The C Terminus of the Ribosomal-Associated Protein LrtA Is an Intrinsically Disordered Oligomer

 ,
,  and
and 
Abstract
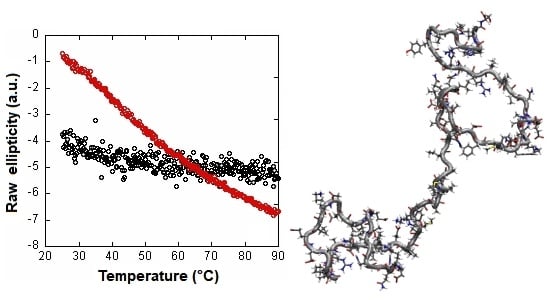
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Isolated C-LrtA Was Intrinsically Disordered in Solution
2.2. Isolated C-LrtA Was Involved in Self-Association Equilibria in Solution
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. Fluorescence
4.4. CD
4.5. NMR Spectroscopy
4.6. Blue-Native PAGE (BN-PAGE)
4.7. Glutaraldehyde Cross-Linking
4.8. SEC
4.9. ITC
4.10. SAXS
4.11. Molecular Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| β-ME | β-mercaptoethanol |
| BN | Blue native |
| CD | Circular dichroism |
| C-LrtA | C-terminal half of LrtA protein (comprising residues 102–191) |
| DOSY | Diffusion ordered spectroscopy |
| GdmCl | Guanidine hydrochloride |
| HPF | Hibernating promoting factor |
| IDP | Intrinsically disordered protein |
| IMAC | Immobilized affinity chromatography |
| IPTG | Isopropyl-β-d-1-tiogalactopyranoside |
| ITC | Isothermal titration calorimetry |
| MD | Molecular dynamics |
| N-LrtA | N-terminal half (residues 1–101) of LrtA protein |
| NMR | Nuclear Magnetic Resonance |
| PAGE | Polyacrylamide gel electrophoresis |
| PFG | Pulse field gradient |
| RaiA | Ribosome associate inhibitor A |
| RNase | Ribonuclease |
| SAXS | Small-angle X-ray scattering |
| SDS | Sodium dodecyl sulphate |
| SEC | Size exclusion chromatography |
| UV | Ultraviolet |
References
- Tan, X.; Varughese, M.; Widger, W.R. A light-repressed transcript found in Synechococcus sp. PCC 7002 is similar to a chloroplast-specific small subunit ribosomal protein and to a transcription modulator protein associated with sigma 54. J. Biol. Chem. 1994, 269, 20905–20912. [Google Scholar] [PubMed]
- Samartzidou, H.; Widger, W.R. Transcriptional and post-transcriptional control of mRNA from lrtA, a light-repressed transcript in Synechococcus sp. PC 7002. Plant Physiol. 1998, 117, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Galmozzi, C.V.; Florencio, F.J.; Muro-Pastor, M.I. The cyanobacterial ribosomal-associated protein LrtA is involved in post-stress survival in Synechocystis sp. PCC 6803. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Wada, A. The 100S ribosome: Ribosomal hibernation induced by stress. Wiley Interdiscipl. Rev. RNA 2014, 5, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Khusainov, I.; Vicens, Q.; Ayupov, R.; Usachev, K.; Myasnikov, A.; Simonetti, A.; Validov, S.; Kieffer, B.; Yuspova, G.; Yusupov, M.; et al. Structures and dynamics of hibernating ribosomes from Staphylococcus aureus mediated by intermolecular interactions of HPF. EMBO J. 2017, 36, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Starosta, A.L.; Lasak, J.; Jung, K.; Wilson, D.N. The bacterial translation stress response. FEMS Microbiol. Rev. 2014, 38, 1172–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agafonov, D.E.; Spirin, A.S. The ribosome-associated inhibitor A reduces translation errors. Biochem. Biophys. Res. Commun. 2004, 320, 354–358. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Blaha, G.M.; Steitz, T.A. How hibernation factors RMF, HPF and YfiA turn off protein synthesis. Science 2012, 336, 915–918. [Google Scholar] [CrossRef]
- Ueta, M.; Yoshida, H.; Wada, C.; Baba, T.; Mori, H.; Wada, A. Ribosome binding proteins YHbH and YfiA have opposite functions during 100S formation in the stationary phase of Escherichia coli. Genes Cells 2005, 10, 1103–1112. [Google Scholar] [CrossRef]
- Ueta, M.; Ohniwa, R.L.; Yoshida, H.; Maki, Y.; Wada, C.; Wada, A. Role of HPF (hibernation promoting factor) in translational activity in Escherichia coli. J. Biochem. 2008, 143, 425–433. [Google Scholar] [CrossRef]
