High Throughput Identification of Novel Conotoxins from the Vermivorous Oak Cone Snail (Conus quercinus) by Transcriptome Sequencing

 ,
,
Abstract
:
1. Introduction
2. Results
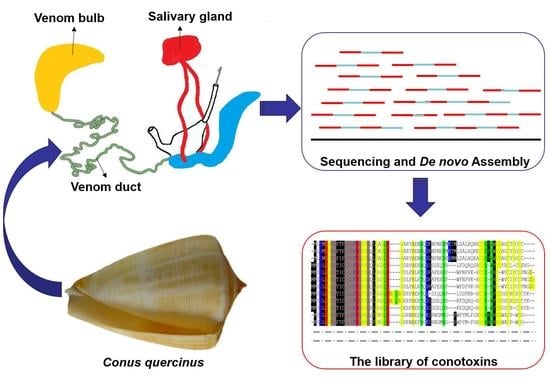
2.1. Assembly of Transcriptome Sequences
2.2. Summary of Conotoxins in the Three Transcriptomes
2.3. Comparison of Conotoxins in the Three Transcriptomes
2.4. Differential Transcription of Conotoxins in Different Organs
2.5. Diversity of Conotoxin Superfamilies
2.6. Phylogeny of the Superfamily Signal Sequences
3. Discussion
4. Materials and Methods
4.1. Sample Collection and RNA Extraction
4.2. Sequence Analysis and Assembling
4.3. Functional Annotation of Transcripts
4.4. Prediction and Identification of Conotoxins
4.5. Classification of Gene Superfamilies
4.6. Alignment and Phylogenetic Analysis
4.7. Availability of Supporting Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prashanth, J.R.; Dutertre, S.; Jin, A.H.; Lavergne, V.; Hamilton, B.; Cardoso, F.C.; Griffin, J.; Venter, D.J.; Alewood, P.F.; Lewis, R.J. The role of defensive ecological interactions in the evolution of conotoxins. Mol. Ecol. 2016, 25, 598–615. [Google Scholar] [CrossRef]
- Himaya, S.W.; Jin, A.H.; Dutertre, S.; Giacomotto, J.; Mohialdeen, H.; Vetter, I.; Alewood, P.F.; Lewis, R.J. Comparative venomics reveals the complex prey capture strategy of the piscivorous cone snail Conus catus. J. Proteome Res. 2015, 14, 4372–4381. [Google Scholar] [CrossRef]
- Macrander, J.; Panda, J.; Janies, D.; Daly, M.; Reitzel, A.M. Venomix: A simple bioinformatic pipeline for identifying and characterizing toxin gene candidates from transcriptomic data. Peer J. 2018, 6, e5361. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef]
- Kumar, P.S.; Kumar, D.S.; Umamaheswari, S. A perspective on toxicology of Conus venom peptides. Asian Pac. J. Trop. Med. 2015, 8, 337–351. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Brust, A.; Jin, A.H.; Alewood, P.F.; Dutertre, S.; Lewis, R.J. Cone snail venomics: From novel biology to novel therapeutics. Future Med. Chem. 2014, 6, 1659–1675. [Google Scholar] [CrossRef]
- Vetter, I.; Lewis, R.J. Therapeutic potential of cone snail venom peptides (conopeptides). Curr. Top. Med. Chem. 2012, 12, 1546–1552. [Google Scholar] [CrossRef]
- Rigo, F.K.; Dalmolin, G.D.; Trevisan, G.; Tonello, R.; Silva, M.A.; Rossato, M.F.; Klafke, J.Z.; Cordeiro Mdo, N.; Castro Junior, C.J.; Montijo, D.; et al. Effect of omega-conotoxin MVIIA and Phalpha1beta on paclitaxel-induced acute and chronic pain. Pharmacol. Biochem. Behav. 2013, 114, 16–22. [Google Scholar] [CrossRef]
- Eisapoor, S.S.; Jamili, S.; Shahbazzadeh, D.; Ghavam Mostafavi, P.; Pooshang Bagheri, K. A New, High yield, rapid, and cost-effective protocol to deprotection of cysteine-rich conopeptide, omega-conotoxin MVIIA. Chem. Biol. Drug Des. 2016, 87, 687–693. [Google Scholar] [CrossRef]
- Olivera, B.M.; Seger, J.; Horvath, M.P.; Fedosov, A.E. Prey-capture strategies of fish-hunting cone snails: Behavior, neurobiology and evolution. Brain Behav. Evol. 2015, 86, 58–74. [Google Scholar] [CrossRef]
