Activation of Bone Marrow-Derived Cells Angiotensin (Ang) II Type 1 Receptor by Ang II Promotes Atherosclerotic Plaque Vulnerability

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Physiological Parameters of Transplanted 2K1C ApoE−/− Mice
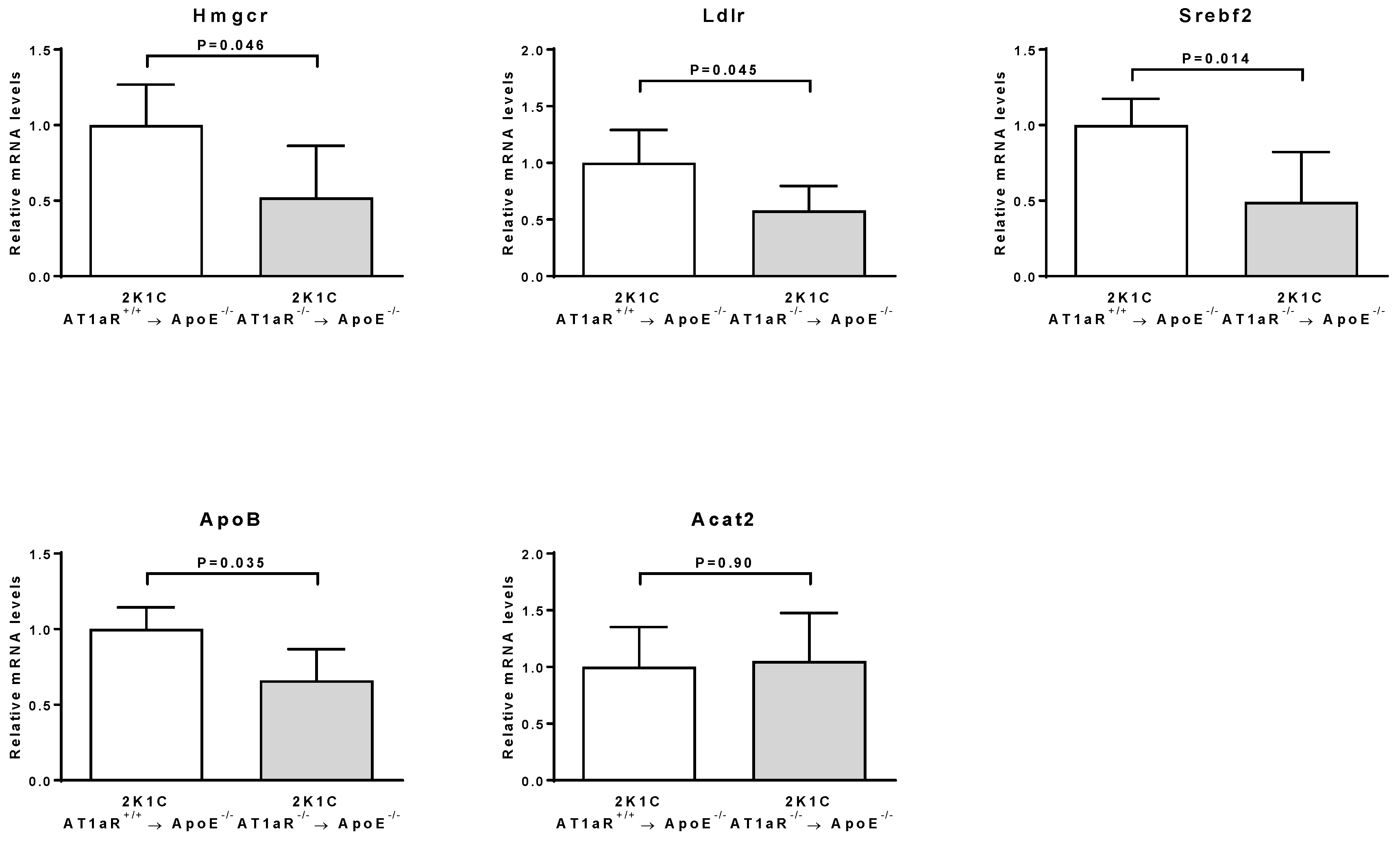
