Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)?

Abstract
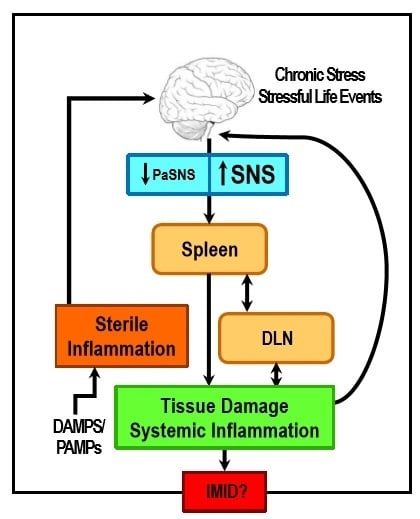
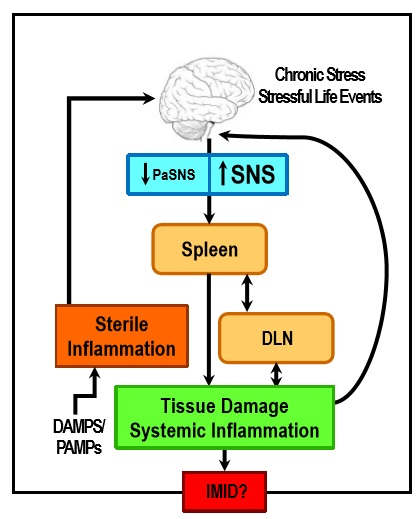
:
1. Introduction
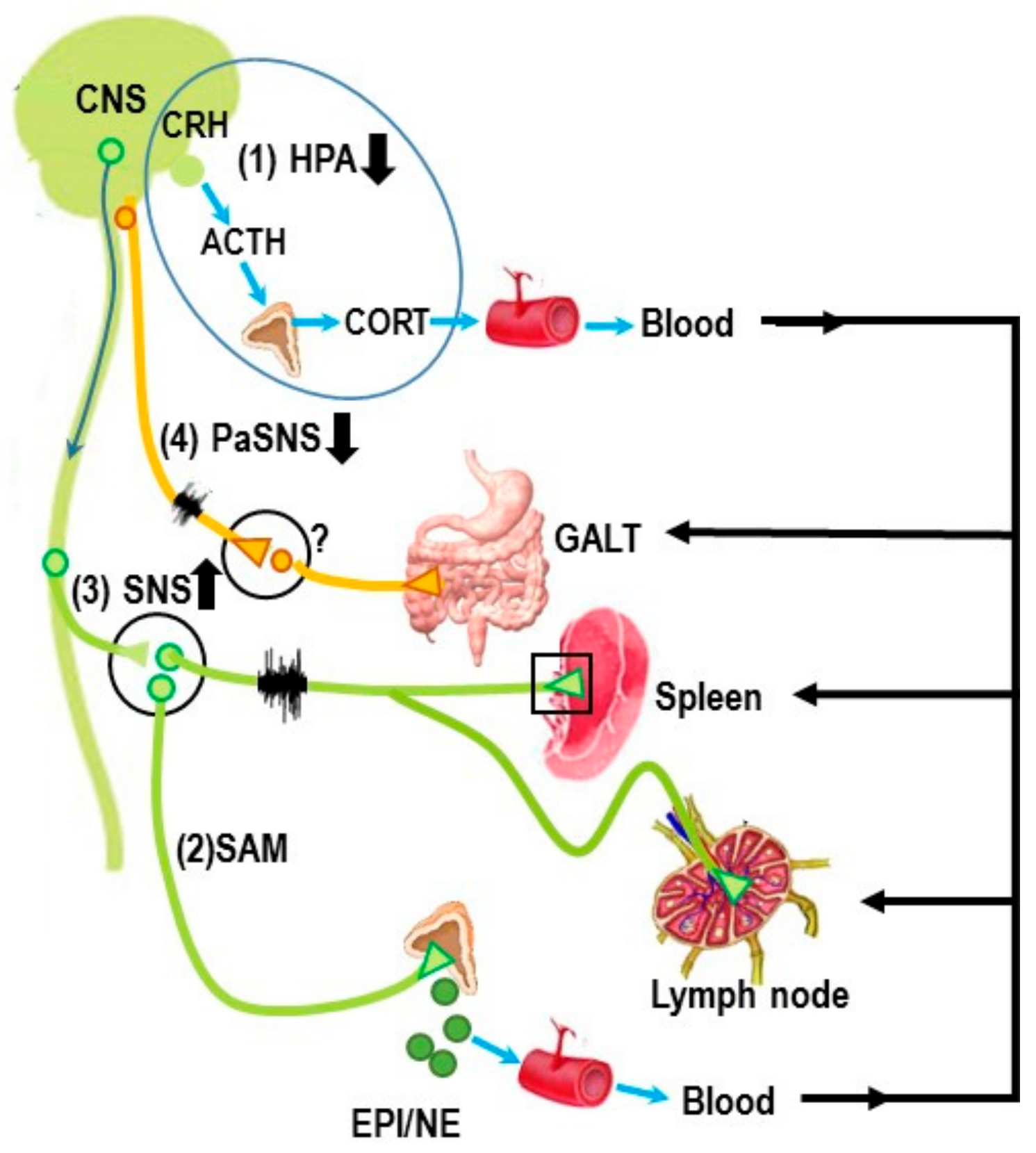
2. Stress and ANS Dysregulation in IMIDs
3. Autonomic Innervation of the Spleen and Systemic Inflammation
Autonomic Circuitry to the Spleen: Anti-Inflammatory Pathways
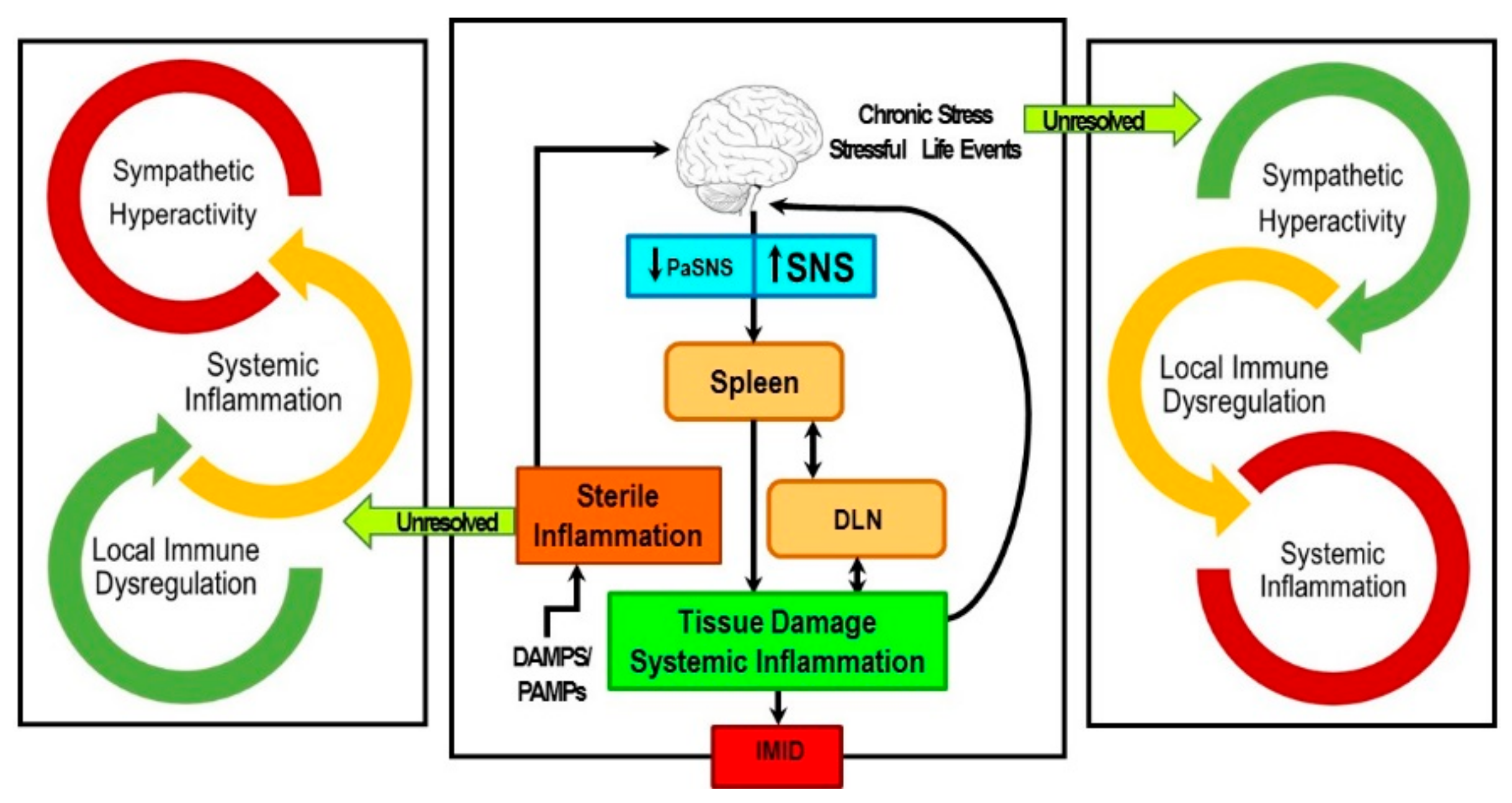
4. Sympathetic Innervation of the Splenic T Cells Is an Important Mediator of Chronic Inflammation in the IMID, Inflammatory Arthritis
5. IMIDs Induce SNS Pathology in Lymphoid Organs
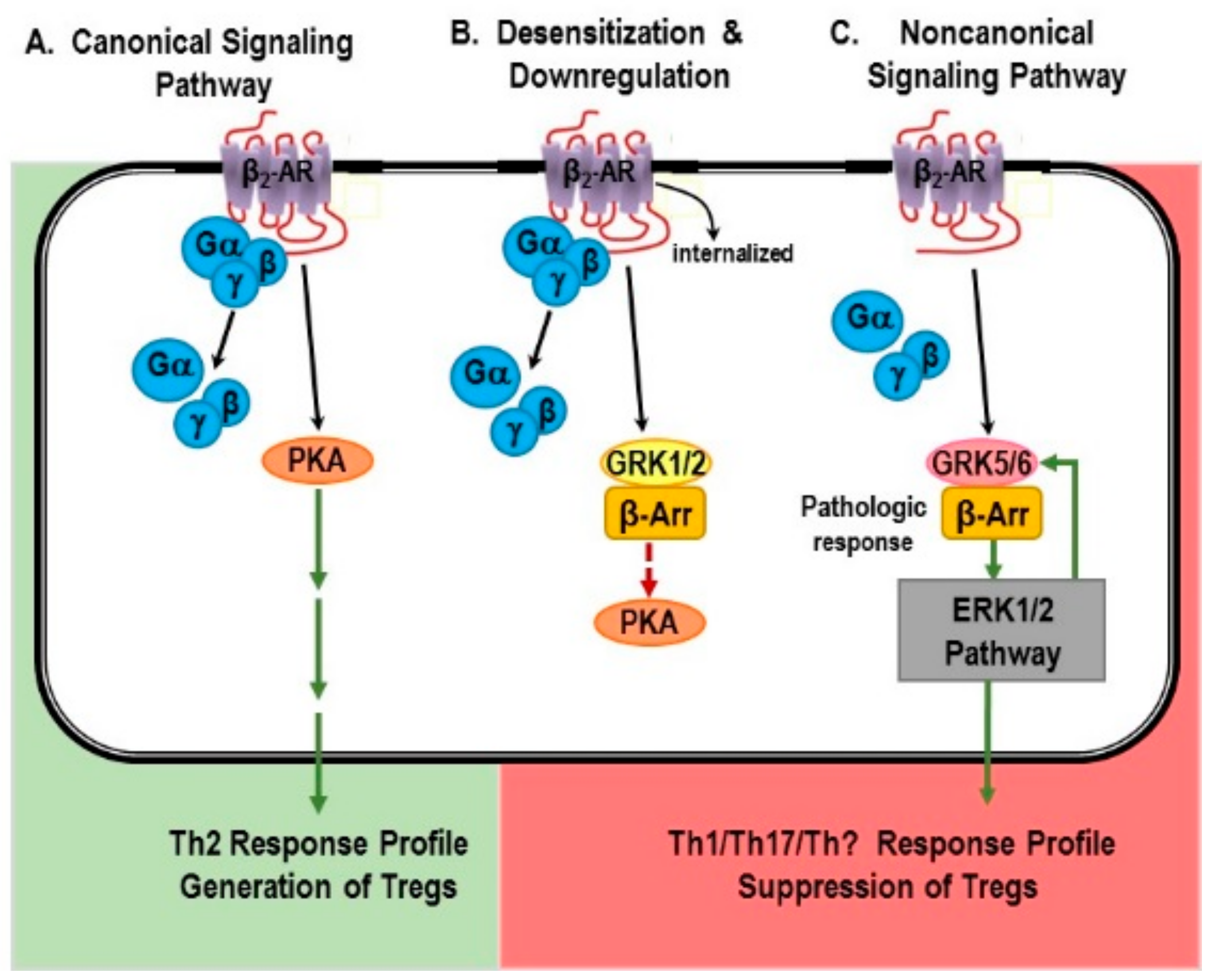
6. β2-Adrenergic Receptors: “Friend or Foe” in IMIDs?
