Gene Expression Profiling Reveals Potential Players of Left-Right Asymmetry in Female Chicken Gonads

 ,
,
Abstract
:
1. Introduction
2. Results
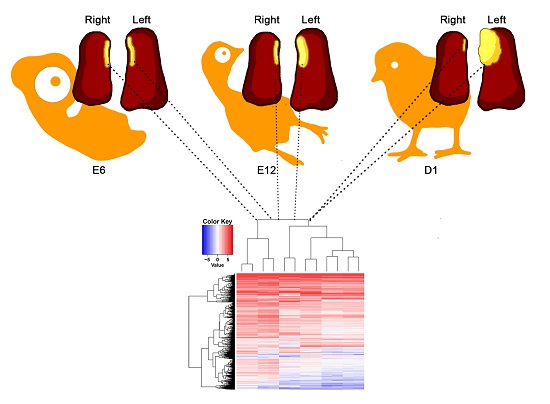
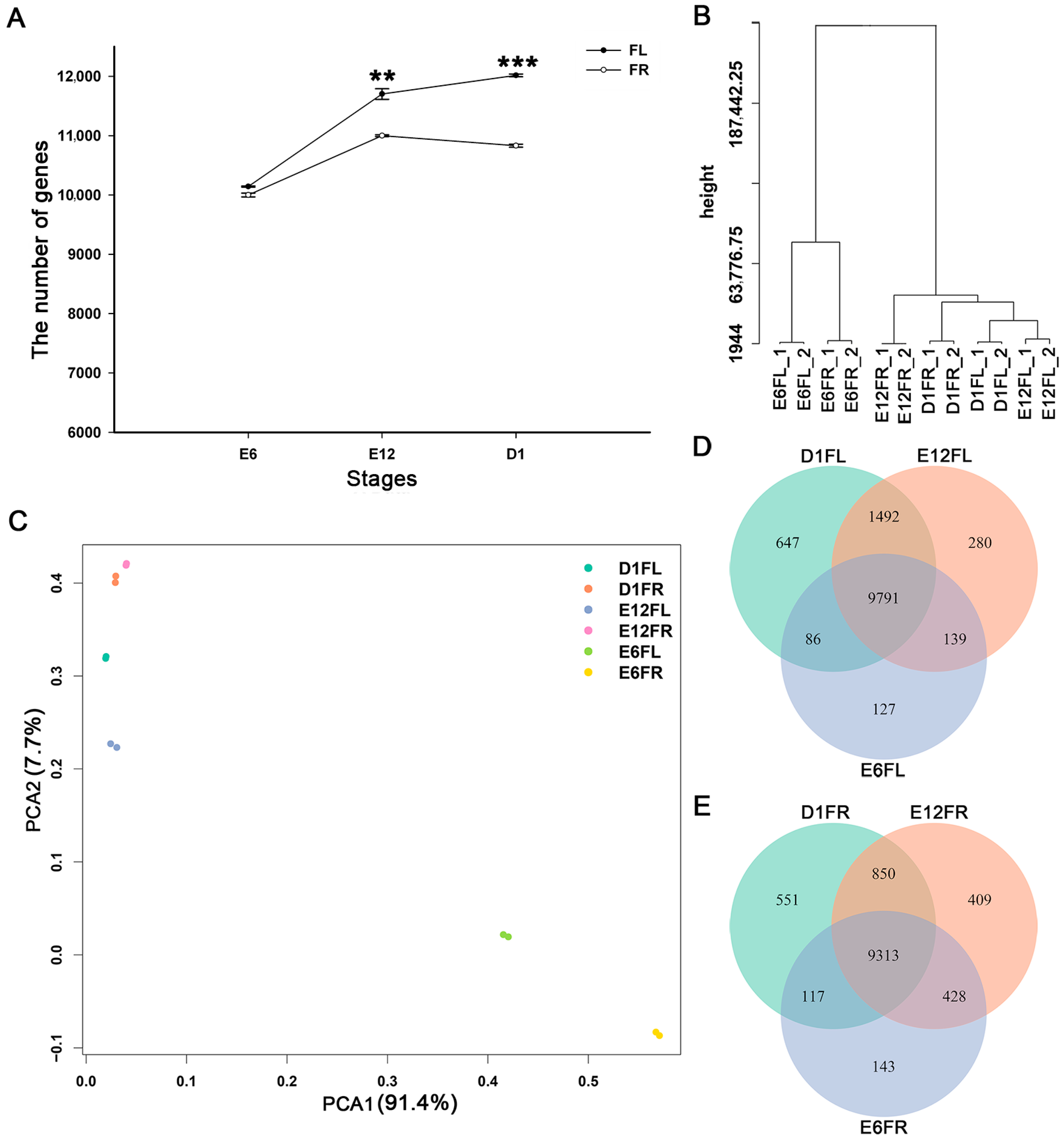
2.1. General Analysis of Gene Expression Profiles among Different Samples
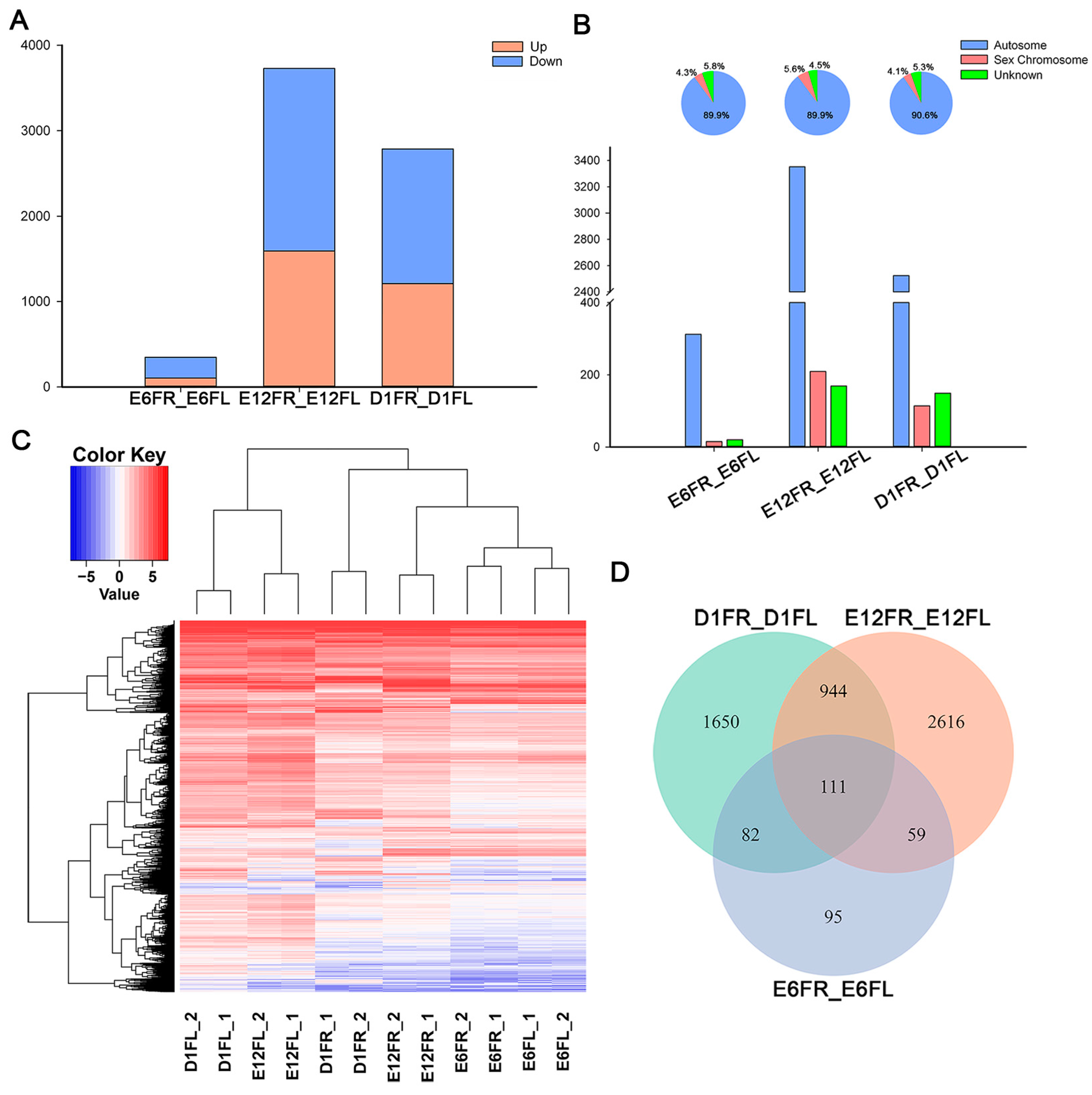
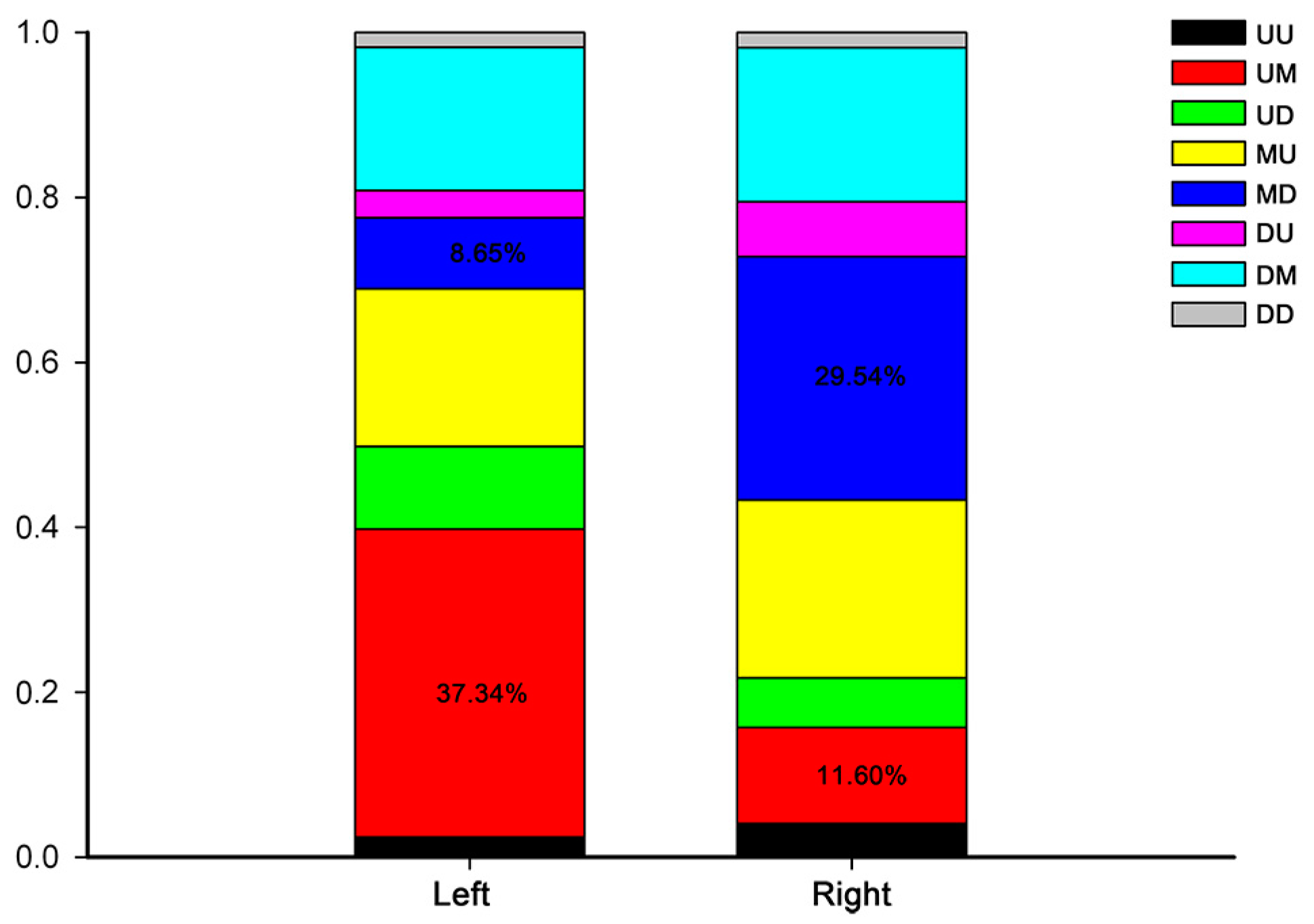
2.2. Right-Left Expression Profile
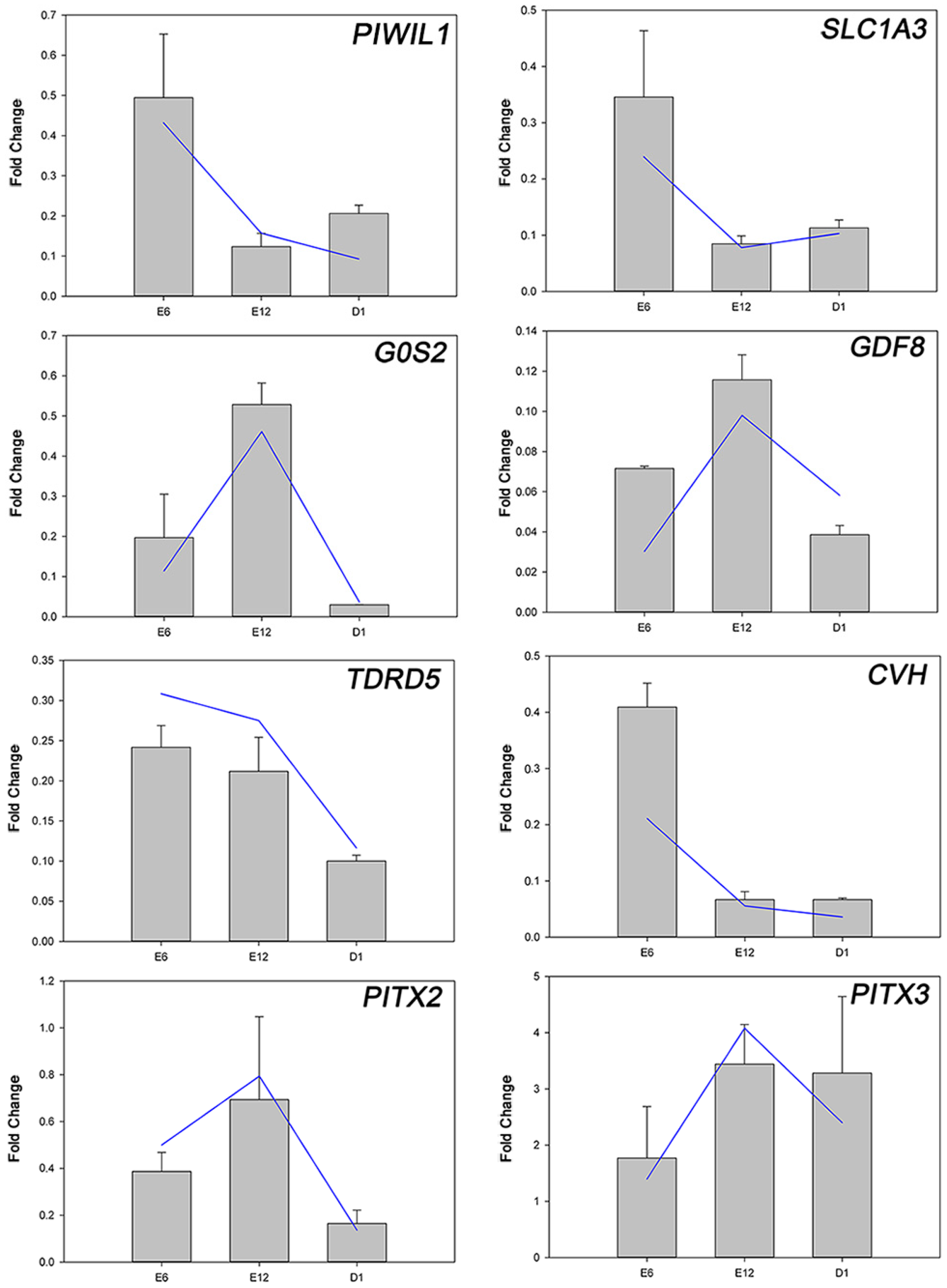
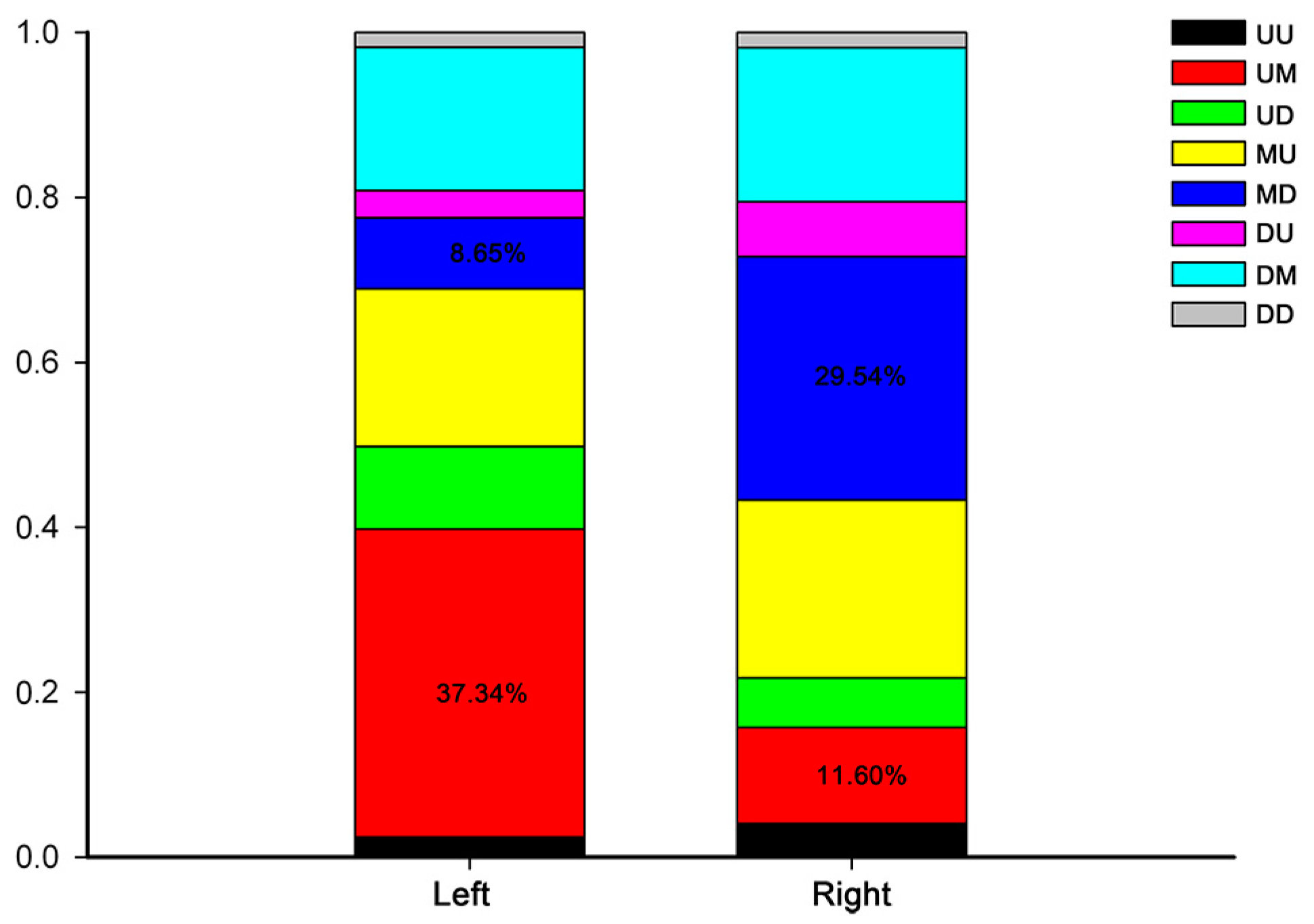
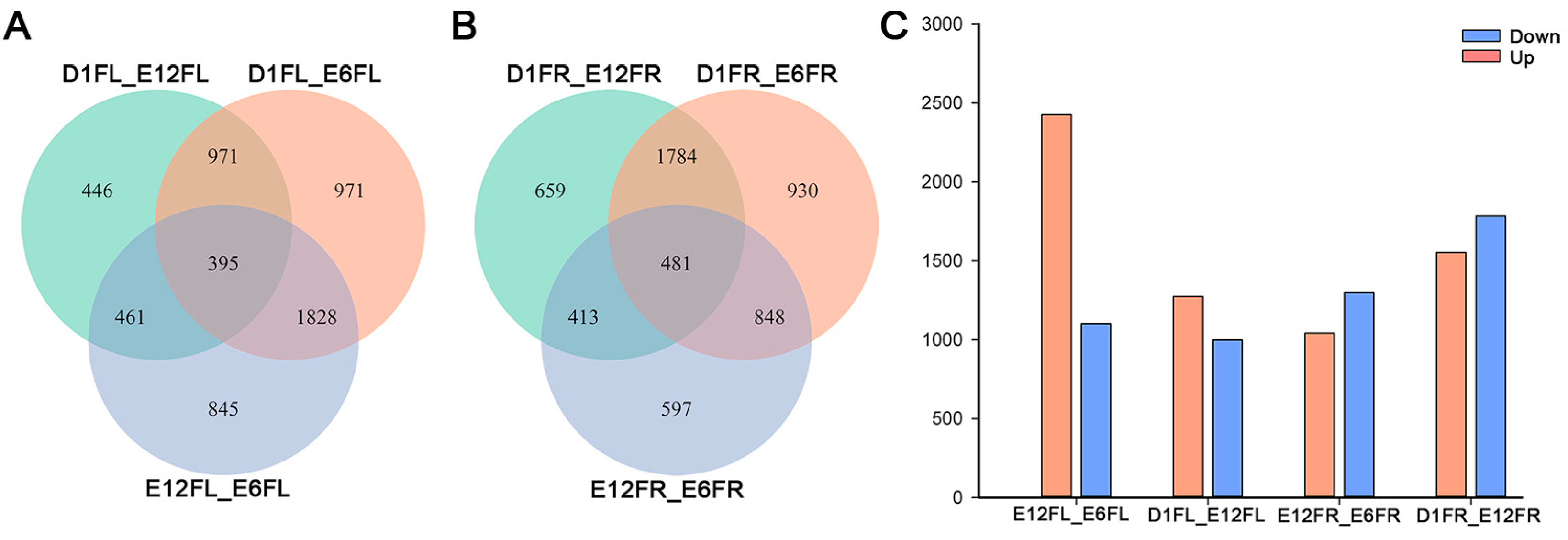
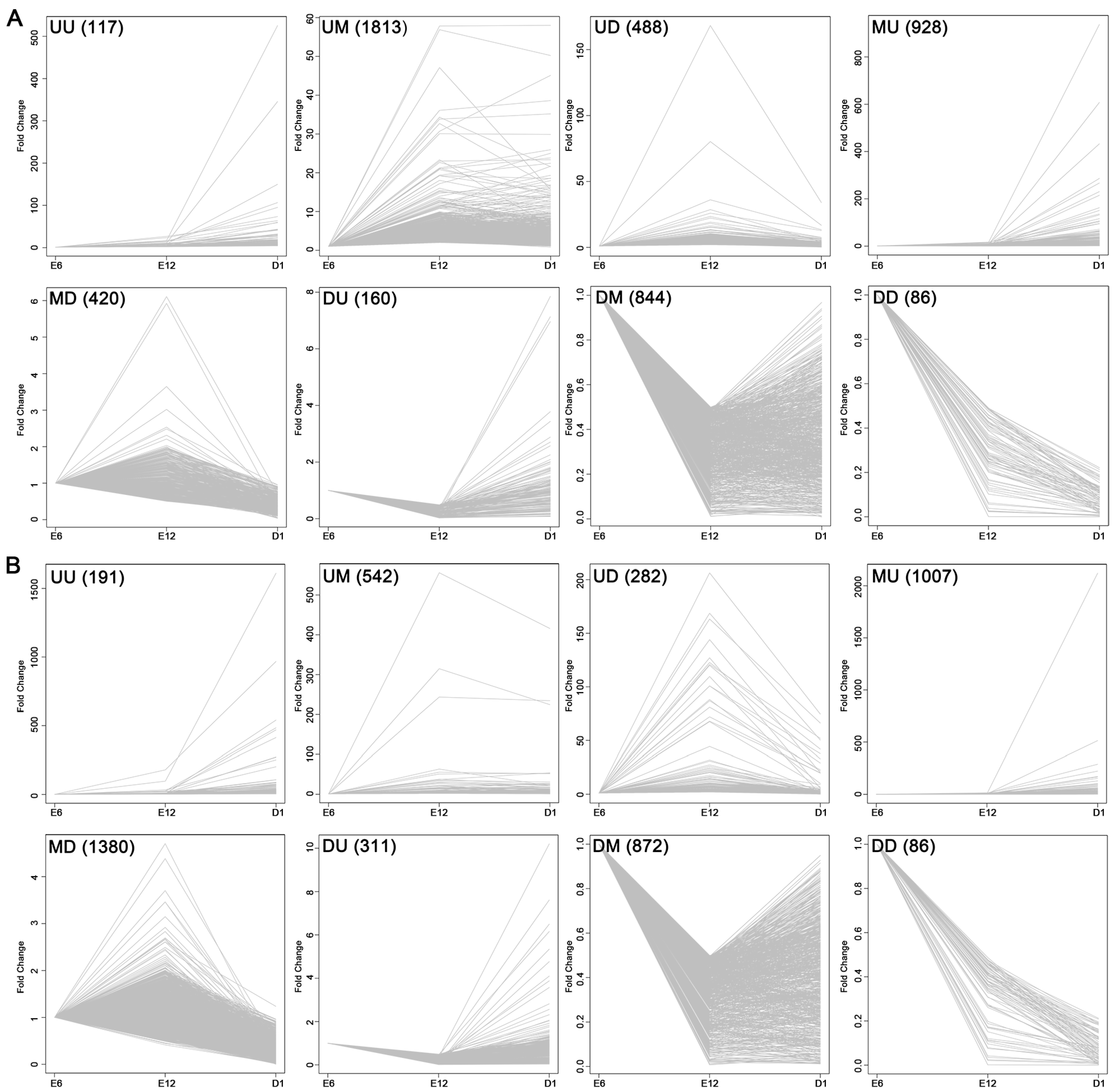
2.3. Development-Dependent Gene Expression Patterns
3. Discussion
4. Materials and Methods
4.1. Embryo Incubation and Tissue Collection
4.2. Library Construction and Sequencing
4.3. Data Filtering and Alignment
4.4. Gene Expression Analysis
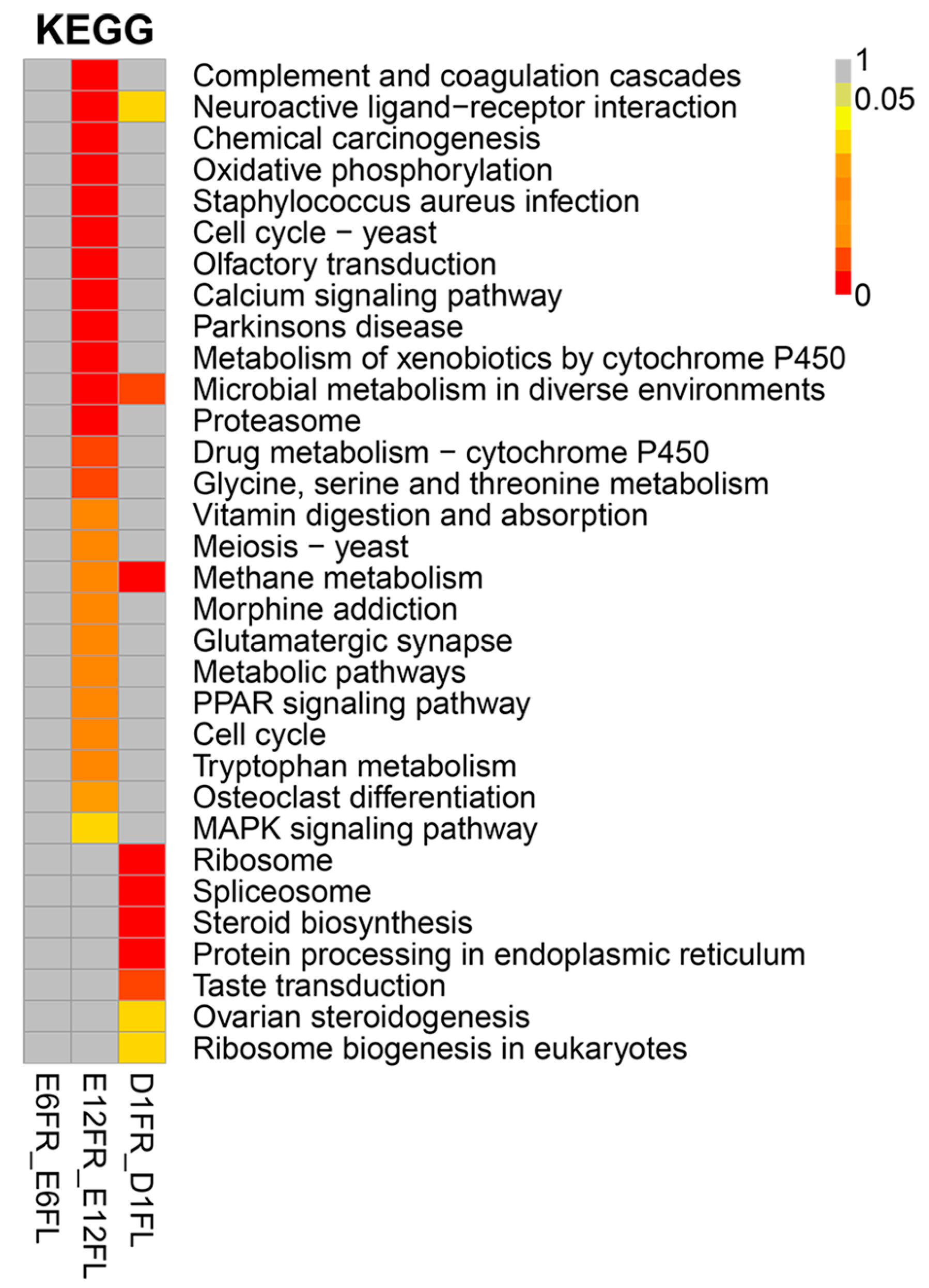
4.5. Functional Annotation and Enrichment Analysis
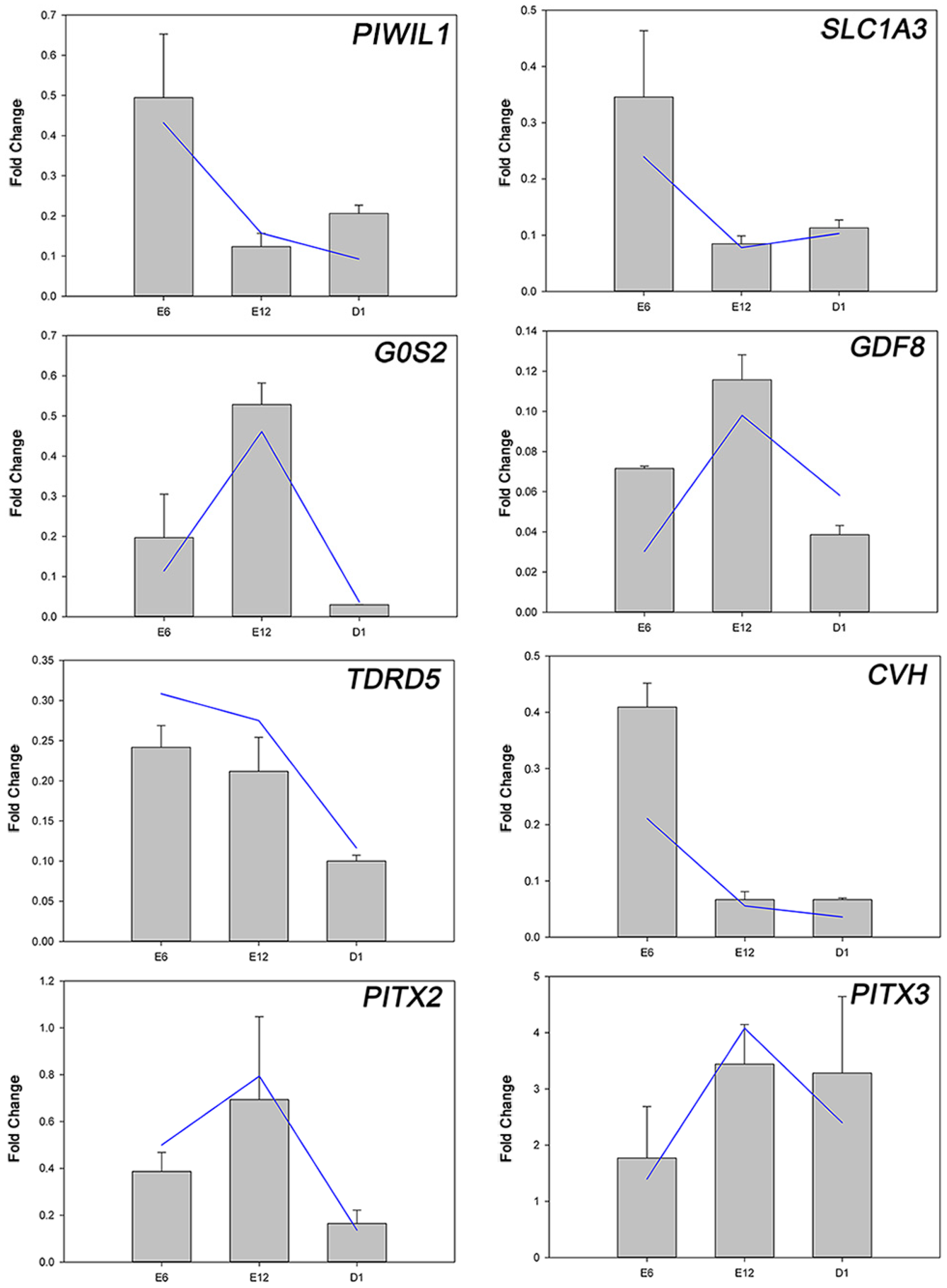
4.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.7. Analysis of Development-Dependent Gene Expression Patterns
4.8. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| E6 | Embryonic day 6 |
| E12 | Embryonic day 12 |
| D1 | Post-hatching day 1 |
| DEGs | Differentially expressed genes |
| FC | Fold change |
| FDR | False discovery rate |
| GO | Gene ontology |
| BP | Biological process |
| CC | Cellular component |
| MF | Molecular function |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| RPKM | Reads per kilobase million mapped reads |
| qRT-PCR | Quantitative real-time RT-PCR |
| SD | Standard deviation |
| UU | Up-up |
| UM | Up-maintain |
| UD | Up-down |
| MU | Maintain-up |
| MD | Maintain-down |
| DU | Down-up |
| DM | Down-maintain |
| DD | Down-down |