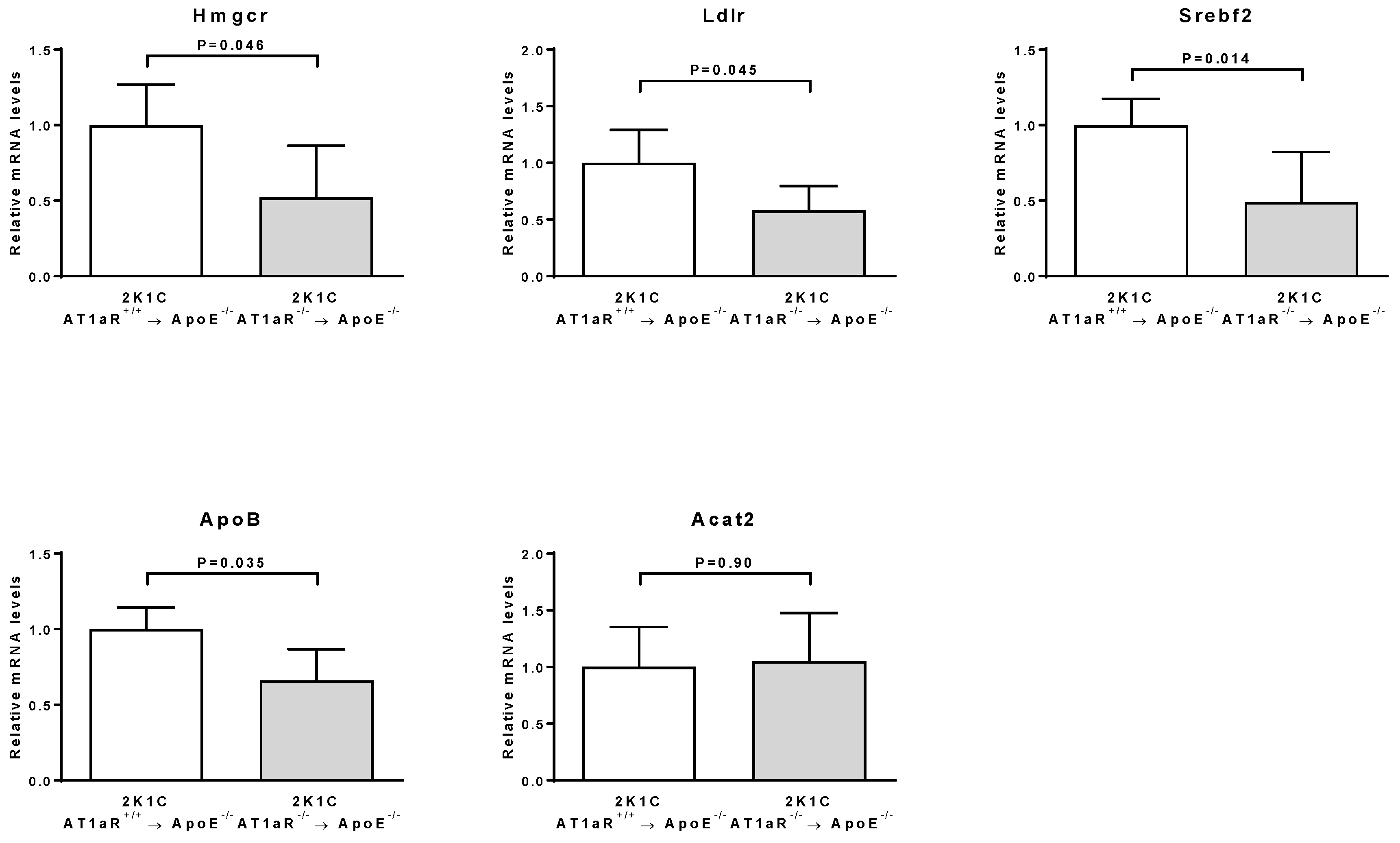
2.2. AT1aR Deficiency in BM-Derived Cells Modulates Cholesterol Metabolism
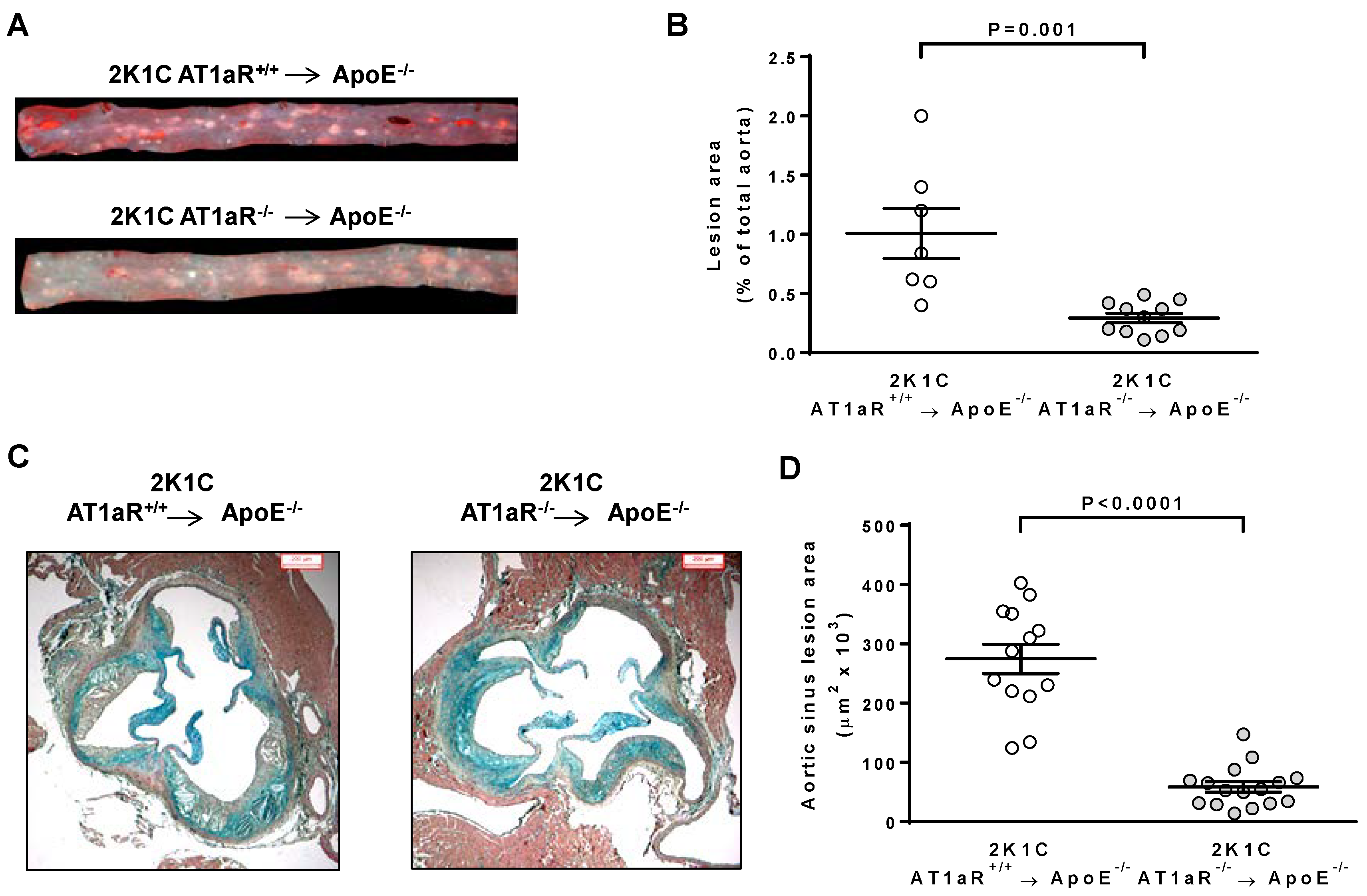