6.1. Canonical Signaling Pathway: “Friend”
6.1.1. Non-Canonical Signaling Pathway: “Friend or Foe”?
6.1.2. The β-AR Switch to Non-Canonical MAPK Pathways
6.1.3. Mechanisms for Receptor “Switching”
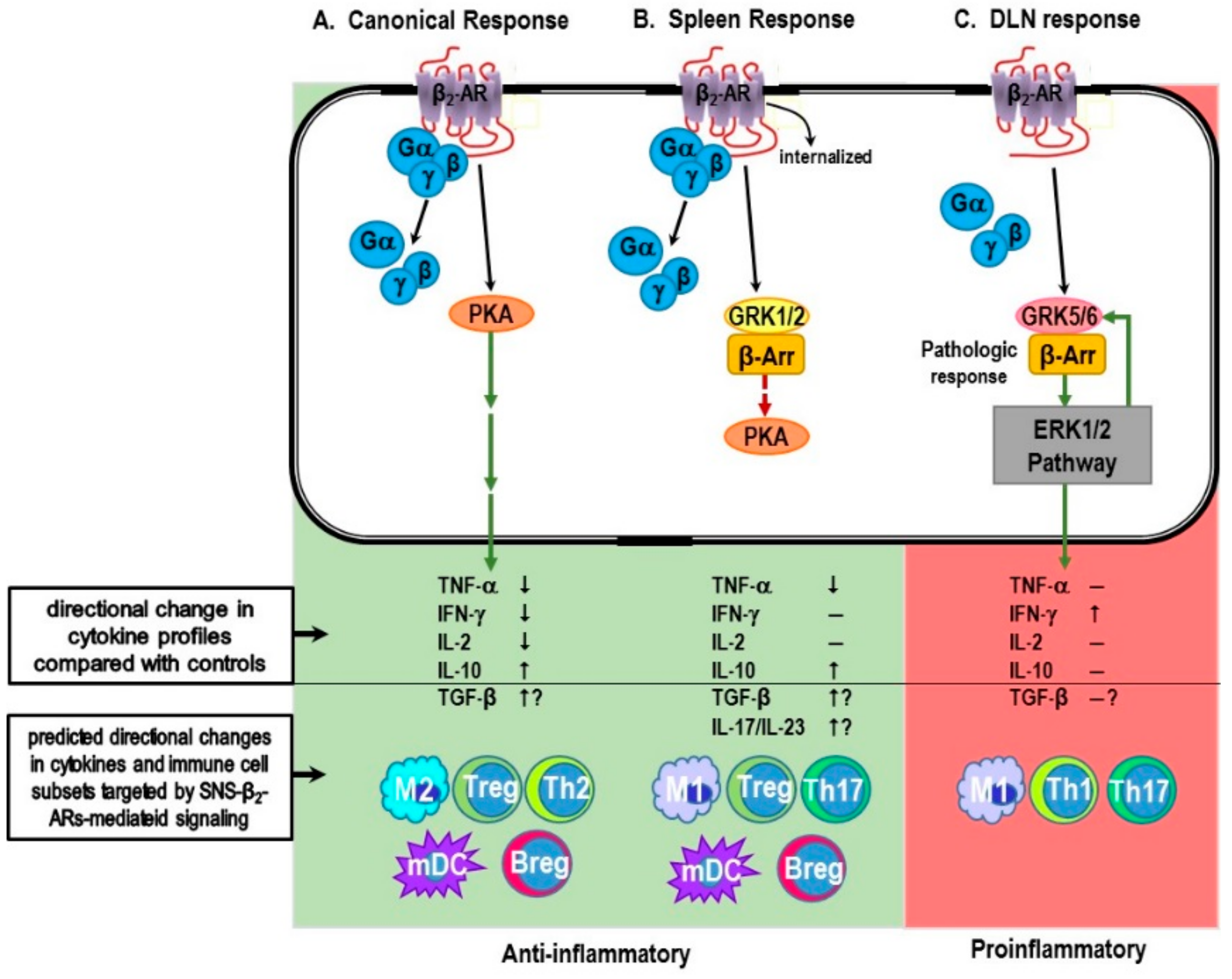
6.2. Evidence for Non-Canonical Pathway Signaling in Immune Cells
6.2.1. Myeloid Cells and Innate Immunity
6.2.2. T Cells and Adaptive Immunity
7. β-AR Promiscuity in the Autoimmune IMID, RA: A Consequence of High SNS Tone in the SNS-Splenic Axis?
8. Concluding Remarks and Potential Significance
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rahman, P.; Inman, R.; El-Gabalawy, H.; Krause, D.O. Pathophysiology and pathogenesis of immune-mediated inflammatory diseases: Commonalities and differences. J. Rheumatol. Suppl. 2010, 85, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D. Autonomic regulation of cellular immune function. Auton. Neurosci. 2014, 182, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.E.; Miyan, J.A. Neural immunoregulation: Emerging roles for nerves in immune homeostasis and disease. Immunol. Today 2000, 21, 281–289. [Google Scholar] [CrossRef]
- McEwen, B.S. Sex, stress and the hippocampus: Allostasis, allostatic load and the aging process. Neurobiol. Aging 2002, 23, 921–939. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Iezzoni, D.G.; Daly, A.; Harris, A.G.; Chrousos, G.P. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation 2005, 12, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Rampton, D.S. The influence of stress on the development and severity of immune-mediated diseases. J. Rheumatol. Suppl. 2011, 88, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Adlan, A.M.; Lip, G.Y.; Paton, J.F.; Kitas, G.D.; Fisher, J.P. Autonomic function and rheumatoid arthritis: A systematic review. Semin. Arthritis Rheum. 2014, 44, 283–304. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M.; Cruser, D.; Podawiltz, A.; Mummert, D.I.; Jones, H.; Mummert, M.E. Psychological Stress and the Cutaneous Immune Response: Roles of the HPA Axis and the Sympathetic Nervous System in Atopic Dermatitis and Psoriasis. Dermatol. Res. Pract. 2012, 2012, 403908. [Google Scholar] [CrossRef] [PubMed]
- Koopman, F.A.; Stoof, S.P.; Straub, R.H.; Van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. Restoring the balance of the autonomic nervous system as an innovative approach to the treatment of rheumatoid arthritis. Mol. Med. 2011, 17, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Schaible, H.G.; Straub, R.H. Function of the sympathetic supply in acute and chronic experimental joint inflammation. Auton. Neurosci. 2014, 182, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B. Inflammatory bowel diseases: A dysfunction of brain–gut interactions? Minerva Gastroenterol. Dietol. 2013, 59, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Boissé, L.; Chisholm, S.P.; Lukewich, M.K.; Lomax, A.E. Clinical and experimental evidence of sympathetic neural dysfunction during inflammatory bowel disease. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Kasselman, L.J.; Sideris, A.; Bruno, C.; Perez, W.R.; Cai, N.; Nicoletti, J.N.; Wiegand, S.J.; Croll, S.D. BDNF: A missing link between sympathetic dysfunction and inflammatory disease? J. Neuroimmunol. 2006, 175, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Van Gestel, A.J.; Steier, J. Autonomic dysfunction in patients with chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 2010, 2, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Van Gestel, A.J.; Kohler, M.; Clarenbach, C.F. Sympathetic overactivity and cardiovascular disease in patients with chronic obstructive pulmonary disease (COPD). Discov. Med. 2012, 14, 359–368. [Google Scholar] [PubMed]
- Stasi, C.; Orlandelli, E. Role of the brain-gut axis in the pathophysiology of Crohn’s disease. Dig. Dis. 2008, 26, 156–166. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, S.J.; van Middendorp, H.; Stormink, C.; Kraaimaat, F.W.; Joosten, I.; Radstake, T.R.; de Jong, E.M.; Schalkwijk, J.; Donders, A.R.; Eijsbouts, A.; et al. Immune responses to stress in rheumatoid arthritis and psoriasis. Rheumatology 2014, 53, 1844–1848. [Google Scholar] [CrossRef] [PubMed]
- Syngle, A.; Verma, I.; Garg, N.; Krishan, P. Autonomic dysfunction in psoriatic arthritis. Clin. Rheumatol. 2013, 32, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Martínez–Martínez, L.A.; Mora, T.; Vargas, A.; Fuentes–Iniestra, M.; Martínez–Lavín, M. Sympathetic nervous system dysfunction in fibromyalgia, chronic fatigue syndrome, irritable bowel syndrome, and interstitial cystitis: A review of case–control studies. J. Clin. Rheumatol. 2014, 20, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Steinkraus, V.; Steinfath, M.; Stöve, L.; Körner, C.; Abeck, D.; Mensing, H. Beta–adrenergic receptors in psoriasis: Evidence for down–regulation in lesional skin. Arch. Dermatol. Res. 1993, 285, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Issa, M.; Barboi, A.; Jaradeh, S.; Zadvornova, Y.; Skaros, S.; Johnson, K.; Otterson, M.F.; Binion, D.G. Impact of autonomic dysfunction on inflammatory bowel disease. J. Clin. Gastroenterol. 2010, 44, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Mravec, B. Autonomic dysfunction in autoimmune diseases: Consequence or cause? Lupus 2007, 16, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Haghighat, S.; Fatemi, A.; Andalib, S. The autonomic dysfunction in patients with lupus disease: An electrophysiological study. Adv. Biomed. Res. 2016, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Stojanovich, L.; Milovanovich, B.; de Luka, S.R.; Popovich–Kuzmanovich, D.; Bisenich, V.; Djukanovich, B.; Randjelovich, T.; Krotin, M. Cardiovascular autonomic dysfunction in systemic lupus, rheumatoid arthritis, primary Sjögren syndrome and other autoimmune diseases. Lupus 2007, 16, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Jiménez–Dalmaroni, M.J.; Gershwin, M.E.; Adamopoulos, I.E. The critical role of toll–like receptors—From microbial recognition to autoimmunity: A comprehensive review. Autoimmun. Rev. 2016, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Danzer, C.; Mattner, J. Impact of microbes on autoimmune diseases. Arch. Immunol. Ther. Exp. (Warsz) 2013, 61, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Ortega–Hernandez, O.D.; Levin, N.A.; Altman, A.; Shoenfeld, Y. Infectious agents in the pathogenesis of primary biliary cirrhosis. Dis. Markers 2010, 29, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T.; Akira, S. Recognition of nucleic acids by pattern–recognition receptors and its relevance in autoimmunity. Immunol. Rev. 2011, 243, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Agostini, L.; Meylan, E.; Tschopp, J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr. Biol. 2004, 14, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, M.; Saikku, P. Evidence for infectious agents in cardiovascular disease and atherosclerosis. Lancet Infect. Dis. 2002, 2, 11–17. [Google Scholar] [CrossRef]
- Vilagut, L.; Parés, A.; Viñas, O.; Vila, J.; Jiménez de Anta, M.T.; Rodés, J. Antibodies to mycobacterial 65–kD heat shock protein cross–react with the main mitochondrial antigens in patients with primary biliary cirrhosis. Eur. J. Clin. Investig. 1997, 27, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Bellinger, D.L.; Schaller, J.A.; Shewmaker, E.; Osredkar, T.; Lubahn, C. Altered sympathetic–to–immune cell signaling via β2–adrenergic receptors in adjuvant arthritis. Clin. Dev. Immunol. 2013, 2013, 764395. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying β–adrenergic receptor–mediated cross–talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef] [PubMed]
- Killeen, M.E.; Ferris, L.; Kupetsky, E.A.; Falo, L., Jr.; Mathers, A.R. Signaling through purinergic receptors for ATP induces human cutaneous innate and adaptive Th17 responses: Implications in the pathogenesis of psoriasis. J. Immunol. 2013, 190, 4324–4336. [Google Scholar] [CrossRef] [PubMed]