References
- Van Limborgh, J.; van Faassen, F. The asymmetry of the gonads in duck embryos experimentally turned on their right side. Acta Morphol. Neerl. Scand. 1960, 3, 79–91. [Google Scholar] [PubMed]
- Van Limborgh, J. The origin of the primary asymmetry of bird gonads. Ned. Tijdschr. Geneeskd. 1960, 104, 2442–2443. [Google Scholar] [PubMed]
- Smith, C.A.; Sinclair, A.H. Sex determination: Insights from the chicken. Bioessays 2004, 26, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Romanoff, A.L. The avian embryo: Structural and functional development. Q. Rev. Biol. 1960, 35, 342. [Google Scholar]
- Hamburger, V.; Hamilton, H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951, 88, 49–92. [Google Scholar] [CrossRef] [PubMed]
- Van Limborgh, J. The first sign of sexual differentiation of the gonads in chick embryos. Arch Anat. Microsc. Morphol. Exp. 1968, 57, 79–90. [Google Scholar] [PubMed]
- Vallisneri, M.; Quaglia, A.; Stagni, A.M.; Zaccanti, F. Differences between male and female protogonia in chick embryos before sex differentiation of the gonads. Boll. Soc. Ital. Biol. Sper. 1990, 66, 91–98. [Google Scholar] [PubMed]
- Guioli, S.; Nandi, S.; Zhao, D.; Burgess-Shannon, J.; Lovell-Badge, R.; Clinton, M. Gonadal asymmetry and sex determination in birds. Sex Dev. 2014, 8, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.E.; Smith, C.A.; Sinclair, A.H. Sites of Estrogen Receptor and Aromatase Expression in the Chicken Embryo. Gen. Comp. Endocrinol. 1997, 108, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Koide, M.; Ono, T.; Yasugi, S. Sex-specific and left-right asymmetric expression pattern of Bmp7 in the gonad of normal and sex-reversed chicken embryos. Dev. Growth Differ. 