- De Bari, H.; Berry, E.A. Structure of Vibrio cholerae ribosome hibernation factor. Acta Cryst. Sect. F 2013, 69, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.M.; Sevilla, P.; Cámara-Artigas, A.; Hernández-Cifre, J.G.; Rizzuti, B.; Florencio, F.J.; Muro-Pastor, M.I.; García de la Torre, J.; Neira, J.L. The Cyanobacterial ribosomal-associated protein LrtA from Synechocystis sp. PCC 6803 is an oligomeric protein in solution with chameleonic sequence properties. Int. J. Mol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W. Circular dichroism. Methods Enzymol. 1995, 246, 34–71. [Google Scholar] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Chemes, L.B.; Alonso, L.G.; Noval, M.G.; de Prat-Gay, G. Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods Mol. Biol. 2012, 895, 387–404. [Google Scholar] [PubMed]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analysis from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–w673. [Google Scholar] [CrossRef]
- Receveur-Bréchot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins 2006, 62, 24–45. [Google Scholar]
- Neira, J.L. Fluorescence, circular dichroism and mass spectrometry as tools to study virus structure. Subcell. Biochem. 2013, 68, 177–202. [Google Scholar]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry; W. H. Freeman: New York, NY, USA, 1980. [Google Scholar]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Rizzuti, B.; Iovanna, J.L. Determinants of the pKa values of ionizable residues in an intrinsically disordered protein. Arch. Biochem. Biophys. 2016, 598, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Cozza, C.; Neira, J.L.; Florencio, F.J.; Muro-Pastor, M.I.; Rizzuti, B. Intrinsically disordered inhibitor of glutamine synthetase is a functional protein with random-coil-like pKa values. Protein Sci. 2017, 26, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Cavanagh, J.F.; Wayne, J.; Palmer, A.G., III; Skelton, N.J. Protein NMR Spectroscopy: Principles and Practice, 1st ed.; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Giudici, A.M.; Molina, M.L.; Ayala, J.L.; Montoya, E.; Renart, M.L.; Fernandez, A.M.; Encinar, J.A.; Ferrer-Montiel, A.V.; Poveda, J.A.; Gonzalez-Ros, J.M. Detergent-labile, supramolecular assemblies of KcsA: Relative abundance and interactions involved. Biochim. Biophys. Acta 2013, 1828, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittig, I.; Beckhaus, T.; Wumaier, Z.; Karas, M.; Schägger, H. Mass estimation of native proteins by blue native electrophoresis: Principles and practical hints. Mol. Cell Proteom. 2010, 9, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Yap, M.F.N. Ribosome hibernation factor promotes Staphylococcal survival and diferentially represses translation. Nucleic Acids Res. 2016, 44, 4881–4893. [Google Scholar] [CrossRef]
- Sigalov, A.B.; Aivazian, D.; Stern, L. Homo-oligomerization of the cytoplasmic domain of the T-cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049–2061. [Google Scholar] [CrossRef]
- Sigalov, A.B.; Zhurauleva, A.V.; Orekhov, V.Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 2007, 89, 419–421. [Google Scholar] [CrossRef] [Green Version]
- Nourse, A.; Mittag, T. The cytoplasmic domain of the T-cell receptor zeta subunit does not form disordered dimers. J. Mol. Biol. 2014, 426, 62–70. [Google Scholar] [CrossRef]
- Rivera-Nájera, L.Y.; Saab-Rincón, G.; Battaglia, M.; Amero, C.; Pulido, N.O.; García-Hernández, E.; Solórzano, R.M.; Reyes, J.L.; Covarrubias, A.A. A group 6 late embryogenesis abundant protein from common bean is a disordered protein with extended helical structure and oligomer-forming properties. J. Biol. Chem. 2014, 289, 31995–32009. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.M.; Sousa, F.J.R.; Mohana-Borges, R.; Walker, G.C. Regulation of Escherichia coli SOS mutagenesis by dimeric intrinsically disordered umuD gene products. Proc. Natl. Acad. Sci USA 2008, 105, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Martínez-Rodríguez, S.; Hernández-Cifre, J.G.; Cámara-Artigas, A.; Clemente, P.; Peralta, S.; Fernández-Moreno, M.Á.; Garesse, R.; García de la Torre, J.; Rizzuti, B. Human COA3 is an oligomeric highly flexible protein in solution. Biochemistry 2016, 55, 6209–6220. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Román-Trufero, M.; Contreras, L.M.; Prieto, J.; Singh, G.; Barrera, F.N.; Renart, M.L.; Vidal, M. The transcriptional repressor RYBP is a natively unfolded protein which folds upon binding to DNA. Biochemistry 2009, 48, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Batra-Safferling, R.; Abarca-Heidermann, K.; Körscehn, H.G.; Tziatzios, C.; Stoldt, M.; Budyak, I.; Willbold, D.; Schwalbe, H.; Klein-Seetharaman, J.; Kaupp, U.B. Glutamic acid-rich proteins of rod photoreceptors are natively unfolded. J. Biol. Chem. 2006, 281, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; López, M.B.; Sevilla, P.; Rizzuti, B.; Cámara-Artigas, A.; Vidal, M.; Iovanna, J.L. The chromatin nuclear protein NUPR1L is intrinsically disordered and binds to the same proteins as its paralogue. Biochem. J. 2018, 475, 2271–2291. [Google Scholar] [CrossRef] [PubMed]
- Fichó, E.; Reményi, I.; Simon, I.; Mészáros, B. MFIB: A repository of protein complexes with mutual folding induced by binding. Bioinformatics 2017, 33, 3682–3684. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation and intrinsic disorder. Cur. Opin. Struct. Biol. 2017, 44, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends. Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness in protein interactions: A historical perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.J. Initial planning: Determination of total protein concentration. In Protein Purification Methods; Harris, E.L.V., Angal, S., Eds.; Oxford University Press: Oxford, UK, 1995; pp. 10–20. [Google Scholar]
- Neira, J.L.; Hornos, F.; Bacarizo, J.; Cámara-Artigas, A.; Gómez, J. The monomeric species of the regulatory domain of Tyrosine Hydroxylase has a low conformational stability. Biochemistry 2016, 55, 6209–6220. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Plenum Press: New York, NY, USA, 1999. [Google Scholar]
- Piotto, M.; Saudek, V.; Sklenar, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1993, 2, 661–665. [Google Scholar] [CrossRef]
- Schagger, H.; von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar] [CrossRef]
- Schagger, H.; Cramer, W.A.; von Jagow, G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 1994, 217, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Neuhoff, V.; Stamm, R.; Pardowitz, I.; Arold, N.; Ehrhardt, W.; Taube, D. Essential problems in quantification of proteins following colloidal staining with coomassie brilliant blue dyes in polyacrylamide gels and their solution. Electrophoresis 1990, 11, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Hammouda, B. Small angle scattering from branched polymers. Macromol. Theory Simul. 2012, 21, 372–381. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Rizzuti, B.; Bartucci, R.; Sportelli, L.; Guzzi, R. Fatty acid binding into the highest affinity site of human serum albumin observed in molecular dynamics simulation. Arch. Biochem. Biophys. 2015, 579, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Evoli, S.; Mobley, D.L.; Guzzi, R.; Rizzuti, B. Multiple binding modes of ibuprofen in human serum albumin identified by absolute binding free energy calculations. Phys. Chem. Chem. Phys. 2016, 18, 32358–32368. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neira, J.L.; Giudici, A.M.; Hornos, F.; Arbe, A.; Rizzuti, B. The C Terminus of the Ribosomal-Associated Protein LrtA Is an Intrinsically Disordered Oligomer. Int. J. Mol. Sci. 2018, 19, 3902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123902
Neira JL, Giudici AM, Hornos F, Arbe A, Rizzuti B. The C Terminus of the Ribosomal-Associated Protein LrtA Is an Intrinsically Disordered Oligomer. International Journal of Molecular Sciences. 2018; 19(12):3902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123902
Chicago/Turabian StyleNeira, José L., A. Marcela Giudici, Felipe Hornos, Arantxa Arbe, and Bruno Rizzuti. 2018. "The C Terminus of the Ribosomal-Associated Protein LrtA Is an Intrinsically Disordered Oligomer" International Journal of Molecular Sciences 19, no. 12: 3902. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123902