- Gombault, A.; Baron, L.; Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 2013, 3, 414. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [PubMed]
- Von Kügelgen, I.; Allgaier, C.; Schobert, A.; Starke, K. Co-release of noradrenaline and ATP from cultured sympathetic neurons. Neuroscience 1994, 61, 199–202. [Google Scholar] [CrossRef]
- Bennett, M.R.; Farnell, L.; Gibson, W.G.; Lin, Y.Q.; Blair, D.H. Quantal and non-quantal current and potential fields around individual sympathetic varicosities on release of ATP. Biophys. J. 2001, 80, 1311–1328. [Google Scholar] [CrossRef]
- Huston, J.M.; Ochani, M.; Rosas-Ballina, M.; Liao, H.; Ochani, K.; Pavlov, V.A.; Gallowitsch-Puerta, M.; Ashok, M.; Czura, C.J.; Foxwell, B.; et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 2006, 203, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.M.; Wang, H.; Ochani, M.; Ochani, K.; Rosas–Ballina, M.; Gallowitsch–Puerta, M.; Ashok, M.; Yang, L.; Tracey, K.J.; Yang, H. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J. Immunol. 2008, 181, 3535–3539. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Stojanovich, L.; Marisavljevich, D. Stress as a trigger of autoimmune disease. Autoimmun. Rev. 2008, 7, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Stojanovich, L. Autonomic dysfunction in autoimmune rheumatic disease. Autoimmun. Rev. 2009, 8, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Stojanovich, L. Stress and autoimmunity. Autoimmun. Rev. 2010, 9, A271–A276. [Google Scholar] [CrossRef] [PubMed]
- Capellino, S.; Lowin, T.; Angele, P.; Falk, W.; Grifka, J.; Straub, R.H. Increased chromogranin A levels indicate sympathetic hyperactivity in patients with rheumatoid arthritis and systemic lupus erythematosus. J. Rheumatol. 2008, 35, 91–99. [Google Scholar] [PubMed]
- Ciesielczyk, K.; Furgała, A.; Dobrek, Ł.; Juszczak, K.; Thor, P. Altered sympathovagal balance and pain hypersensitivity in TNBS–induced colitis. Arch. Med. Sci. 2017, 13, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Perin, P.C.; Maule, S.; Quadri, R. Sympathetic nervous system, diabetes, and hypertension. Clin. Exp. Hypertens. 2001, 23, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Chida, D.; Hashimoto, O.; Kuwahara, M.; Sagara, H.; Osaka, T.; Tsubone, H.; Iwakura, Y. Increased fat: Carbohydrate oxidation ratio in IL1ra (-/-) mice on a high-fat diet is associated with increased sympathetic tone. Diabetologia 2008, 51, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Maule, S.; Pierangeli, G.; Cevoli, S.; Grimaldi, D.; Gionchetti, P.; Barbara, G.; Rizzello, F.; Stanghellini, V.; Corinaldesi, R.; Campieri, M.; et al. Sympathetic hyperactivity in patients with ulcerative colitis. Clin. Auton. Res. 2007, 17, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Dubost, J.J.; Soubrier, M.; Ristori, J.M.; Guillemot, C.; Bussière, J.L.; Sauvezie, B. Late-onset spondyloarthropathy mimicking reflex sympathetic dystrophy syndrome. Jt. Bone Spine 2003, 70, 226–229. [Google Scholar] [CrossRef]
- Koopman, F.A.; van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. Balancing the autonomic nervous system to reduce inflammation in rheumatoid arthritis. J. Intern. Med. 2017, 282, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Millar, B.A.; Perez, S.; Carter, J.; Wood, C.; ThyagaRajan, S.; Molinaro, C.; Lubahn, C.; Lorton, D. Sympathetic modulation of immunity: Relevance to disease. Cell. Immunol. 2008, 252, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Maestroni, G.J. Sympathetic nervous modulation of the skin innate and adaptive immune response to peptidoglycan but not lipopolysaccharide: Involvement of beta–adrenoceptors and relevance in inflammatory diseases. Brain Behav. Immun. 2008, 22, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Saraswathi, P.V.; Neelambikai, N.; Mahesh, A.; Govindarajan, K. Cardiovascular parasympathetic nervous system dysfunction in female rheumatoid arthritis patients. Indian J. Physiol. Pharmacol. 2013, 57, 23–30. [Google Scholar] [PubMed]
- Scott, G.D.; Fryer, A.D. Role of parasympathetic nerves and muscarinic receptors in allergy and asthma. Chem. Immunol. Allergy 2012, 98, 48–69. [Google Scholar] [CrossRef] [PubMed]
- Kistemaker, L.E.; Gosens, R. Acetylcholine beyond bronchoconstriction: Roles in inflammation and remodeling. Trends Pharmacol. Sci. 2015, 36, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Jaseja, H.; Verma, N. Increased parasympathetic tone as the underlying cause of asthma: A hypothesis. Med. Hypotheses 2010, 74, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.A.; Robinson, A.M.; Jovanovska, V.; Eri, R.; Nurgali, K. Alterations in the distal colon innervation in Winnie mouse model of spontaneous chronic colitis. Cell. Tissue Res. 2015, 362, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Koopman, F.A.; Tang, M.W.; Vermeij, J.; de Hair, M.J.; Choi, I.Y.; Vervoordeldonk, M.J.; Gerlag, D.M.; Karemaker, J.M.; Tak, P.P. Autonomic Dysfunction Precedes Development of Rheumatoid Arthritis: A Prospective Cohort Study. EBioMedicine 2016, 6, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Bywater, E.G. Unilateral rheumatoid arthritis following hemiplegia. Ann. Rheum. Dis. 1962, 21, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Stangenberg, L.; Burzyn, D.; Binstadt, B.A.; Weissleder, R.; Mahmood, U.; Benoist, M.D. Denervation protects limbs from IA via an impact on the microvasculature. Proc. Natl. Acad. Sci. USA 2014, 111, 11419–11424. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.; Farrell, M.; Fitzgerald, O. Mechanism of joint sparing in a patient with unilateral psoriatic arthritis and a longstanding hemiplegia. Br. J. Rheumatol. 1993, 32, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Dolan, A.L. Asymmetric rheumatoid vasculitis in a hemiplegic patient. Ann. Rheum. Dis. 1995, 54, 532. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Sequeira, W. Sparing effect of hemiplegia on scleroderma. Ann. Rheum. Dis. 1990, 49, 999–1000. [Google Scholar] [CrossRef] [PubMed]
- Decaris, E.; Guingamp, C.; Chat, M.; Philippe, L.; Grillasca, J.P.; Abid, A.; Minn, A.; Gillet, P.; Netter, P.; Terlain, B. Evidence for neurogenic transmission inducing degenerative cartilage damage distant from local inflammation. Arthritis Rheum. 1999, 42, 1951–1960. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.; Engan, C.; Schaller, J.; Felten, D.L.; Bellinger, D.L. Local application of capsaicin into the draining lymph nodes attenuates expression of adjuvant-induced arthritis. Neuroimmunomodulation 2000, 7, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Shenker, N.; Haigh, R.; Roberts, E.; Mapp, P.; Harris, N.; Blake, D. A review of contralateral responses to a unilateral inflammatory lesion. Rheumatology 2003, 42, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.R.; Leite, P.E. The Involvement of Parasympathetic and Sympathetic Nerve in the Inflammatory Reflex. J. Cell. Physiol. 2016, 231, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Martelli, D.; McKinley, M.J.; McAllen, R.M. The cholinergic anti–inflammatory pathway: A critical review. Auton. Neurosci. 2014, 182, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Drummond, P.D. Alpha–1 adrenoceptor stimulation triggers axon–reflex vasodilatation in human skin. Auton. Neurosci. 2009, 151, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zhang, W.; Zhao, X.; Chen, X.; Xiao, W.; Zhang, L.; Chen, Y.; Zhu, W. Phenylephrine promotes cardiac fibroblast proliferation through calcineurin–NFAT pathway. Front. Biosci. 2016, 21, 502–513. [Google Scholar]
- Balligand, J.L. Beta3–adrenoreceptors in cardiovascular diseases: New roles for an “old” receptor. Curr. Drug Deliv. 2013, 10, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Perlini, S.; Palladini, G.; Ferrero, I.; Tozzi, R.; Fallarini, S.; Facoetti, A.; Nano, R.; Clari, F.; Busca, G.; Fogari, R.; et al. Sympathectomy or doxazosin, but not propranolol, blunt myocardial interstitial fibrosis in pressure–overload hypertrophy. Hypertension 2005, 46, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Ayala–Lopez, N.; Watts, S.W. New actions of an old friend: Perivascular adipose tissue’s adrenergic mechanisms. Br. J. Pharmacol. 2017, 174, 3454–3465. [Google Scholar] [CrossRef] [PubMed]