2005, 47, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Guioli, S.; Lovell-Badge, R. PITX2 controls asymmetric gonadal development in both sexes of the chick and can rescue the degeneration of the right ovary. Development 2007, 134, 4199–4208. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, O.; Kikuchi, H.; Kikuchi, T.; Mizuno, S. Differential expression of genes for aromatase and estrogen receptor during the gonadal development in chicken embryos. J. Mol. Endocrinol. 1998, 20, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Scheider, J.; Afonso-Grunz, F.; Hoffmeier, K.; Horres, R.; Groher, F.; Rycak, L.; Oehlmann, J.; Winter, P. Gene expression of chicken gonads is sex- and side-specific. Sex Dev. 2014, 8, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Ayers, K.L.; Davidson, N.M.; Demiyah, D.; Roeszler, K.N.; Grützner, F.; Sinclair, A.H.; Oshlack, A.; Smith, C.A. RNA sequencing reveals sexually dimorphic gene expression before gonadal differentiation in chicken and allows comprehensive annotation of the W-chromosome. Genome Biol. 2013, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ayers, K.L.; Lambeth, L.S.; Davidson, N.M.; Sinclair, A.H.; Oshlack, A.; Smith, C.A. Identification of candidate gonadal sex differentiation genes in the chicken embryo using RNA-seq. BMC Genom. 2015, 16, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Smith, M.J.; Sinclair, A.H. Gene expression during gonadogenesis in the chicken embryo. Gene 1999, 234, 395–402. [Google Scholar] [CrossRef]
- Mazaud, S.; Oréal, E.; Guigon, C.J.; Carréeusèbe, D.; Magre, S. Lhx9 expression during gonadal morphogenesis as related to the state of cell differentiation. Gene Exp. Patterns 2002, 2, 373–377. [Google Scholar] [CrossRef]