- Mir, R.; Karim, S.; Kamal, M.A.; Wilson, C.M.; Mirza, Z. Conotoxins: Structure, therapeutic potential and pharmacological applications. Curr. Pharm. Des. 2016, 22, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Ansorge, W.J. Next-generation DNA sequencing techniques. Nat. Biotechnol. 2009, 25, 195–203. [Google Scholar] [CrossRef]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Characterization of the Conus bullatus genome and its venom-duct transcriptome. BMC Genom. 2011, 12, 60. [Google Scholar] [CrossRef]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Elucidation of the molecular envenomation strategy of the cone snail Conus geographus through transcriptome sequencing of its venom duct. BMC Genom. 2012, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Terrat, Y.; Biass, D.; Dutertre, S.; Favreau, P.; Remm, M.; Stocklin, R.; Piquemal, D.; Ducancel, F. High-resolution picture of a venom gland transcriptome: Case study with the marine snail Conus consors. Toxicon 2012, 59, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Dutertre, S.; Jin, A.H.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the Conus marmoreus venom duct transcriptome with ConoSorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 2013, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Harliwong, I.; Jones, A.; Miller, D.; Taft, R.J.; Alewood, P.F. Optimized deep-targeted proteo-transcriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. USA 2015, 112, E3782–E3791. [Google Scholar] [CrossRef]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS ONE 2014, 9, e87648. [Google Scholar] [CrossRef]
- Lluisma, A.O.; Milash, B.A.; Moore, B.; Olivera, B.M.; Bandyopadhyay, P.K. Novel venom peptides from the cone snail Conus pulicarius discovered through next-generation sequencing of its venom duct transcriptome. Mar. Genom. 2012, 5, 43–51. [Google Scholar] [CrossRef]
- Jin, A.H.; Dutertre, S.; Kaas, Q.; Lavergne, V.; Kubala, P.; Lewis, R.J.; Alewood, P.F. Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity. Mol. Cell Proteom. 2013, 12, 3824–3833. [Google Scholar] [CrossRef]
- Jin, A.H.; Vetter, I.; Himaya, S.W.; Alewood, P.F.; Lewis, R.J.; Dutertre, S. Transcriptome and proteome of Conus planorbis identify the nicotinic receptors as primary target for the defensive venom. Proteomics 2015, 15, 4030–4040. [Google Scholar] [CrossRef]
- Barghi, N.; Concepcion, G.P.; Olivera, B.M.; Lluisma, A.O. High conopeptide diversity in Conus tribblei revealed through analysis of venom duct transcriptome using two high-throughput sequencing platforms. Mar. Biotechnol. (NY) 2015, 17, 81–98. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, Y.; Wang, L.; Liang, A.; Chen, S.; Xu, A.J. Identifying novel conopepetides from the venom ducts of Conus litteratus through integrating transcriptomics and proteomics. J. Proteom. 2018. Available online: https://europepmc.org/abstract/med/30267875 (accessed on 26 September 2018). [CrossRef]
- Peng, C.; Yao, G.; Gao, B.M.; Fan, C.X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi- transcriptome sequencing. GigaScience 2016, 5, 17. [Google Scholar] [CrossRef]
- Huang, Y.; Peng, C.; Yi, Y.; Gao, B.; Shi, Q. A Transcriptomic survey of ion channel-based conotoxins in the Chinese tubular cone snail (Conus betulinus). Mar. Drugs 2017, 15, 228. [Google Scholar] [CrossRef]
- Ye, M.; Hong, J.; Zhou, M.; Huang, L.; Shao, X.; Yang, Y.; Sigworth, F.J.; Chi, C.; Lin, D.; Wang, C. A novel conotoxin, qc16a, with a unique cysteine framework and folding. Peptides 2011, 32, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Chen, W.; Han, Y.; Sanders, T.; Chew, G.; Liu, J.; Hawrot, E.; Chi, C.; Wang, C. Characterization of a novel α4/4-conotoxin, Qc1.2, from vermivorous Conus quercinus. Acta Biochim. Biophys. Sin. (Shanghai) 2009, 41, 858–864. [Google Scholar] [CrossRef]