- Preite, N.Z.; Nascimento, B.P.; Muller, C.R.; Américo, A.L.; Higa, T.S.; Evangelista, F.S.; Lancellotti, C.L.; Henriques, F.S.; Batista, M.L., Jr.; Bianco, A.C.; et al. Disruption of beta3 adrenergic receptor increases susceptibility to DIO in mouse. J. Endocrinol. 2016, 231, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From inflammation to cancer. Immun. Ageing 2018, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Aging, inflammation and the environment. Exp. Gerontol. 2018, 105, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, J.F.; Brown, E.J. The role of the spleen in resistance to infection. Annu. Rev. Med. 1986, 37, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kraal, G.; Mebius, R. New insights into the cell biology of the marginal zone of the spleen. Int. Rev. Cytol. 2006, 250, 175–215. [Google Scholar] [CrossRef] [PubMed]
- Lopes–Carvalho, T.; Foote, J.; Kearney, J.F. Marginal zone B cells in lymphocyte activation and regulation. Curr. Opin. Immunol. 2005, 17, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Zandvoort, A.; Timens, W. The dual function of the splenic marginal zone: Essential for initiation of anti–TI–2 responses but also vital in the general first–line defense against blood–borne antigens. Clin. Exp. Immunol. 2002, 130, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Girard, J.P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Buettner, M.; Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clin. Exp. Immunol. 2012, 169, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.J.; Weiss, M.L.; Haywood, J.R. The paraventricular nucleus: An important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol. Scand. 2003, 177, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Krebs, L.T.; Stobb, J.W.; Harding, J.W. The angiotensin IV system: Functional implications. Front. Neuroendocrinol. 1995, 16, 23–52. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, R.L.; Guyenet, P.G. C1 neurons: A nodal point for stress? Exp. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.C.; Levesque, J.P.; Ruitenberg, M.J. It takes nerve to fight back: The significance of neural innervation of the bone marrow and spleen for immune function. Semin. Cell Dev. Biol. 2017, 61, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D.; Felten, S.Y.; Felten, D.L. Innervation of lymphoid organs and implications in development, aging, and autoimmunity. Int. J. Immunopharmacol. 1992, 14, 329–344. [Google Scholar] [CrossRef]
- Bellinger, D.L.; Felten, S.Y.; Lorton, D.; Felten, D.L. Origin of noradrenergic innervation of the spleen in rats. Brain Behav. Immun. 1989, 3, 291–311. [Google Scholar] [CrossRef]
- MacNeil, B.J.; Jansen, A.H.; Janz, L.J.; Greenberg, A.H.; Nance, D.M. Peripheral endotoxin increases splenic sympathetic nerve activity via central prostaglandin synthesis. Am. J. Physiol. 1997, 273, R609–R614. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The cholinergic antiinflammatory pathway: A missing link in neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [PubMed]
- Rosas-Ballina, M.; Tracey, K.J. Cholinergic control of inflammation. J. Intern. Med. 2009, 265, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Martelli, D.; Farmer, D.G.; Yao, S.T. The splanchnic anti-inflammatory pathway: Could it be the efferent arm of the inflammatory reflex? Exp. Physiol. 2016, 101, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Nardi, D.; Zhang, M.; Yang, H.; Ombrellino, M.; Tracey, K.J. Role of vagus nerve signaling in CNI–1493–mediated suppression of acute inflammation. Auton. Neurosci. 2000, 85, 141–147. [Google Scholar] [CrossRef]
- Bernik, T.R.; Friedman, S.G.; Ochani, M.; DiRaimo, R.; Ulloa, L.; Yang, H.; Sudan, S.; Czura, C.J.; Ivanova, S.M.; Tracey, K.J. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J. Exp. Med. 2002, 195, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Ochani, M.; Gallowitsch–Puerta, M.; Ochani, K.; Huston, J.M.; Czura, C.J.; Al–Abed, Y.; Tracey, K.J. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc. Natl. Acad. Sci. USA 2006, 103, 5219–5223. [Google Scholar] [CrossRef] [PubMed]
- Bratton, B.O.; Martelli, D.; McKinley, M.J.; Trevaks, D.; Anderson, C.R.; McAllen, R.M. Neural regulation of inflammation: No neural connection from the vagus to splenic sympathetic neurons. Exp. Physiol. 2012, 97, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Vida, G.; Pena, G.; Deitch, E.A.; Ulloa, L. α7–cholinergic receptor mediates vagal induction of splenic norepinephrine. J. Immunol. 2011, 186, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, P.S.; Katz, D.A.; Rosas–Ballina, M.; Levine, Y.A.; Ochani, M.; Valdés–Ferrer, S.I.; Pavlov, V.A.; Tracey, K.J.; Chavan, S.S. α7 nicotinic acetylcholine receptor (α7nAChR) expression in bone marrow–derived non–T cells is required for the inflammatory reflex. Mol. Med. 2012, 18, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D.; Hamill, R.W.; Felten, S.Y.; Felten, D.L. Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: Lack of evidence for cholinergic innervation. Brain Behav. Immun. 1993, 7, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Nance, D.M.; Sanders, V.M. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav. Immun. 2007, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Si, M.L.; Lee, T.J. Presynaptic alpha7–nicotinic acetylcholine receptors mediate nicotine–induced nitric oxidergic neurogenic vasodilation in porcine basilar arteries. J. Pharmacol. Exp. Ther. 2001, 298, 122–128. [Google Scholar] [PubMed]
- Mravec, B. Role of catecholamine-induced activation of vagal afferent pathways in regulation of sympathoadrenal system activity: Negative feedback loop of stress response. Endocr. Regul. 2011, 45, 37–41. [Google Scholar] [PubMed]
- Saper, C.B.; Romanovsky, A.A.; Scammell, T.E. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat. Neurosci. 2012, 15, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, J.M.; Hanly, E.J.; Aurora, A.R.; De Maio, A.; Talamini, M.A. Anesthesia–specific protection from endotoxic shock is not mediated through the vagus nerve. Surgery 2005, 138, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Sweeney, S.; Major, A.; Lindquist, C.A.; Schaller, J.; Washington, C.; Bellinger, D.L. Differences in the injury/sprouting response of splenic noradrenergic nerves in Lewis rats with adjuvant–induced arthritis compared with rats treated with 6–hydroxydopamine. Brain Behav. Immun. 2009, 23, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Lindquist, C.A.; Schaller, J.; Washington, C.; Bellinger, D.L. Changes in the density and distribution of sympathetic nerves in spleens from Lewis rats with adjuvant–induced arthritis suggest that an injury and sprouting response occurs. J. Comp. Neurol. 2005, 489, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Felten, S.Y.; Bellinger, D. Norepinephrine content in primary and secondary lymphoid organs is altered in rats with adjuvant–induced arthritis. Mech. Ageing Dev. 1997, 94, 145–163. [Google Scholar] [CrossRef]
- Miller, L.E.; Jüsten, H.P.; Schölmerich, J.; Straub, R.H. The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J. 2000, 14, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Mundinger, T.O.; Mei, Q.; Foulis, A.K.; Fligner, C.L.; Hull, R.L.; Taborsky, J., Jr. Human Type 1 Diabetes Is Characterized by an Early, Marked, Sustained, and Islet-Selective Loss of Sympathetic Nerves. Diabetes 2016, 65, 2322–2330. [Google Scholar] [CrossRef] [PubMed]
- Taborsky, G.J., Jr.; Mei, Q.; Hackney, D.J.; Mundinger, T.O. The search for the mechanism of early sympathetic islet neuropathy in autoimmune diabetes. Diabetes Obes. Metab. 2014, 16 (Suppl. 1), 96–101. [Google Scholar] [CrossRef] [PubMed]