- Oreal, E.; Mazaud, S.; Picard, J.Y.; Magre, S.; Carre-Eusebe, D. Different patterns of anti-Mullerian hormone expression, as related to DMRT1, SF-1, WT1, GATA-4, Wnt-4, and Lhx9 expression, in the chick differentiating gonads. Dev. Dyn. 2002, 225, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Witschi, E. Origin of asymmetry in the reproductive system of birds. Am. J. Anat. 2005, 56, 119–141. [Google Scholar] [CrossRef]
- Dubois, R.; Cuminge, D. Primary asymmetry in the distribution of primordial germ cells during colonization of gonadal buds in chick embryo. C. R. Acad. Sci. Hebd. Séances. Acad. Sci. D 1978, 286, 535–538. [Google Scholar] [PubMed]
- Tsunekawa, N.; Naito, M.; Sakai, Y.; Nishida, T.; Noce, T. Isolation of chicken vasa homolog gene and tracing the origin of primordial germ cells. Development 2000, 127, 2741–2750. [Google Scholar] [PubMed]
- Cox, D.; Chao, A.; Lin, H. Piwi encodes a nucleoplasmic factor whose activity modulates the number and division rate of germline stem cells. Development 2000, 127, 503–514. [Google Scholar] [PubMed]
- Rengaraj, D.; Zheng, Y.H.; Kang, K.S.; Park, K.J.; Lee, B.R.; Lee, S.I.; Choi, J.W.; Han, J.Y. Conserved expression pattern of chicken DAZL in primordial germ cells and germ-line cells. Theriogenology 2010, 74, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Thomson, T.; Lasko, P. Tudor and its domains: Germ cell formation from a Tudor perspective. Cell Res. 2005, 15, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Chuma, S. piRNAs and their involvement in male germline development in mice. Dev. Growth Differ. 2012, 54, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Hosokawa, M.; Vagin, V.V.; Reuter, M.; Hayashi, E.; Mochizuki, A.L.; Kitamura, K.; Yamanaka, H.; Kondoh, G.; Okawa, K. Tudor domain containing 7 (Tdrd7) is essential for dynamic ribonucleoprotein (RNP) remodeling of chromatoid bodies during spermatogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 10579–10584. [Google Scholar] [CrossRef] [PubMed]