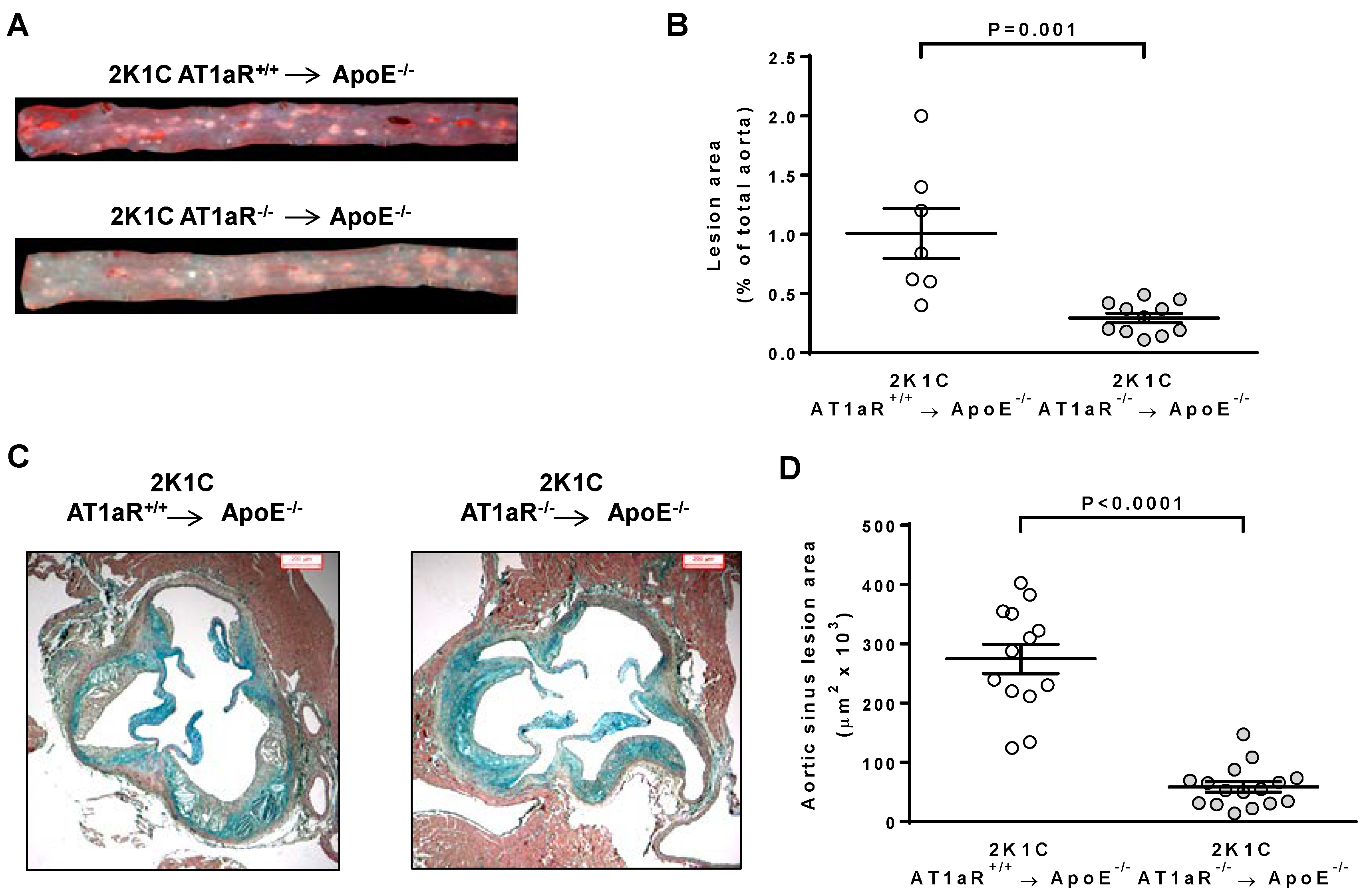
2.3. AT1aR Deficiency in BM-Derived Cells Prevents Ang II-Induced ATS
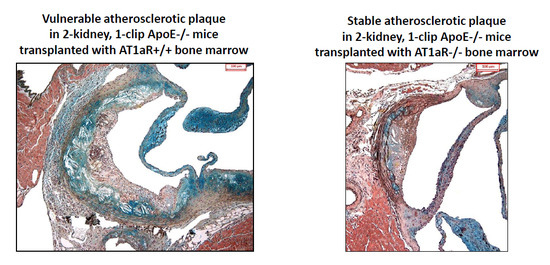