- Taborsky, G.J., Jr.; Mei, Q.; Bornfeldt, K.E.; Hackney, D.J.; Mundinger, T.O. The p75 neurotrophin receptor is required for the major loss of sympathetic nerves from islets under autoimmune attack. Diabetes 2014, 63, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Lubahn, C.; Klein, N.; Schaller, J.; Bellinger, D.L. Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant–induced arthritis. Brain Behav. Immun. 1999, 13, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Pinho, D.; Morato, M.; Couto, M.R.; Marques-Lopes, J.; Tavares, I.; Albino-Teixeira, A. Does chronic pain alter the normal interaction between cardiovascular and pain regulatory systems? Pain modulation in the hypertensive-monoarthritic rat. J. Pain 2011, 12, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Lubahn, C.L.; Schaller, J.A.; Bellinger, D.L.; Sweeney, S.; Lorton, D. The importance of timing of adrenergic drug delivery in relation to the induction and onset of adjuvant–induced arthritis. Brain Behav. Immun. 2004, 18, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Rauch, L.; Fassold, A.; Lowin, T.; Pongratz, G. Neuronally released sympathetic neurotransmitters stimulate splenic interferon–gamma secretion from T cells in early type II collagen–induced arthritis. Arthritis Rheum. 2008, 58, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Härle, P.; Möbius, D.; Carr, D.J.; Schölmerich, J.; Straub, R.H. An opposing time–dependent immune–modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen–induced arthritis. Arthritis Rheum. 2005, 52, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Härle, P.; Pongratz, G.; Albrecht, J.; Tarner, I.H.; Straub, R.H. An early sympathetic nervous system influence exacerbates collagen–induced arthritis via CD4+CD25+ cells. Arthritis Rheum. 2008, 58, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Lubahn, C.L.; Lorton, D.; Schaller, J.A.; Sweeney, S.J.; Bellinger, D.L. Targeting α– and β–Adrenergic Receptors Differentially Shifts Th1, Th2, and Inflammatory Cytokine Profiles in Immune Organs to Attenuate Adjuvant Arthritis. Front. Immunol. 2014, 5, 346. [Google Scholar] [CrossRef] [PubMed]
- Ebbinghaus, M.; Gajda, M.; Boettger, M.K.; Schaible, H.G.; Bräuer, R. The anti–inflammatory effects of sympathectomy in murine antigen–induced arthritis are associated with a reduction of Th1 and Th17 responses. Ann. Rheum. Dis. 2012, 71, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Mapp, P.I.; Walsh, D.A.; Garrett, N.E.; Kidd, B.L.; Cruwys, S.C.; Polak, J.M.; Blake, D.R. Effect of three animal models of inflammation on nerve fibres in the synovium. Ann. Rheum. Dis. 1994, 53, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Niissalo, S.; Hukkanen, M.; Imai, S.; Törnwall, J.; Konttinen, Y.T. Neuropeptides in experimental and degenerative arthritis. Ann. N. Y. Acad. Sci. 2002, 966, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Velayos, E.E.; Cohen, B.S. The effect of stroke on well–established rheumatoid arthritis. Md. State Med. J. 1972, 21, 38–42. [Google Scholar] [PubMed]
- Syngle, V.; Syngle, A.; Garg, N.; Krishan, P.; Verma, I. Predictors of autonomic neuropathy in rheumatoid arthritis. Auton. Neurosci. 2016, 201, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, V.; Allen, S.C. The effects of age, seropositivity and disease duration on autonomic cardiovascular reflexes in patients with rheumatoid arthritis. Int. J. Clin. Pract. 2004, 58, 740–745. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, B.J.; Jansen, A.H.; Greenberg, A.H.; Nance, D.M. Activation and selectivity of splenic sympathetic nerve electrical activity response to bacterial endotoxin. Am. J. Physiol. 1996, 270, R264–R270. [Google Scholar] [CrossRef] [PubMed]
- Breneman, S.M.; Moynihan, J.A.; Grota, L.J.; Felten, D.L.; Felten, S.Y. Splenic norepinephrine is decreased in MRL–lpr/lpr mice. Brain Behav. Immun. 1993, 7, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Del Rey, A.; Kabiersch, A.; Petzoldt, S.; Besedovsky, H.O. Sympathetic abnormalities during autoimmune processes: Potential relevance of noradrenaline-induced apoptosis. Ann. N. Y. Acad. Sci. 2003, 992, 158–167. [Google Scholar] [PubMed]
- Del Rey, A.; Roggero, E.; Kabiersch, A.; Schäfer, M.; Besedovsky, H.O. The role of noradrenergic nerves in the development of the lymphoproliferative disease in Fas–deficient, lpr/lpr mice. J. Immunol. 2006, 176, 7079–7086. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, J.; Meeus, M.; Derom, E.; Da Silva, H.; Calders, P. Evidence for Autonomic Function and Its Influencing Factors in Subjects With COPD: A Systematic Review. Respir. Care 2015, 60, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.G.; Marsh, F.; Waterhouse, J.C.; Howard, P. Autonomic nerve dysfunction in COPD as assessed by the acetylcholine sweat–spot test. Eur. Respir. J. 1994, 7, 1090–1095. [Google Scholar] [PubMed]
- Schulte, G.; Levy, F.O. Novel aspects of G–protein–coupled receptor signaling—Different ways to achieve specificity. Acta Physiol. 2007, 190, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W. Adenylyl cyclase–A–kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol. Pharmacol. 2009, 76, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.S. Lipid rafts: Now you see them, now you don’t. Nat. Immunol. 2006, 7, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Seven transmembrane receptors: Something old, something new. Acta. Physiol. 2007, 190, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; Sanders, V.M. Norepinephrine and β2–adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol. Rev. 2001, 53, 487–525. [Google Scholar] [PubMed]
- Baillie, G.S.; Houslay, M.D. Arrestin times for compartmentalised cAMP signalling and phosphodiesterase–4 enzymes. Curr. Opin. Cell Biol. 2005, 17, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Shirshev, S.V. Role of Epac proteins in mechanisms of cAMP–dependent immunoregulation. Biochemistry 2011, 76, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.T.; Sallese, M.; Ambrosini, G.; Parruti, G.; de Blasi, A. High expression of β–adrenergic receptor kinase in peripheral human blood leukocytes. J. Biol. Chem. 1992, 267, 6886–6892. [Google Scholar] [PubMed]
- Peng, Y.X.; Shan, J.; Qi, X.Y.; Zhang, S.J.; Ma, S.P.; Wang, N.; Li, J.P.; Xue, H.; Wu, M. The catecholamine–β–adrenoreceptor–cAMP system and prediction of cardiovascular events in hypertension. Clin. Exp. Pharmacol. Physiol. 2006, 33, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Bernardin, G.; Strosberg, A.D.; Bernard, A.; Mattei, M.; Marullo, S. β–Adrenergic receptor–dependent and –independent stimulation of adenylate cyclase is impaired during severe sepsis in humans. Intensive Care Med. 1998, 24, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Notterman, D.A.; Metakis, L. Tumor necrosis factor produces homologous desensitization of lymphocyte β2–adrenergic responses. Circ. Shock 1993, 39, 275–278. [Google Scholar] [PubMed]
- Silverman, H.J.; Lee, N.H.; el–Fakahany, E.E. Effects of canine endotoxin shock on lymphocytic β–adrenergic receptors. Circ. Shock 1990, 32, 293–306. [Google Scholar] [PubMed]
- Baerwald, C.; Graefe, C.; von Wichert, P.; Krause, A. Decreased density of β–adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatoid arthritis. J. Rheumatol. 1992, 19, 204–210. [Google Scholar] [PubMed]
- Baerwald, C.G.; Wahle, M.; Ulrichs, T.; Jonas, D.; von Bierbrauer, A.; von Wichert, P.; Burmester, G.R.; Krause, A. Reduced catecholamine response of lymphocytes from patients with rheumatoid arthritis. Immunobiology 1999, 200, 77–91. [Google Scholar] [CrossRef]
- Wahle, M.; Hanefeld, G.; Brunn, S.; Straub, R.H.; Wagner, U.; Krause, A.; Häntzschel, H.; Baerwald, C.G. Failure of catecholamines to shift T–cell cytokine responses toward a Th2 profile in patients with rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R138. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xue, Q.; Lin, Y.; Wang, L.; Zhu, G.; Zhao, Q.; Chen, Y. Role of β–adrenoceptor at different stages of bronchial asthma. Chin. Med. J. 2001, 114, 1317–1319. [Google Scholar] [PubMed]