- Chuma, S.; Hosokawa, M.; Kitamura, K.; Kasai, S.; Fujioka, M.; Hiyoshi, M.; Takamune, K.; Noce, T.; Nakatsuji, N. Tdrd1/Mtr-1, a tudor-related gene, is essential for male germ-cell differentiation and nuage/germinal granule formation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15894–15899. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Tanaka, T.; Hosokawa, M.; Reuter, M.; Stark, A.; Kato, Y.; Kondoh, G.; Okawa, K.; Chujo, T.; Suzuki, T. The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev. Cell 2009, 17, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, Y.; Ohta, H.; Abe, T.; Kurimoto, K.; Chuma, S.; Saitou, M. TDRD5 is required for retrotransposon silencing, chromatoid body assembly, and spermiogenesis in mice. J. Cell Biol. 2011, 192, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Lee, T.C.; Weeraratne, S.D.; Korzh, V.; Lim, T.M.; Gong, Z. Ziwi, the zebrafish homologue of the Drosophila piwi: Co-localization with vasa at the embryonic genital ridge and gonad-specific expression in the adults. Mech. Dev. 2003, 119 (Suppl. S1), 257–260. [Google Scholar]
- Zhang, D.; Xiong, H.; Shan, J.; Xia, X.; Trudeau, V.L. Functional insight into Maelstrom in the germline piRNA pathway: A unique domain homologous to the DnaQ-H 3′–5′ exonuclease, its lineage-specific expansion/loss and evolutionarily active site switch. Biol. Direct. 