- Peng, C.; Tang, S.; Pi, C.; Liu, J.; Wang, F.; Wang, L.; Zhou, W.; Xu, A. Discovery of a novel class of conotoxin from Conus litteratus, lt14a, with a unique cysteine pattern. Peptides 2006, 27, 2174–2181. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, L.; Wu, Y.; Zhu, X.; Feng, Y.; Chen, Z.; Li, Y.; Sun, D.; Ren, Z.; Xu, A. Characterizing the evolution and functions of the M-superfamily conotoxins. Toxicon 2013, 76, 150–159. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puillandre, N.; Watkins, M.; Olivera, B.M. Evolution of Conus peptide genes: Duplication and positive selection in the A-superfamily. J. Mol. Evol. 2010, 70, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.B.; Ortiz, E.; Kaas, Q.; López-Vera, E.; Becerril, B.; Possani, L.D.; de la Cotera, E.P. Precursor De13.1 from Conus delessertii defines the novel G gene superfamily. Peptides 2013, 41, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, R.P. Novel M-Superfamily and T-Superfamily conotoxins and contryphans from the vermivorous snail Conus figulinus. J. Pept. Sci. 2015, 21, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Tae, H.S.; Xu, S.; Shao, X.; Adams, D.J.; Wang, C. Identification of a Novel O-Conotoxin Reveals an Unusual and Potent Inhibitor of the Human α9α10 Nicotinic Acetylcholine Receptor. Mar. Drugs 2017, 15, 170. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.C.; Craig, A.G.; Watkins, M.; Hillyard, D.R.; Gray, W.R.; Gulyas, J.; Rivier, J.E.; Cruz, L.J.; Olivera, B.M. Bromocontryphan: Post-translational bromination of tryptophan. Biochemistry 1997, 36, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Vijayasarathy, M.; Balaram, P. Mass spectrometric identification of bromotryptophan containing conotoxin sequences from the venom of C. amadis. Toxicon 2018, 144, 68–74. [Google Scholar] [CrossRef]
- Zhangsun, D.; Luo, S.; Wu, Y.; Zhu, X.; Hu, Y.; Xie, L. Novel O-superfamily conotoxins identified by cDNA cloning from three vermivorous Conus species. Chem. Biol. Drug Des. 2006, 68, 256–265. [Google Scholar] [CrossRef]
- Zamora-Bustillos, R.; Aguilar, M.B.; Falcón, A. Identification, by molecular cloning, of a novel type of I2-superfamily conotoxin precursor and two novel I2-conotoxins from the worm-hunter snail Conus spurius from the Gulf of México. Peptides 2010, 31, 384–393. [Google Scholar] [CrossRef]
- Phuong, M.A.; Mahardika, G.N. Targeted sequencing of venom genes from cone snail genomes improves understanding of conotoxin molecular evolution. Mol. Biol. Evol. 2018, 35, 1210–1224. [Google Scholar] [CrossRef]
- Robinson, S.D.; Norton, R.S. Conotoxin gene superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Ren, Z.; Chen, Y.; Xu, A. Discovery of two P-superfamily conotoxins, lt9a and lt9b, with different modifications on voltage-sensitive sodium channels. Toxicon 2017, 134, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.W.A.; Lewis, R.J. Venomics-Accelerated Cone Snail Venom Peptide Discovery. Int. J. Mol. Sci. 2018, 19, 788. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; Kelley, W.P.; Rubakhin, S.S.; Bingham, J.P.; Sweedler, J.V.; Gilly, W.F. Anatomical correlates of venom production in Conus californicus. Biol. Bull. 2002, 203, 27–41. [Google Scholar] [CrossRef]