- Hataoka, I.; Okayama, M.; Sugi, M.; Inoue, H.; Takishima, T.; Shirato, K. Decrease in β–adrenergic receptors of lymphocytes in spontaneously occurring acute asthma. Chest 1993, 104, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Yoshie, Y.; Nakazawa, T. Hormone–sensitive adenylate cyclase system in lymphocytes from asthmatic patients: Possible defects at the postreceptor sites. Ann. Allergy 1991, 66, 167–172. [Google Scholar] [PubMed]
- Krause, A.; Henrich, A.; Beckh, K.H.; Von Wichert, P.; Baerwald, C. Correlation between density of beta 2–adrenergic receptors on peripheral blood mononuclear cells and serum levels of soluble interleukin–2 receptors in patients with chronic inflammatory diseases. Eur. J. Clin. Investig. 1992, 22 (Suppl. 1), 47–51. [Google Scholar]
- Wahle, M.; Kölker, S.; Krause, A.; Burmester, G.R.; Baerwald, C.G. Impaired catecholaminergic signalling of B lymphocytes in patients with chronic rheumatic diseases. Ann. Rheum. Dis. 2001, 60, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Baerwald, C.; Graefe, C.; Muhl, C.; Von Wichert, P.; Krause, A. Beta 2–adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatic diseases. Eur. J. Clin. Investig. 1992, 22 (Suppl. 1), 42–46. [Google Scholar]
- Takahashi, H.; Kinouchi, M.; Tamura, T.; Iizuka, H. Decreased beta 2–adrenergic receptor–mRNA and loricrin–mRNA, and increased involucrin–mRNA transcripts in psoriatic epidermis: Analysis by reverse transcription–polymerase chain reaction. Br. J. Dermatol. 1996, 134, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Liao, Y.; Tang, Q.; Chen, X.Y.; Zhang, L.J.; Liu, X.X.; Zhong, M. Role of β2–adrenoceptor–β–arrestin2–nuclear factor–κB signal transduction pathway and intervention effects of oxymatrine in ulcerative colitis. Chin. J. Integr. Med. 2012, 18, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta–arrestin: A protein that regulates β–adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β–arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; Ménard, L.; Barak, L.S.; Koch, W.J.; Colapietro, A.M.; Caron, M.G. Role of phosphorylation in agonist–promoted β2–adrenergic receptor sequestration. Rescue of a sequestration–defective mutant receptor by βARK1. J. Biol. Chem. 1995, 270, 24782–24789. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. Beta–arrestin–dependent, G protein–independent ERK1/2 activation by the β2–adrenergic receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Trester–Zedlitz, M.; Burlingame, A.; Kobilka, B.; von Zastrow, M. Mass spectrometric analysis of agonist effects on posttranslational modifications of the β2–adrenoceptor in mammalian cells. Biochemistry 2005, 44, 6133–6143. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β2–adrenergic receptor establish a barcode that encodes differential functions of β–arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 2010, 62, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Loudon, R.P.; Perussia, B.; Benovic, J.L. Differentially regulated expression of the G-protein-coupled receptor kinases, betaARK and GRK6, during myelomonocytic cell development in vitro. Blood 1996, 88, 4547–4557. [Google Scholar] [PubMed]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; LaPorte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. β–arrestin 2: A receptor–regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal–regulated kinases by β–arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. β–Arrestin–dependent formation of β2 adrenergic receptor–Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. β–Arrestin2 is critically involved in CXCR4–mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Park, D.H.; Wessel, T.C.; Song, B.; Wagner, J.A.; Joh, T.H. A dual role for the cAMP–dependent protein kinase in tyrosine hydroxylase gene expression. Proc. Natl. Acad. Sci. USA 1993, 90, 3471–3475. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.; Lohse, M.J.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Desensitization of the isolated β2–adrenergic receptor by beta–adrenergic receptor kinase, cAMP–dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry 1992, 31, 3193–3197. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.M.; Friedman, J.; Qunaibi, E.; Baameur, F.; Moore, R.H.; Clark, R.B. Characterization of agonist stimulation of cAMP–dependent protein kinase and G protein–coupled receptor kinase phosphorylation of the β2–adrenergic receptor using phosphoserine–specific antibodies. Mol. Pharmacol. 2004, 65, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Millman, E.E.; Rosenfeld, J.L.; Vaughan, D.J.; Nguyen, J.; Dai, W.; Alpizar–Foster, E.; Clark, R.B.; Knoll, B.J.; Moore, R.H. Endosome sorting of β2–adrenoceptors is GRK5 independent. Br. J. Pharmacol. 2004, 141, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Szelenyi, J.; Selmeczy, Z.; Brozik, A.; Medgyesi, D.; Magocsi, M. Dual β–adrenergic modulation in the immune system. Stimulus–dependent effect of isoproterenol on MAPK activation and inflammatory mediator production in macrophages. Neurochem. Int. 2006, 49, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Lee, H.T.; Hirshman, C.A.; Xu, D.; Emala, C.W. Lipopolysaccharide–induced sensitization of adenylyl cyclase activity in murine macrophages. Am. J. Physiol. Cell Physiol. 2006, 290, C143–C151. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, Y.; Li, Y.; Wang, J.; Fan, L.; Scott, M.J.; Xiao, G.; Li, S.; Billiar, T.R.; Wilson, M.A.; Fan, J. TLR4 Signaling augments monocyte chemotaxis by regulating G protein–coupled receptor kinase 2 translocation. J. Immunol. 2013, 191, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Loniewski, K.; Shi, Y.; Pestka, J.; Parameswaran, N. Toll–like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol. Immunol. 2008, 45, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Clark, R.B.; Friedman, J.; Dixon, R.A.; Strader, C.D. Identification of a specific domain in the β–adrenergic receptor required for phorbol ester–induced inhibition of catecholamine–stimulated adenylyl cyclase. Mol. Pharmacol. 1990, 38, 289–293. [Google Scholar] [PubMed]
- Yuan, N.; Friedman, J.; Whaley, B.S.; Clark, R.B. cAMP–dependent protein kinase and protein kinase C consensus site mutations of the β–adrenergic receptor. Effect on desensitization and stimulation of adenylyl cyclase. J. Biol. Chem. 1994, 269, 23032–23038. [Google Scholar] [PubMed]
- Carmena, M.J.; García–Paramio, P.; Solano, R.M.; Prieto, J.C. Protein kinase C regulation of the adenylyl cyclase system in rat prostatic epithelium. Prostate 1995, 27, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Deiss, K.; Kisker, C.; Lohse, M.J.; Kristina Lorenz, K. Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein–coupled receptor kinase (GRK) 2. J. Biol. Chem. 2012, 287, 23407–23417. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf–1 to GRK–2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef] [PubMed]
- De Blasi, A.; Parruti, G.; Sallese, M. Regulation of G protein–coupled receptor kinase subtypes in activated T lymphocytes. Selective increase of beta–adrenergic receptor kinase 1 and 2. J. Clin. Investig. 1995, 95, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Benovic, J.L. Regulation of the G protein–coupled receptor kinase GRK5 by protein kinase C. J. Biol. Chem. 1997, 272, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF–κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, M.; Chaudhari, S.; Ding, Y.; Yuan, J.; Stankowska, D.; He, S.; Krishnamoorthy, R.; Cunningham, J.T.; Ma, R. Nuclear factor κB mediates suppression of canonical transient receptor potential 6 expression by reactive oxygen species and protein kinase C in kidney cells. J. Biol. Chem. 2013, 288, 12852–12865. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kim, M.S.; Rhew, J.H.; Park, R.W.; de Crombrugghe, B.; Kim, I.S. Transcriptional regulation of fibronectin gene by phorbol myristate acetate in hepatoma cells: A negative role for NF–κB. J. Cell. Biochem. 2000, 76, 437–451. [Google Scholar] [CrossRef]