2008, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Kotaja, N.; Sassone-Corsi, P. The chromatoid body: A germ-cell-specific RNA-processing centre. Nat. Rev. Mol. Cell Biol. 2007, 8, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Gruidl, M.E.; Smith, P.A.; Kuznicki, K.A.; Mccrone, J.S.; Kirchner, J.; Roussell, D.L.; Strome, S.; Bennett, K.L. Multiple potential germ-line helicases are components of the germ-line-specific P granules of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1996, 93, 13837–13842. [Google Scholar] [CrossRef] [PubMed]
- Intarapat, S.; Stern, C.D. Sexually dimorphic and sex-independent left-right asymmetries in chicken embryonic gonads. PLoS ONE 2013, 8, e69893. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Zhang, X.; Xie, X.; Liu, J. The G0/G1 switch gene 2 (G0S2): Regulating metabolism and beyond. Biochim. Biophys. Acta 2012, 1831, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Choi, J.W.; Park, K.J.; Lee, Y.M.; Kim, T.M.; Sohn, S.H.; Lim, J.M.; Han, J.Y. Development of a pheasant interspecies primordial germ cell transfer to chicken embryo: Effect of donor cell sex on chimeric semen production. Theriogenology 2009, 72, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Kendziorski, C.; Irizarry, R.A.; Chen, K.S.; Haag, J.D.; Gould, M.N. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 4252–4257. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Planet, E.; Attolini, C.S.; Reina, O.; Flores, O.; Rossell, D. htSeqTools: High-throughput sequencing quality control, processing and visualization in R. Bioinformatics 2012, 28, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2015, 11, 1–12. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Clean Reads | Mapped Reads | Mapping Rate (%) | Uniq-Mapped Reads | Uniq-Mapping Rate (%) |
|---|---|---|---|---|---|