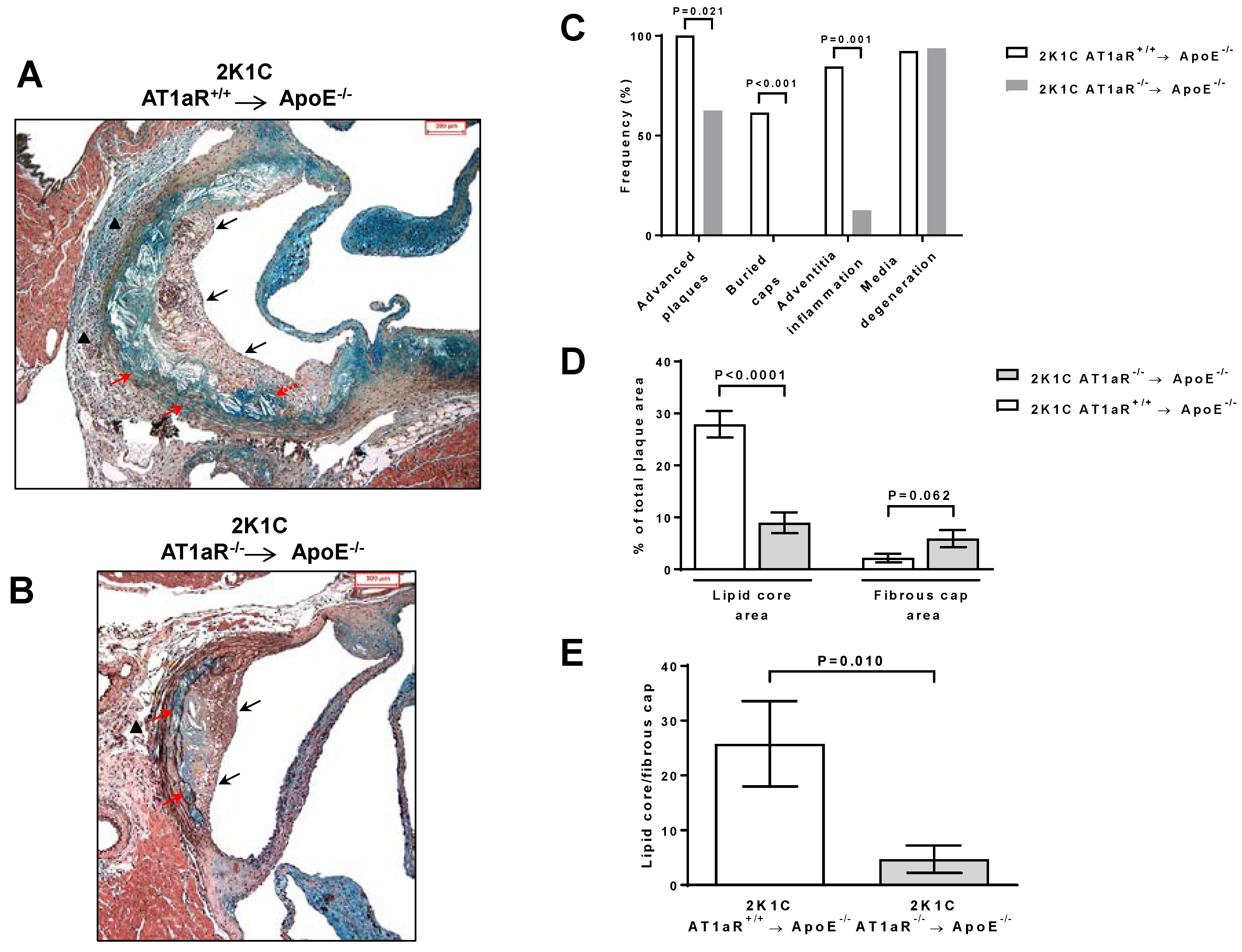
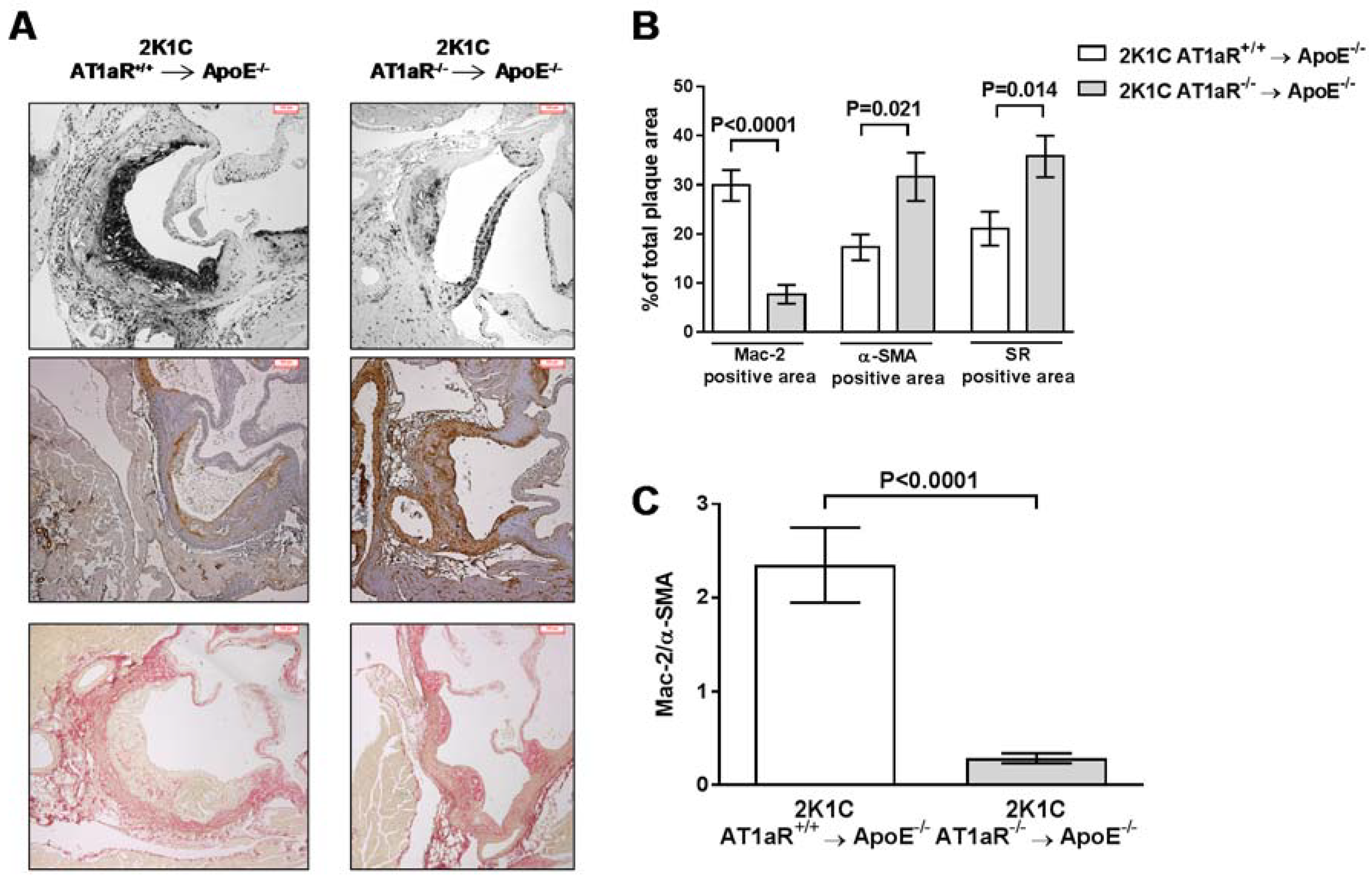
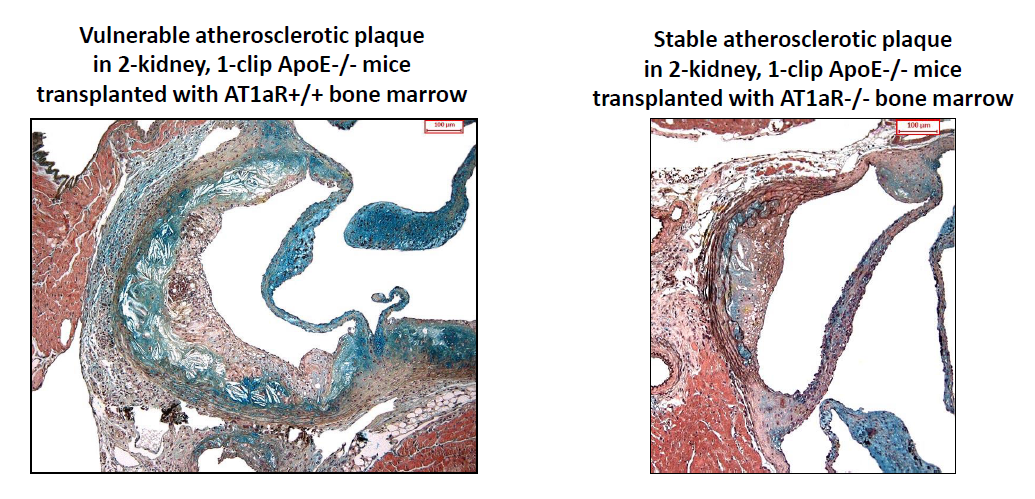
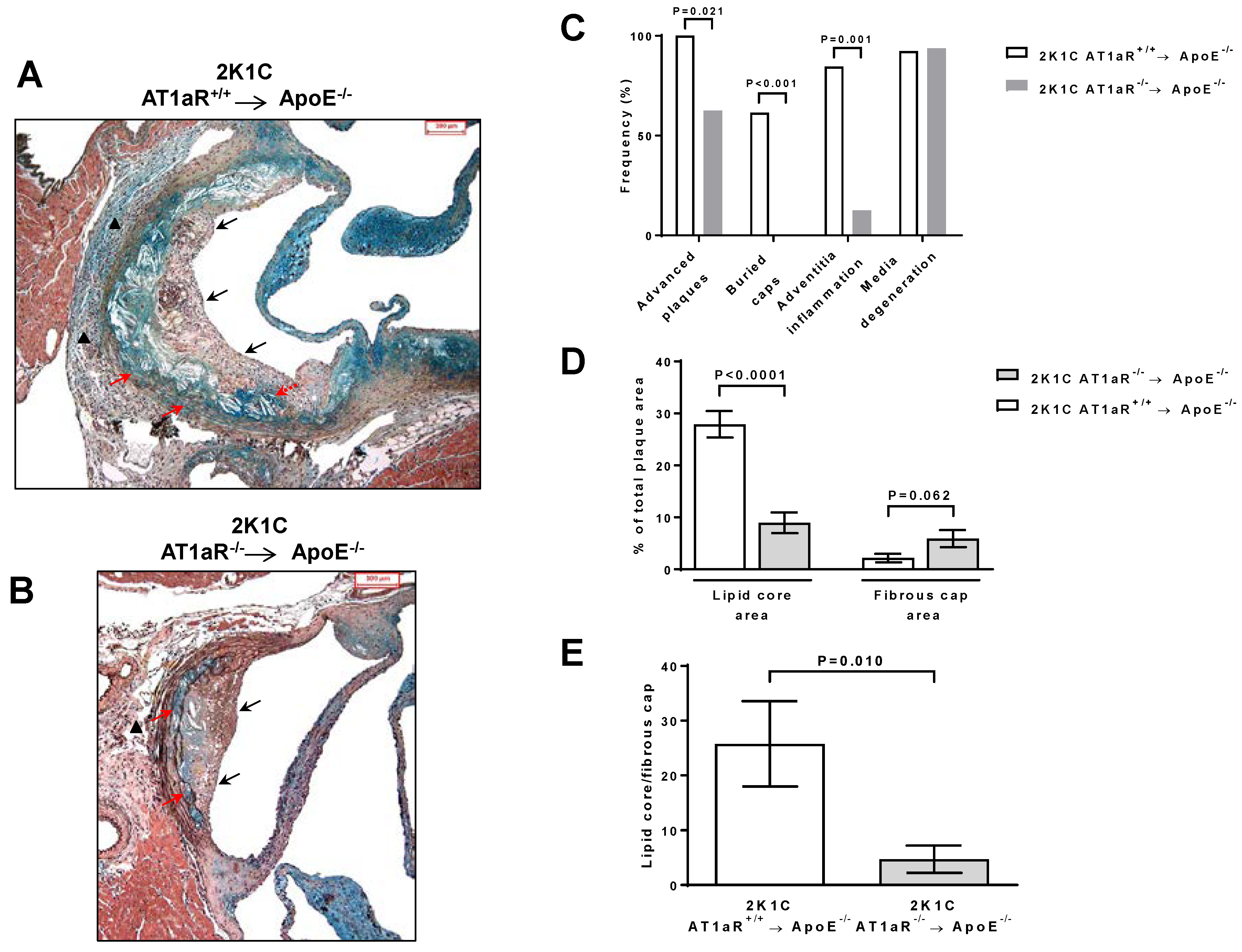
