.; Cozzi, S.J.; Boyle, G.M.; Morris, M.; McAlpine, D.; Johns, J.; Scott, T.M.; Sutherland, K.P.; et al. Antitumor activity of 3-ingenyl angelate: Plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004, 64, 2833–2839. [Google Scholar] [CrossRef] [PubMed]
- Serova, M.; Ghoul, A.; Benhadji, K.A.; Faivre, S.; Le Tourneau, C.; Cvitkovic, E.; Lokiec, F.; Lord, J.; Ogbourne, S.M.; Calvo, F.; et al. Effects of protein kinase C modulation by PEP005, a novel ingenol angelate, on mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling in cancer cells. Mol. Cancer Ther. 2008, 7, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Manley, P.W.; Cowan-Jacob, S.W.; Hochhaus, A.; Griffin, J.D. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer 2007, 7, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Wang, Y.; Cai, D.; von Bubnoff, N.; Paschka, P.; Muller-Brusselbach, S.; Ottmann, O.G.; Duyster, J.; Hochhaus, A.; Neubauer, A. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 2005, 19, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Tolentino, J.H.; Hazlehurst, L.A. Role of STAT3 in transformation and drug resistance in CML. Front. Oncol. 2012, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Mencalha, A.L.; Correa, S.; Abdelhay, E. Role of calcium-dependent protein kinases in chronic myeloid leukemia: combined effects of PKC and BCR-ABL signaling on cellular alterations during leukemia development. OncoTargets Ther. 2014, 7, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Hampson, P.; Chahal, H.; Khanim, F.; Hayden, R.; Mulder, A.; Assi, L.K.; Bunce, C.M.; Lord, J.M. PEP005, a selective small-molecule activator of protein kinase C, has potent antileukemic activity mediated via the delta isoform of PKC. Blood 2005, 106, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Kedei, N.; Lundberg, D.J.; Toth, A.; Welburn, P.; Garfield, S.H.; Blumberg, P.M. Characterization of the interaction of ingenol 3-angelate with protein kinase C. Cancer Res. 2004, 64, 3243–3255. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Deininger, M.W.N.; Eide, C.A.; Clackson, T.; Druker, B.J. Targeting the BCR-ABL signaling pathway in therapy-resistant philadelphia chromosome-positive leukemia. Am. Assoc. Cancer Res. 2011, 17, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Coppo, P.; Flamant, S.; Mas, V.D.; Jarrier, P.; Guillier, M.; Bonnet, M.-L.; Lacout, C.; Guilhot, F.; Vainchenker, W.; Turhan, A.G. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Free Energy (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| VDW | EEL | EGB | ESURF | TΔS | ΔG | |
| AAI | −30.69 (0.39) | −15.80 (0.68) | 25.14 (0.41) | −3.95 (0.04) | −20.39 (2.86) | −4.91 (2.54) |
| ingenol mebutate | −40.51 (0.34) | −19.20 (0.48) | 27.91 (0.27) | −4.46 (0.02) | −20.31 (2.71) | −15.95 (2.31) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Chen, F.; Yu, R.; Zhang, W.; Han, M.; Liu, F.; Wu, J.; Zhao, X.; Miao, J. Synthesis and Cytotoxicity against K562 Cells of 3-O-Angeloyl-20-O-acetyl Ingenol, a Derivative of Ingenol Mebutate. Int. J. Mol. Sci. 2016, 17, 1348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081348
Liu M, Chen F, Yu R, Zhang W, Han M, Liu F, Wu J, Zhao X, Miao J. Synthesis and Cytotoxicity against K562 Cells of 3-O-Angeloyl-20-O-acetyl Ingenol, a Derivative of Ingenol Mebutate. International Journal of Molecular Sciences. 2016; 17(8):1348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081348
Chicago/Turabian StyleLiu, Ming, Fangling Chen, Rilei Yu, Weiyi Zhang, Mei Han, Fei Liu, Jing Wu, Xingzeng Zhao, and Jinlai Miao. 2016. "Synthesis and Cytotoxicity against K562 Cells of 3-O-Angeloyl-20-O-acetyl Ingenol, a Derivative of Ingenol Mebutate" International Journal of Molecular Sciences 17, no. 8: 1348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081348