| E6FR_1 | 17482191 | 15924839 | 91 | 14374889 | 82 |
| E6FR_2 | 17482191 | 15932034 | 91 | 14389796 | 82 |
| E6FL_1 | 16816894 | 15089322 | 90 | 13945497 | 83 |
| E6FL_2 | 17199818 | 15466729 | 90 | 14299262 | 83 |
| E12FR_1 | 16848050 | 14545265 | 86 | 14104709 | 84 |
| E12FR_2 | 17179086 | 14818688 | 86 | 14358014 | 84 |
| E12FL_1 | 17059514 | 15157661 | 89 | 14570665 | 85 |
| E12FL_2 | 16845550 | 14823214 | 88 | 14364057 | 85 |
| D1FR_1 | 17983758 | 15664230 | 87 | 15239760 | 85 |
| D1FR_2 | 16637905 | 14418212 | 87 | 14093197 | 85 |
| D1FL_1 | 16795901 | 14538377 | 87 | 13981293 | 83 |
| D1FL_2 | 16717969 | 14463322 | 87 | 14001712 | 84 |
| Group | GO:Term | Description | Gene_Num De | Gene_Num Back | p |
|---|---|---|---|---|---|
| E6FR-E6FL | Biological process | ||||
| GO:0034587 | piRNA metabolic process | 7 | 14 | 9.4 × 10−9 | |
| GO:0043046 | DNA methylation involved in gamete generation | 6 | 10 | 2.7 × 10−8 | |
| Cellular component | |||||
| GO:0045495 | pole plasm | 9 | 12 | 3.4 × 10−13 | |
| GO:0060293 | germ plasm | 8 | 11 | 1.1 × 10−11 | |
| GO:0043186 | P granule | 8 | 11 | 1.1 × 10−11 | |
| E12FR-E12FL | Biological process | ||||
| GO:0022402 | cell cycle process | 293 | 845 | 5.5 × 10−10 | |
| GO:0007049 | cell cycle | 381 | 1159 | 2.4 × 10−9 | |
| GO:0022412 | cellular process involved in reproduction in multicellular organism | 62 | 136 | 2.9 × 10−7 | |
| Cellular component | |||||
| GO:0005694 | chromosome | 197 | 558 | 1.5 × 10−8 | |
| GO:0044427 | chromosomal part | 170 | 478 | 8.3 × 10−8 | |
| GO:0000775 | chromosome, centromeric region | 61 | 139 | 7.4 × 10−7 | |
| Molecular function | |||||
| GO:0016491 | oxidoreductase activity | 204 | 587 | 2.3 × 10−7 | |
| GO:0004386 | helicase activity | 58 | 130 | 1.7 × 10−6 | |