2.4. AT1aR Deficiency in BM-Derived Cells Prevents Ang II-Mediated Plaque Vulnerability
2.5. AT1aR Deficiency in BM-Derived Cells Does Not Modulate Monocyte Recruitment
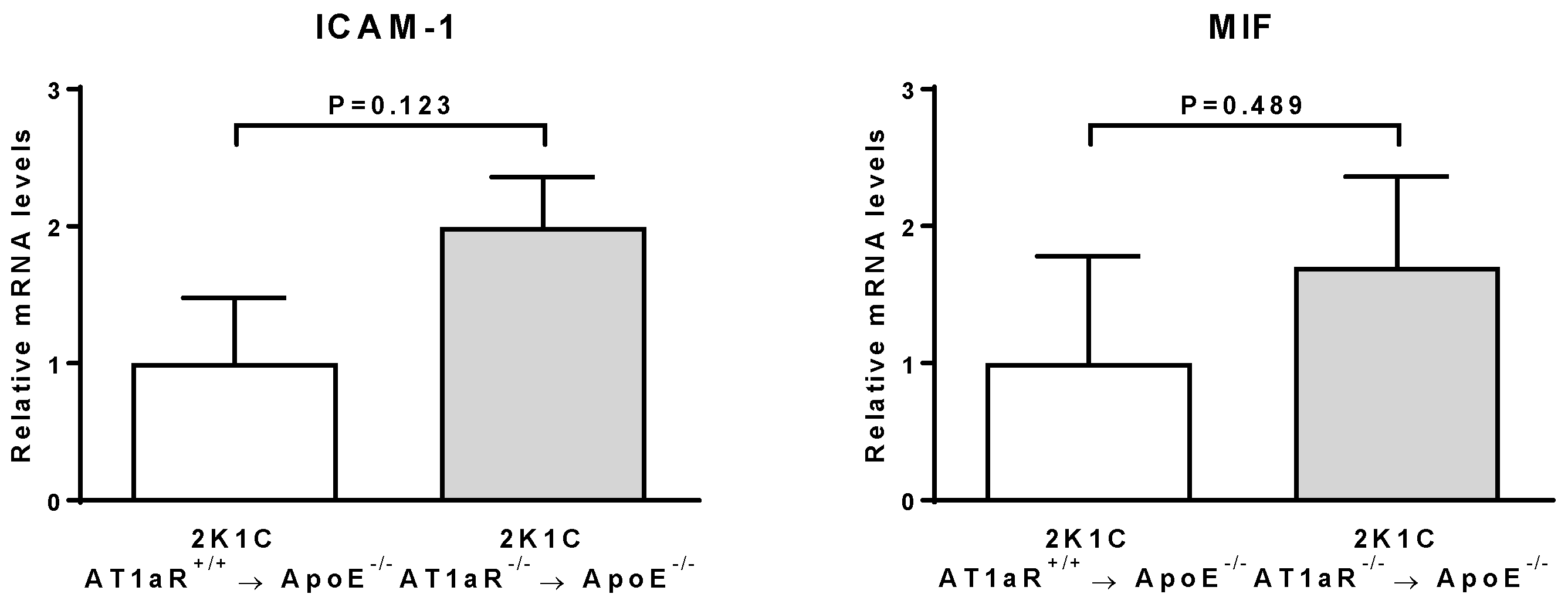
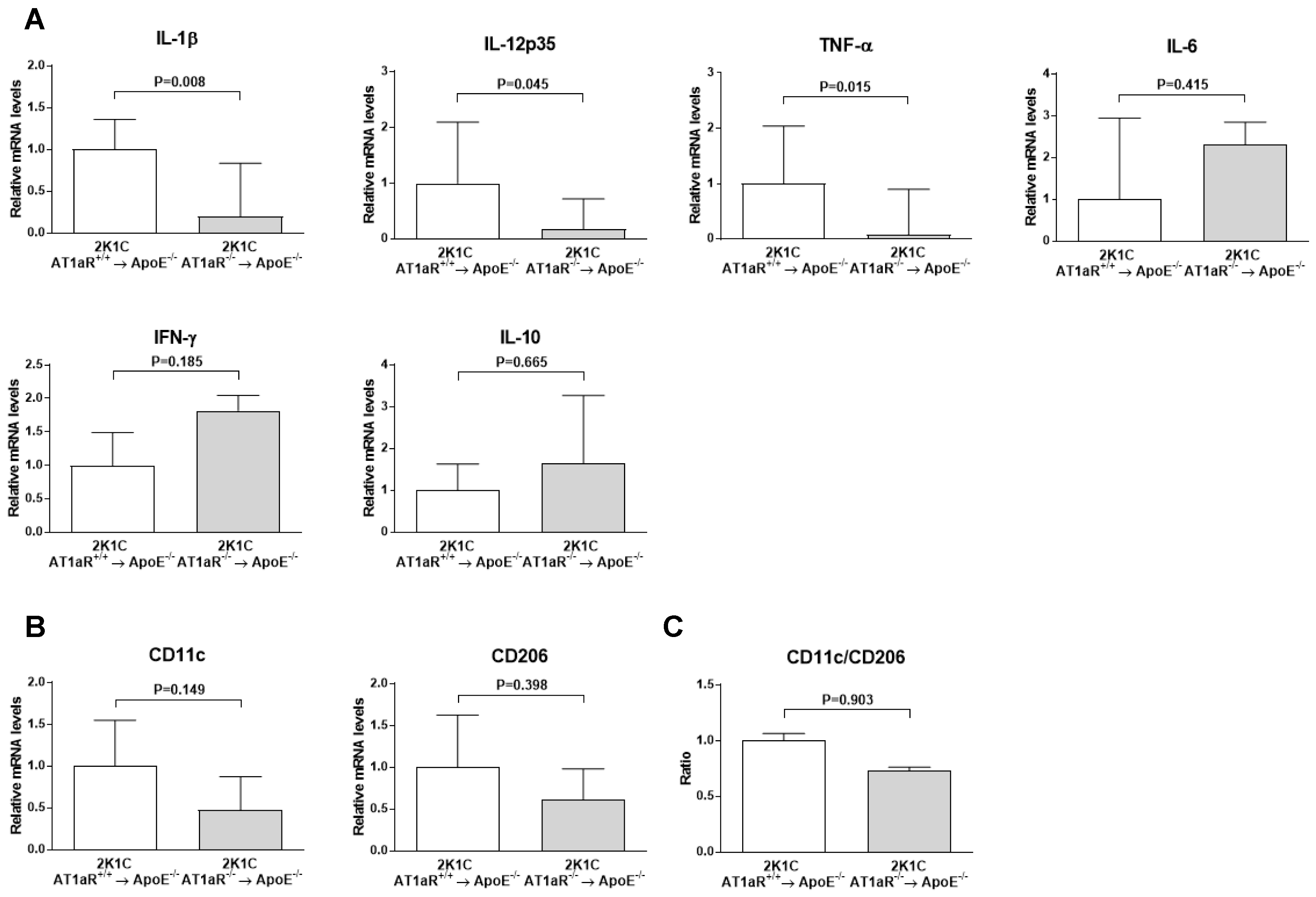
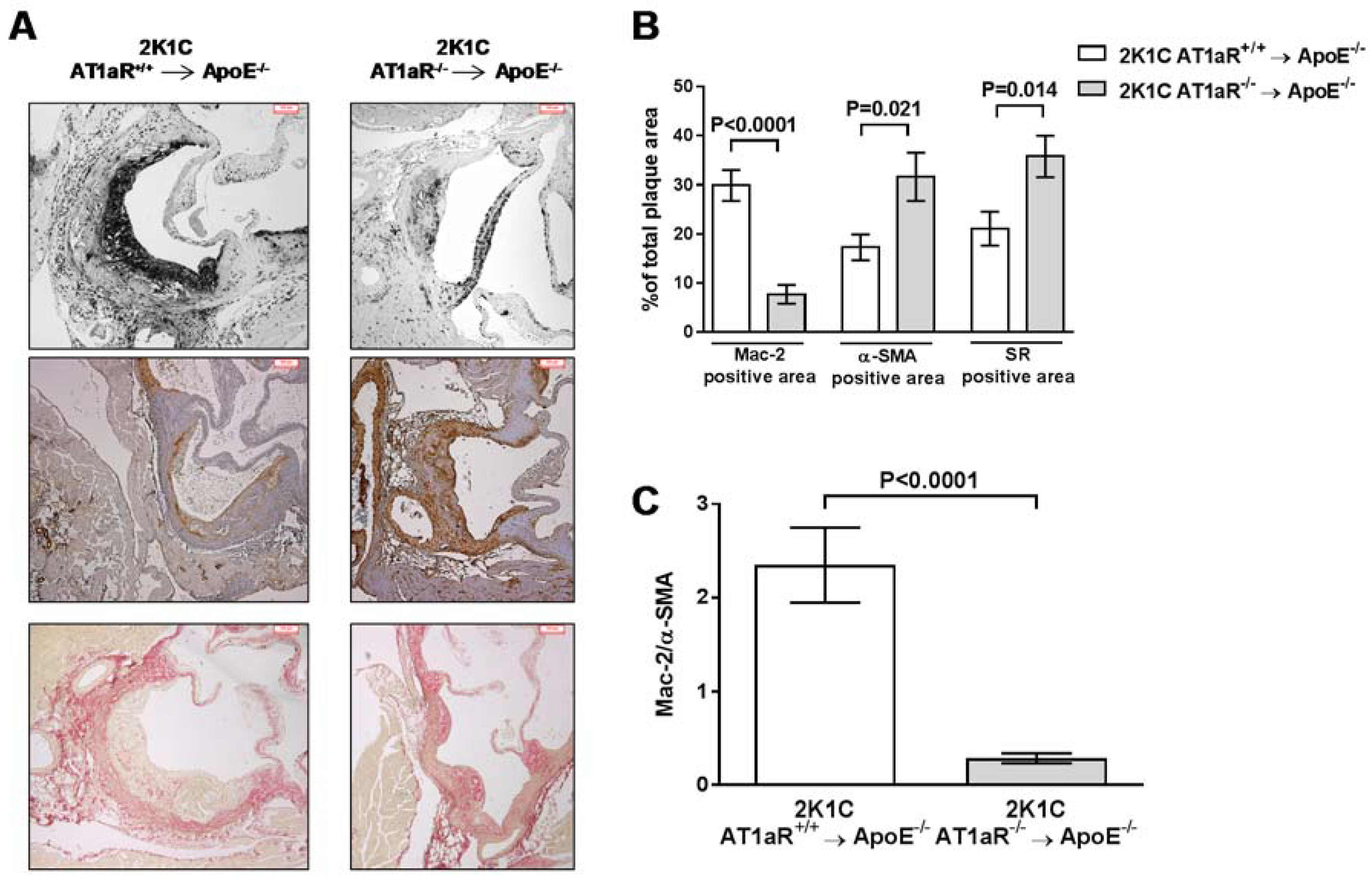