- Safavi-Hemami, H.; Young, N.D.; Williamson, N.A.; Purcell, A.W. Proteomic interrogation of venom delivery in marine cone snails: Novel insights into the role of the venom bulb. J. Proteome Res. 2010, 9, 5610–5619. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Dutertre, S.; Lewis, R.J. Pharmacology of predatory and defensive venom peptides in cone snails. Mol. Biosyst. 2017, 13, 2453–2465. [Google Scholar] [CrossRef]
- Biggs, J.S.; Olivera, B.M.; Kantor, Y.I. α-Conopeptides specifically expressed in the salivary gland of Conus pulicarius. Toxicon 2008, 52, 101–105. [Google Scholar] [CrossRef]
- Shimek, R.L. The morphology of the buccal apparatus of Oenopota levidensis (Gastropoda, Turridae). Zoomorphology 1975, 80, 59–96. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Chen, Q.; Zhang, J.; Shi, Q. Mitochondrial genome sequencing of a vermivorous cone snail Conus quercinus supports the correlative analysis between phylogenetic relationships and dietary types of Conus species. PLoS ONE 2018, 13, e0193053. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2016, 45, D190–D199. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laht, S.; Koua, D.; Kaplinski, L.; Lisacek, F.; Stöcklin, R.; Remm, M. Identification and classification of conopeptides using profile Hidden Markov Models. Biochim. Biophys. Acta 2012, 1824, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Diego, D.; Guillermo, L.; Taboada, R.D.; David, P. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar]
- Alexandros, S. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | VD | SG | VB |
|---|---|---|---|
| Raw data | |||
| Total Reads | 29,451,202 | 30,184,621 | 30,775,070 |
| Total length (bp) | 4,417,680,300 | 4,527,693,150 | 4,616,260,500 |
| Read length (bp) | 150 | 150 | 150 |
| Clean data | |||
| Total Reads | 28,865,798 | 28,664,492 | 29,290,756 |
| Total length (bp) | 4,041,211,720 | 4,013,028,880 | 4,100,705,840 |
| Read length (bp) | 140 | 140 | 140 |
| Clean data ratio | 91.48% | 88.63% | 88.83% |
| Contigs | |||
| Total Number | 171,606 | 225,404 | 124,936 |
| Total Length (bp) | 80,150,026 | 111,578,962 | 61,180,077 |
| Mean Length (bp) | 467 | 495 | 489 |
| N50 (bp) | 586 | 669 | 651 |
| N70 (bp) | 331 | 353 | 349 |
| N90 (bp) | 212 | 214 | 215 |
| GC Content | 43.70% | 44.29% | 44.32% |
| Unigenes | |||
| Total Number | 91,392 | 113,472 | 66,549 |
| Total Length (bp) | 53,668,190 | 72,145,553 | 41,085,333 |
| Mean Length (bp) | 587 | 635 | 617 |
| N50 (bp) | 800 | 934 | 884 |
| N70 (bp) | 436 | 485 | 464 |
| N90 (bp) | 254 | 262 | 259 |
| GC Content | 44.21% | 44.87% | 44.72% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, B.; Peng, C.; Zhu, Y.; Sun, Y.; Zhao, T.; Huang, Y.; Shi, Q. High Throughput Identification of Novel Conotoxins from the Vermivorous Oak Cone Snail (Conus quercinus) by Transcriptome Sequencing. Int. J. Mol. Sci. 2018, 19, 3901. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123901
Gao B, Peng C, Zhu Y, Sun Y, Zhao T, Huang Y, Shi Q. High Throughput Identification of Novel Conotoxins from the Vermivorous Oak Cone Snail (Conus quercinus) by Transcriptome Sequencing. International Journal of Molecular Sciences. 2018; 19(12):3901. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123901
Chicago/Turabian StyleGao, Bingmiao, Chao Peng, Yabing Zhu, Yuhui Sun, Tian Zhao, Yu Huang, and Qiong Shi. 2018. "High Throughput Identification of Novel Conotoxins from the Vermivorous Oak Cone Snail (Conus quercinus) by Transcriptome Sequencing" International Journal of Molecular Sciences 19, no. 12: 3901. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123901