| GO:0004364 | glutathione transferase activity | 13 | 16 | 4.9 × 10−6 | |
| D1FR-D1FL | Biological process | ||||
| GO:0044699 | single-organism process | 1861 | 9535 | 2.2 × 10−16 | |
| GO:0007275 | multicellular organismal development | 753 | 3374 | 2.4 × 10−13 | |
| GO:0032501 | multicellular organismal process | 962 | 4477 | 2.9 × 10−13 | |
| Cellular component | |||||
| GO:0071944 | cell periphery | 742 | 3418 | 1.6 × 10−10 | |
| GO:0031224 | intrinsic component of membrane | 876 | 4140 | 4.5 × 10−10 | |
| GO:0005886 | plasma membrane | 723 | 3341 | 6.3 × 10−10 | |
| Molecular function | |||||
| GO:0005215 | transporter activity | 250 | 1009 | 1.4 × 10−9 | |
| GO:0022857 | transmembrane transporter activity | 203 | 800 | 8.2 × 10−9 | |
| GO:0022892 | substrate-specific transporter activity | 210 | 853 | 6.0 × 10−8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Z.; Lu, Y.; Rui, L.; Yu, X.; Yang, F.; Tu, C.; Li, Z. Gene Expression Profiling Reveals Potential Players of Left-Right Asymmetry in Female Chicken Gonads. Int. J. Mol. Sci. 2017, 18, 1299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061299
Wan Z, Lu Y, Rui L, Yu X, Yang F, Tu C, Li Z. Gene Expression Profiling Reveals Potential Players of Left-Right Asymmetry in Female Chicken Gonads. International Journal of Molecular Sciences. 2017; 18(6):1299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061299
Chicago/Turabian StyleWan, Zhiyi, Yanan Lu, Lei Rui, Xiaoxue Yu, Fang Yang, Chengfang Tu, and Zandong Li. 2017. "Gene Expression Profiling Reveals Potential Players of Left-Right Asymmetry in Female Chicken Gonads" International Journal of Molecular Sciences 18, no. 6: 1299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061299