- Kin, N.W.; Sanders, V.M. It takes nerve to tell T and B cells what to do. J. Leukoc. Biol. 2006, 79, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.A.; Lee, W.T.; Sanders, V.M. IFN–gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J. Immunol. 2001, 166, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Green, P.G.; Miao, F.J.; Strausbaugh, H.; Heller, P.; Jänig, W.; Levine, J.D. Endocrine and vagal controls of sympathetically dependent neurogenic inflammation. Ann. N. Y. Acad. Sci. 1998, 840, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Bertoli, S.; Seravalle, G. Sympathetic nervous system: Role in hypertension and in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Seravalle, G.; Dell’Oro, R.; Mancia, G. Sympathetic mechanisms, organ damage, and antihypertensive treatment. Curr. Hypertens. Rep. 2011, 13, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.C.; Ganda, A.; Lin, J.; Onat, D.; Harxhi, A.; Iyasere, J.E.; Uriel, N.; Cotter, G. Inflammatory activation: Cardiac, renal, and cardio–renal interactions in patients with the cardiorenal syndrome. Heart Fail. Rev. 2012, 17, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Lambert, E.; Sari, C.I.; Dawood, T.; Nguyen, J.; McGrane, M.; Eikelis, N.; Chopra, R.; Wong, C.; Chatzivlastou, K.; Head, G.; et al. Sympathetic nervous system activity is associated with obesity–induced subclinical organ damage in young adults. Hypertension 2010, 56, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pan, Y.; Gao, L.; Lu, G.; Zhang, J.; Xie, X.; Tong, Z.; Li, B.; Li, G.; Li, W. Dexmedetomidine attenuates pancreatic injury and inflammatory response in mice with pancreatitis by possible reduction of NLRP3 activation and up-regulation of NET expression. Biochem. Biophys. Res. Commun. 2018, 495, 2439–2447. [Google Scholar] [CrossRef] [PubMed]
- Biancardi, V.C.; Bomfim, G.F.; Reis, W.L.; Al-Gassimi, S.; Nunes, K.P. The interplay between Angiotensin II, TLR4 and hypertension. Pharmacol. Res. 2017, 20, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, A.; Li, J.; Wu, C.; Cui, S.; Zhou, Z.; Liu, Y.; Wilcox, C.S.; Hou, F.F. Reno-Cerebral Reflex Activates the Renin-Angiotensin System, Promoting Oxidative Stress and Renal Damage After Ischemia-Reperfusion Injury. Antioxid. Redox Signal. 2017, 27, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Chapleau, M.W.; Harwani, S.C.; Abboud, F.M. The immune system and hypertension. Immunol. Res. 2014, 59, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Rüster, C.; Wolf, G. Adipokines promote chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28 (Suppl. 4), iv8–iv14. [Google Scholar] [CrossRef] [PubMed]
- Panoulas, V.F.; Toms, T.E.; Metsios, G.S.; Stavropoulos-Kalinoglou, A.; Kosovitsas, A.; Milionis, H.J.; Douglas, K.M.; John, H.; Kitas, G.D. Target organ damage in patients with rheumatoid arthritis: The role of blood pressure and heart rate. Atherosclerosis 2010, 209, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Rauch, L.; Rauh, L.; Pongratz, G. Sympathetic inhibition of IL–6, IFN–γ, and KC/CXCL1 and sympathetic stimulation of TGF–β in spleen of early arthritic mice. Brain Behav. Immun. 2011, 25, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Felten, D.L.; Felten, S.Y.; Bellinger, D.L.; Lorton, D. Noradrenergic and peptidergic innervation of secondary lymphoid organs: Role in experimental rheumatoid arthritis. Eur. J. Clin. Investig. 1992, 22 (Suppl. 1), 37–41. [Google Scholar]
- Moreira, M.C.; Pinto, I.S.; Mourão, A.A.; Fajemiroye, J.O.; Colombari, E.; Reis, Â.A.; Freiria–Oliveira, A.H.; Ferreira–Neto, M.L.; Pedrino, G.R. Does the sympathetic nervous system contribute to the pathophysiology of metabolic syndrome? Front. Physiol. 2015, 6, 234. [Google Scholar] [CrossRef] [PubMed]
- Brooks, V.L.; Shi, Z.; Holwerda, S.W.; Fadel, P.J. Obesity–induced increases in sympathetic nerve activity: Sex matters. Auton. Neurosci. 2015, 187, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Straznicky, N.E.; Lambert, G.W.; Lambert, E.A. Neuroadrenergic dysfunction in obesity: An overview of the effects of weight loss. Curr. Opin. Lipidol. 2010, 21, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Oben, J.A.; Yang, S.; Lin, H.; Stafford, E.A.; Soloski, M.J.; Thomas, S.A.; Diehl, A.M. Norepinephrine regulates hepatic innate immune system in leptin–deficient mice with nonalcoholic steatohepatitis. Hepatology 2004, 40, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, N.V.; Pinge-Filho, P.; Panis, C.; Silva, B.R.; Pernomian, L.; Grando, M.D.; Cecchini, R.; Bendhack, L.M.; Martins-Pinge, M.C. Decreased endothelial nitric oxide, systemic oxidative stress, and increased sympathetic modulation contribute to hypertension in obese rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1472–H1480. [Google Scholar] [CrossRef] [PubMed]
- Verschure, D.O.; van Eck-Smit, B.L.; Somsen, G.A.; Verberne, H.J. Cardiac sympathetic activity in hypertrophic cardiomyopathy and Tako–tsubo cardiomyopathy. Clin. Transl. Imaging 2015, 3, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Winklewski, P.J.; Radkowski, M.; Demkow, U. Relevance of immune–sympathetic nervous system interplay for the development of hypertension. Adv. Exp. Med. Biol. 2016, 884, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Thorp, A.A.; Schlaich, M.P. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J. Diabetes Res. 2015, 2015, 341583. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D. Functional sympatholysis in hypertension. Auton. Neurosci. 2015, 188, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez–Iturbe, B.; Pons, H.; Quiroz, Y.; Johnson, R.J. The immunological basis of hypertension. Am. J. Hypertens. 2014, 27, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Aslam, N.; Kedar, A.; Nagarajarao, H.S.; Reddy, K.; Rashed, H.; Cutts, T.; Riely, C.; Abell, T.L. Serum catecholamines and dysautonomia in diabetic gastroparesis and liver cirrhosis. Am. J. Med. Sci. 2015, 350, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Nayak, U.B.; Acharya, V.; Jain, H.; Lenka, S. Clinical assessment of the autonomic nervous system in diabetes mellitus and its correlation with glycemic control. Indian J. Med. Sci. 2013, 67, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Schölmerich, J.; Straub, R.H. Stress and rheumatic diseases. Rheum. Dis. Clin. N. Am. 2000, 26, 737–763. [Google Scholar] [CrossRef]
- Malysheva, O.; Pierer, M.; Wagner, U.; Baerwald, C.G. Stress and rheumatoid arthritis. Z. Rheumatol. 2010, 69, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H. Rheumatoid arthritis: Stress in RA: A trigger of proinflammatory pathways? Nat. Rev. Rheumatol. 2014, 10, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Motivala, S.J.; Khanna, D.; FitzGerald, J.; Irwin, M.R. Stress activation of cellular markers of inflammation in rheumatoid arthritis: Protective effects of tumor necrosis factor alpha antagonists. Arthritis Rheum. 2008, 58, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Van Eden, W.; Wick, G.; Albani, S.; Cohen, I. Stress, heat shock proteins, and autoimmunity: How immune responses to heat shock proteins are to be used for the control of chronic inflammatory diseases. Ann. N. Y. Acad. Sci. 2007, 1113, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, D.C.; Ker, J.A.; Grant, C.C.; Fletcher, L. Autonomic impairment in rheumatoid arthritis. Int. J. Rheum. Dis. 2012, 15, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Vlcek, M.; Rovensky, J.; Eisenhofer, G.; Radikova, Z.; Penesova, A.; Kerlik, J.; Imrich, R. Autonomic nervous system function in rheumatoid arthritis. Cell. Mol. Neurobiol. 2012, 32, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.C.; Geenen, R.; Godaert, G.L.; Bijlsma, J.W.; van Doornen, L.J. Elevated sympathetic nervous system activity in patients with recently diagnosed rheumatoid arthritis with active disease. Clin. Exp. Rheumatol. 2004, 22, 63–70. [Google Scholar] [PubMed]
- Bellinger, D.L.; Silva, D.; Millar, A.B.; Molinaro, C.; Ghamsary, M.; Carter, J.; Perez, S.; Lorton, D.; Lubahn, C.; Araujoa, G.; Thyagarajan, S. Sympathetic nervous system and lymphocyte proliferation in the Fischer 344 rat spleen: A longitudinal study. Neuroimmunomodulation 2008, 15, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.D.; Kozic, B.; Molinaro, C.A.; Thyagarajan, S.; Ghamsary, M.; Lubahn, C.L.; Lorton, D.; Bellinger, D.L. Chronically lowering sympathetic activity protects sympathetic nerves in spleens from aging F344 rats. J. Neuroimmunol. 2012, 247, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. A brief history of G–protein coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2013, 52, 6366–6378. [Google Scholar] [CrossRef] [PubMed]
- Kahsai, A.W.; Xiao, K.; Rajagopal, S.; Ahn, S.; Shukla, A.K.; Sun, J.; Oas, T.G.; Lefkowitz, R.J. Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat. Chem. Biol. 2011, 7, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Barak, L.S.; Xiao, K.; Ahn, S.; Berthouze, M.; Shukla, A.K.; Luttrell, L.M.; Lefkowitz, R.J. Ubiquitination of beta–arrestin links seven–transmembrane receptor endocytosis and ERK activation. J. Biol. Chem. 2007, 282, 29549–29562. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Maudsley, S.; Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, J.; Tuncel, J.; Yamada, H.; Lu, S.; Olofsson, P.; Holmdahl, R. Pristane, a non–antigenic adjuvant, induces MHC class II–restricted, arthritogenic T cells in the rat. J. Immunol. 2006, 176, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Svelander, L.; Müssener, A.; Erlandsson–Harris, H.; Kleinau, S. Polyclonal Th1 cells transfer oil–induced arthritis. Immunology 1997, 91, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Bellinger, D.; Duclos, M.; Felten, S.Y.; Felten, D.L. Application of 6–hydroxydopamine into the fatpads surrounding the draining lymph nodes exacerbates adjuvant–induced arthritis. J. Neuroimmunol. 1996, 64, 103–113. [Google Scholar] [CrossRef]
- Ermirio, R.; Ruggeri, P.; Molinari, C.; Weaver, L.C. Somatic and visceral inputs to neurons of the rostral ventrolateral medulla. Am. J. Physiol. 1993, 265 1 Pt 2, R35–R40. [Google Scholar] [CrossRef] [PubMed]
- Beluli, D.J.; Weaver, L.C. Areas of rostral medulla providing tonic control of renal and splenic nerves. Am. J. Physiol. 1991, 261 6 Pt 2, H1687–H1692. [Google Scholar] [CrossRef] [PubMed]
- Khokhlova, O.N.; Murashev, A.N.; Medvedev, O.S. Role of I(1)–imidazoline receptors and alpha2–adrenoceptors in hemodynamic effects of moxonidine administration into the rostroventrolateral medulla. Bull. Exp. Biol. Med. 2001, 131, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Haxhiu, M.A.; Dreshaj, I.; Schäfer, S.G.; Ernsberger, P. Selective antihypertensive action of moxonidine is mediated mainly by I1–imidazoline receptors in the rostral ventrolateral medulla. J. Cardiovasc. Pharmacol. 1994, 24 (Suppl. 1), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Scalera, A.; Finelli, C. Liver-spleen axis: Intersection between immunity, infections and metabolism. World J. Gastroenterol. 2013, 19, 3534–3542. [Google Scholar] [CrossRef] [PubMed]
- Lykken, J.M.; Candando, K.M.; Tedder, T.F. Regulatory B10 cell development and function. Int. Immunol. 2015, 27, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Gotoh, K.; Seike, M.; Masaki, T.; Honda, K.; Kakuma, T.; Yoshimatsu, H. Role of the spleen in the development of steatohepatitis in high-fat-diet-induced obese rats. Exp. Biol. Med. 2012, 237, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Leite Nde, C.; Monte Oliveira, J.C.; Martins-Pinge, M.C.; Kanunfre, C.C.; Souza, K.L.; Grassiolli, S. Splenectomy attenuates obesity and decreases insulin hypersecretion in hypothalamic obese rats. Metabolism 2015, 64, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, K.; Inoue, M.; Masaki, T.; Chiba, S.; Shimasaki, T.; Matsuoka, K.; Ando, H.; Fujiwara, K.; Fukunaga, N.; et al. Obesity-related chronic kidney disease is associated with spleen-derived IL-10. Nephrol. Dial. Transplant. 2013, 28, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Harmon, E.B.; Porter, J.M.; Porter, J.E. Beta-adrenergic receptor activation in immortalized human urothelial cells stimulates inflammatory responses by PKA-independent mechanisms. Cell. Commun. Signal. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorton, D.; Lubahn, C.; Bellinger, D.L. Potential use of drugs that target neural-immune pathways in the treatment of rheumatoid arthritis and other autoimmune diseases. Curr. Drug Targets Inflamm. Allergy 2003, 2, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Green, A. Projection of Chronic Illness Prevalence and Cost Inflation; RAND Corporation: Santa Monica, CA, USA, October 2000. [Google Scholar]
- The Growing Crisis of Chronic Disease in the United States. Partnership to Fight Chronic Disease. Available online: www.fightchronicdisease.org/sites/default/files/docs/ (accessed on 2 May 2018).
- Thyagarajan, V.; Norman, H.; Alexander, K.A.; Napalkov, P.; Enger, C. Risk of mortality, fatal infection, and fatal malignancy related to use of anti-tumor necrosis factor-α biologics by rheumatoid arthritis patients. Semin. Arthritis Rheum. 2012, 42, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C. Does anti-tumor necrosis factor-α therapy affect risk of serious infection and cancer in patients with rheumatoid arthritis?: A review of longterm data. J. Rheumatol. 2011, 38, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, H.; Amital, H. Anti-TNF therapy: Safety aspects of taking the risk. Autoimmun. Rev. 2011, 10, 563–568. [Google Scholar] [CrossRef] [PubMed]
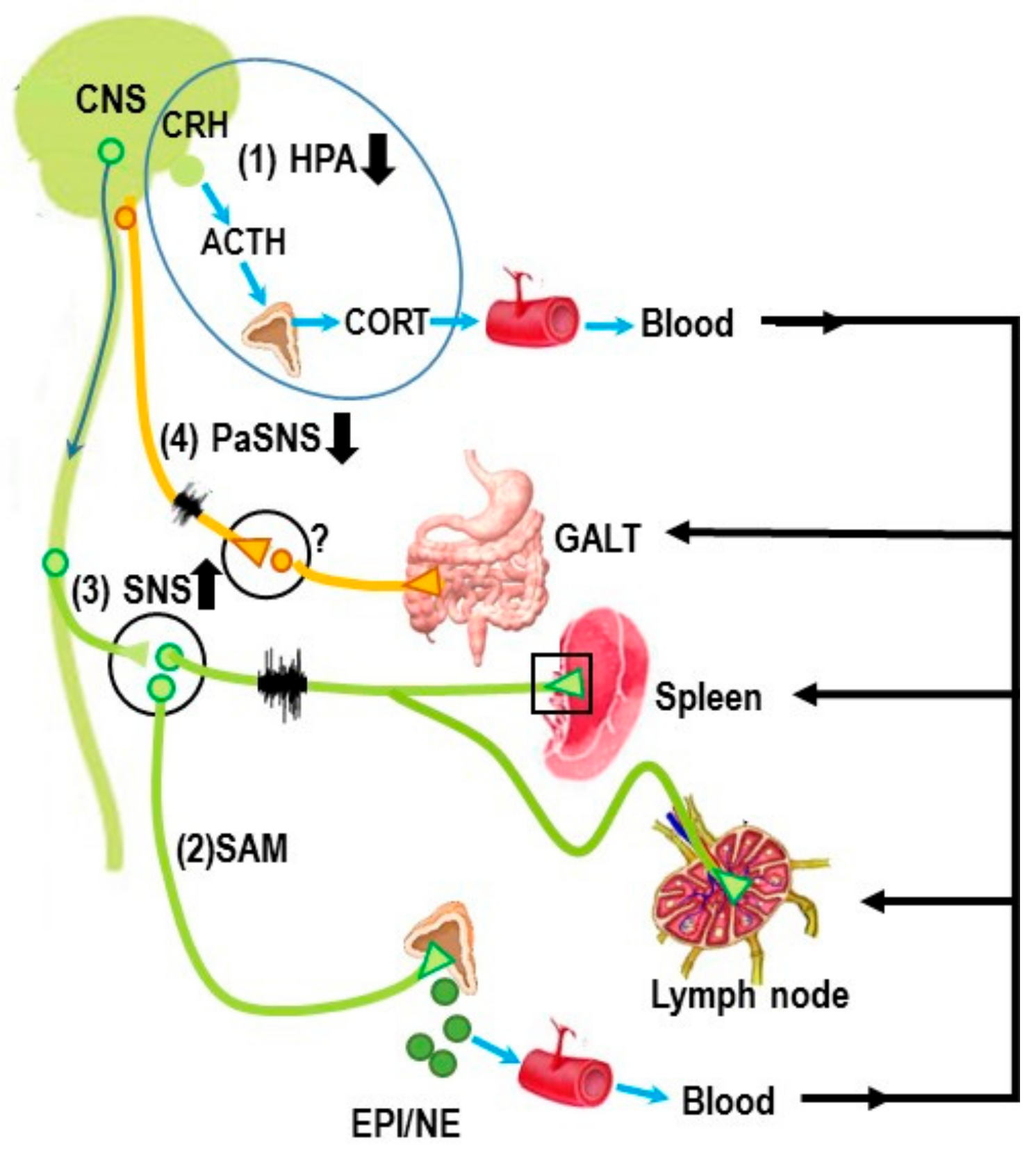



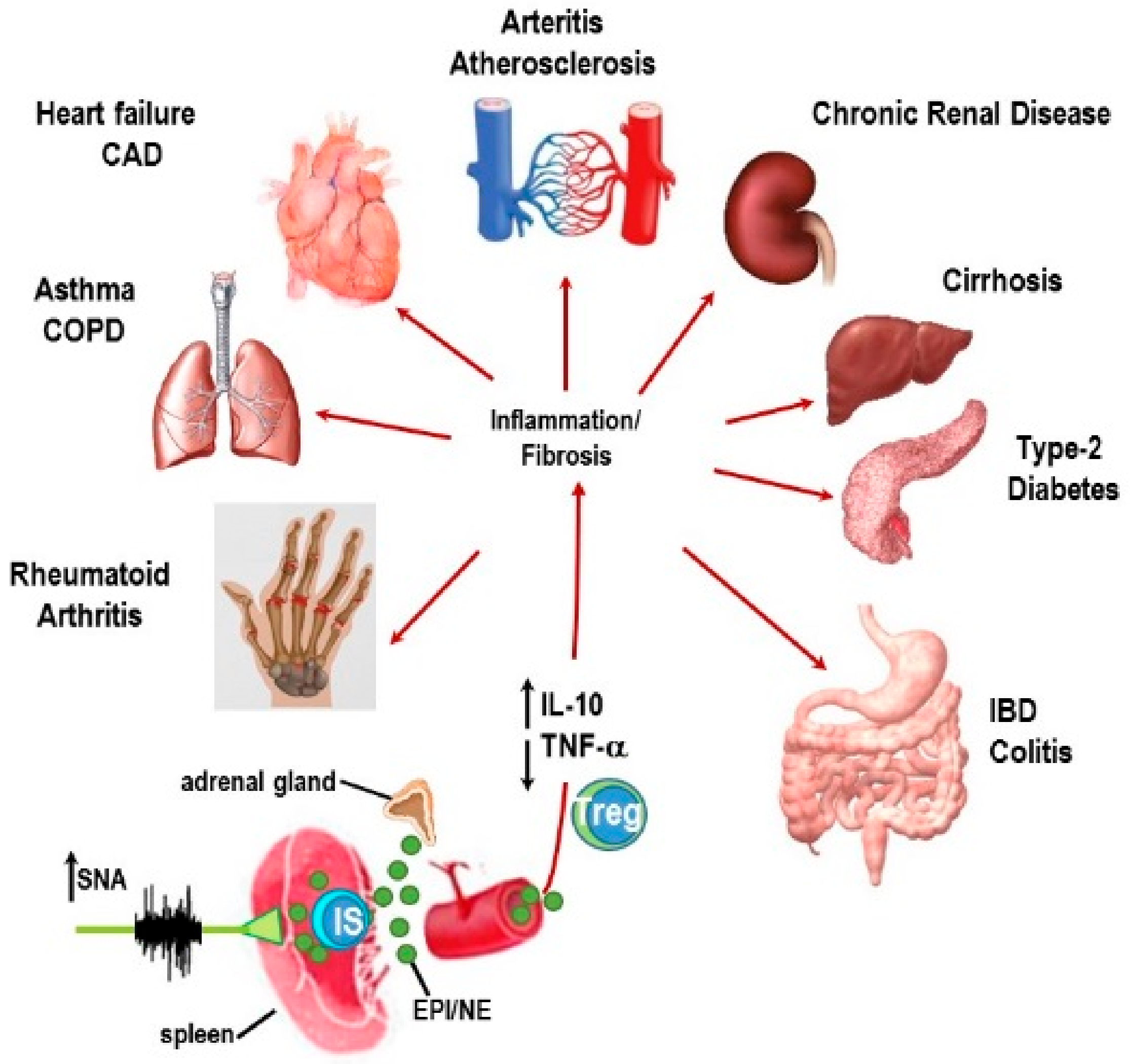

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
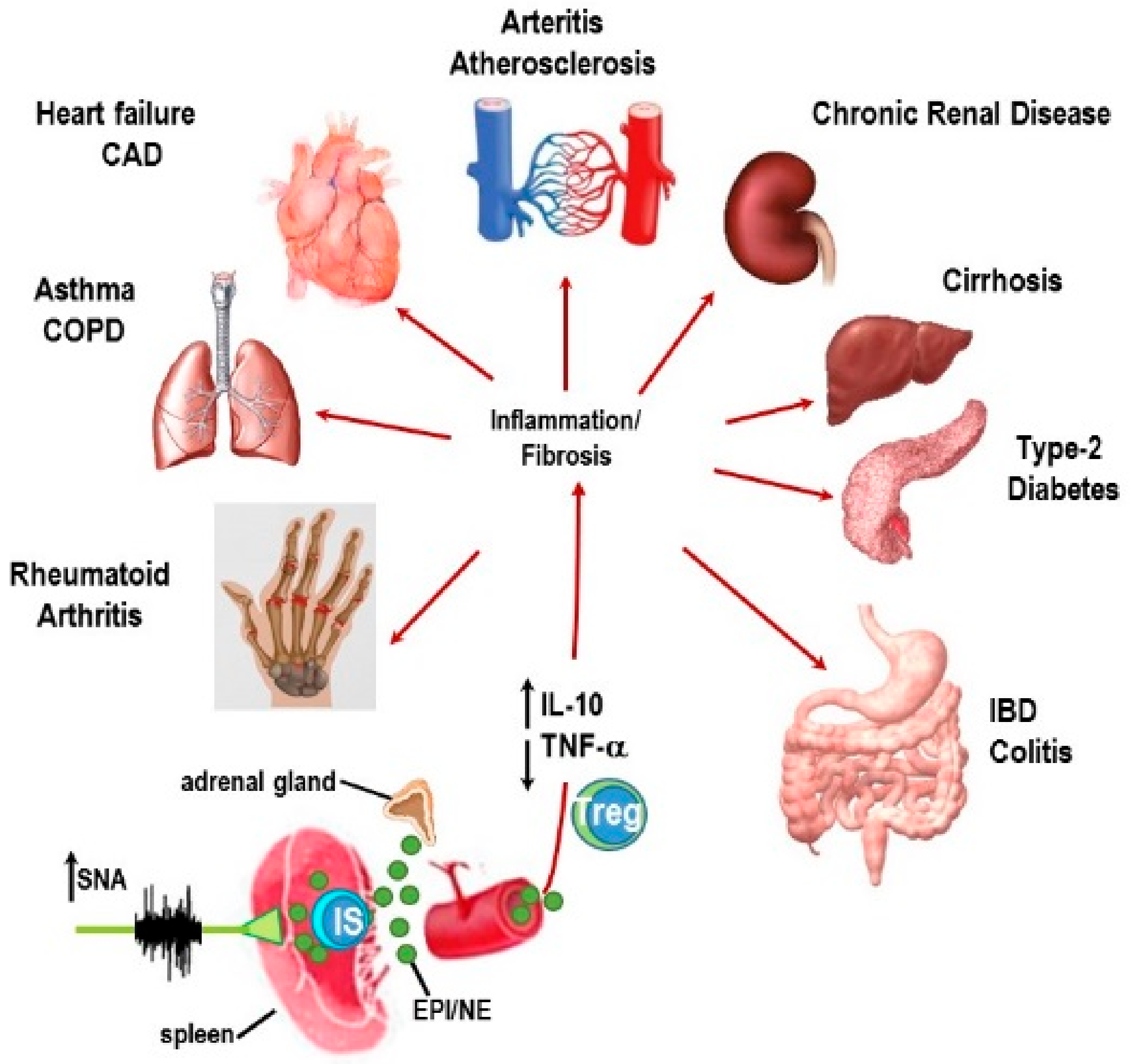{kind=link}
| Examples of IMIDs * | Etiology | Chronic Inflammation | Th Cell Immune | Dysregulation of Stress Pathways | ||
|---|---|---|---|---|---|---|
| Main Target | Systemic | Activation Profile | ANS | HPA | ||
| Ankylosing spondylitis | Autoimmune? | Joints of spine | yes | Th2/Th17 | yes | yes↓ |
| Arteritis/Vasculitis | Autoimmune? | Blood vessels | yes | Th17/Treg | yes | yes↓ |
| Behcet’s disease | Autoimmune? | Blood vessels | yes | Th1/Th17 | yes | yes↓ |
| Inflammatory bowel disease | ||||||
| Crohn’s disease | Unknown | Gastrointestinal tract | yes | Th1/Th17 | yes | yes↓ |
| Ulcerative colitis | Autoimmune? | Colon and rectum | yes | Th1/Th17 | yes | |
| Juvenile idiopathic arthritis | Autoimmune? | Joints | yes | Th1/Th17 | yes | yes↓ |
| Multiple Sclerosis | Autoimmune? | CNS myelin | yes | Th17/Treg | yes | |
| Psoriasis | Autoimmune? | Skin | yes | Th1/Th2 | yes | yes↓ |
| Psoriatic arthritis | Autoimmune? | Skin and joints | yes | Th1/Th17 | yes | yes↓ |
| Rheumatoid arthritis | Autoimmune? | Joints | yes | Th1/Th17 | yes | yes↓ |
| Sarcoidosis | Autoimmune? | Lungs, skin | yes | Th1/Th17 | yes | yes↓ |
| Systemic lupus erythematosus | Autoimmune? | Skin, joints, visceral organs | yes | Th1/Th17 | yes | yes↓ |
| Type 1 diabetes mellitus | Autoimmune? | β-cells in pancreas | yes | Th1/Th17 | yes | – |
| Uveitis | Autoimmune? | Uvea (iris, ciliary body & choroid) | yes | Th17 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellinger, D.L.; Lorton, D. Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)? Int. J. Mol. Sci. 2018, 19, 1188. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041188
Bellinger DL, Lorton D. Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)? International Journal of Molecular Sciences. 2018; 19(4):1188. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041188
Chicago/Turabian StyleBellinger, Denise L., and Dianne Lorton. 2018. "Sympathetic Nerve Hyperactivity in the Spleen: Causal for Nonpathogenic-Driven Chronic Immune-Mediated Inflammatory Diseases (IMIDs)?" International Journal of Molecular Sciences 19, no. 4: 1188. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041188