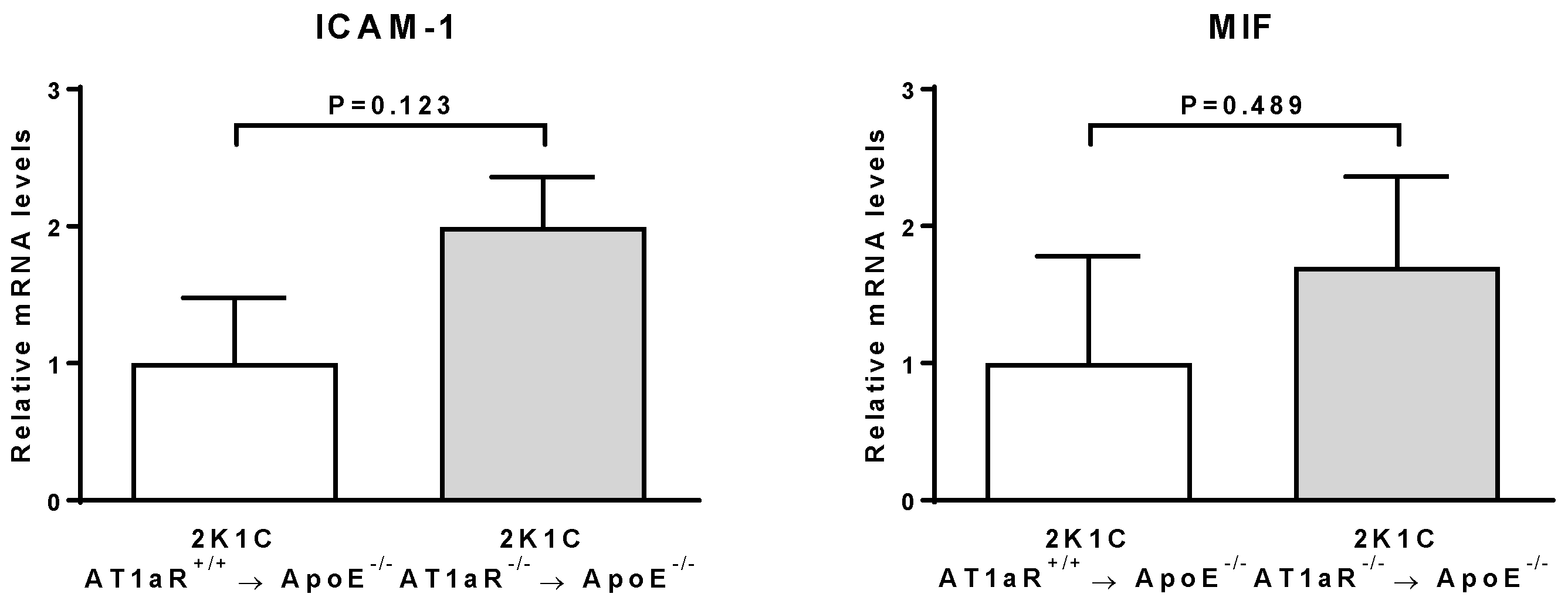
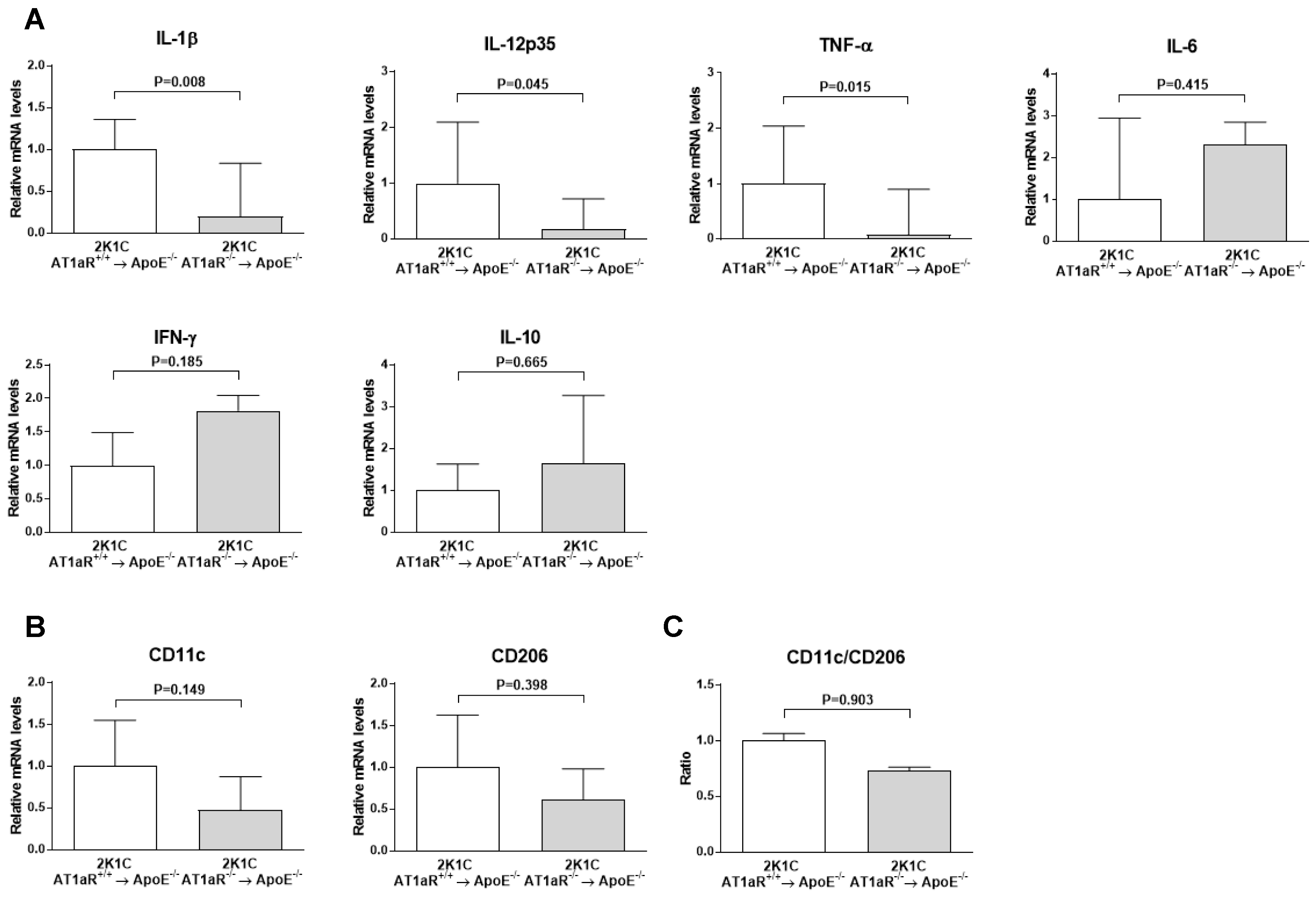
2.6. AT1aR Deficiency in BM-Derived Cells Leads to Decreased Vascular Inflammatory State
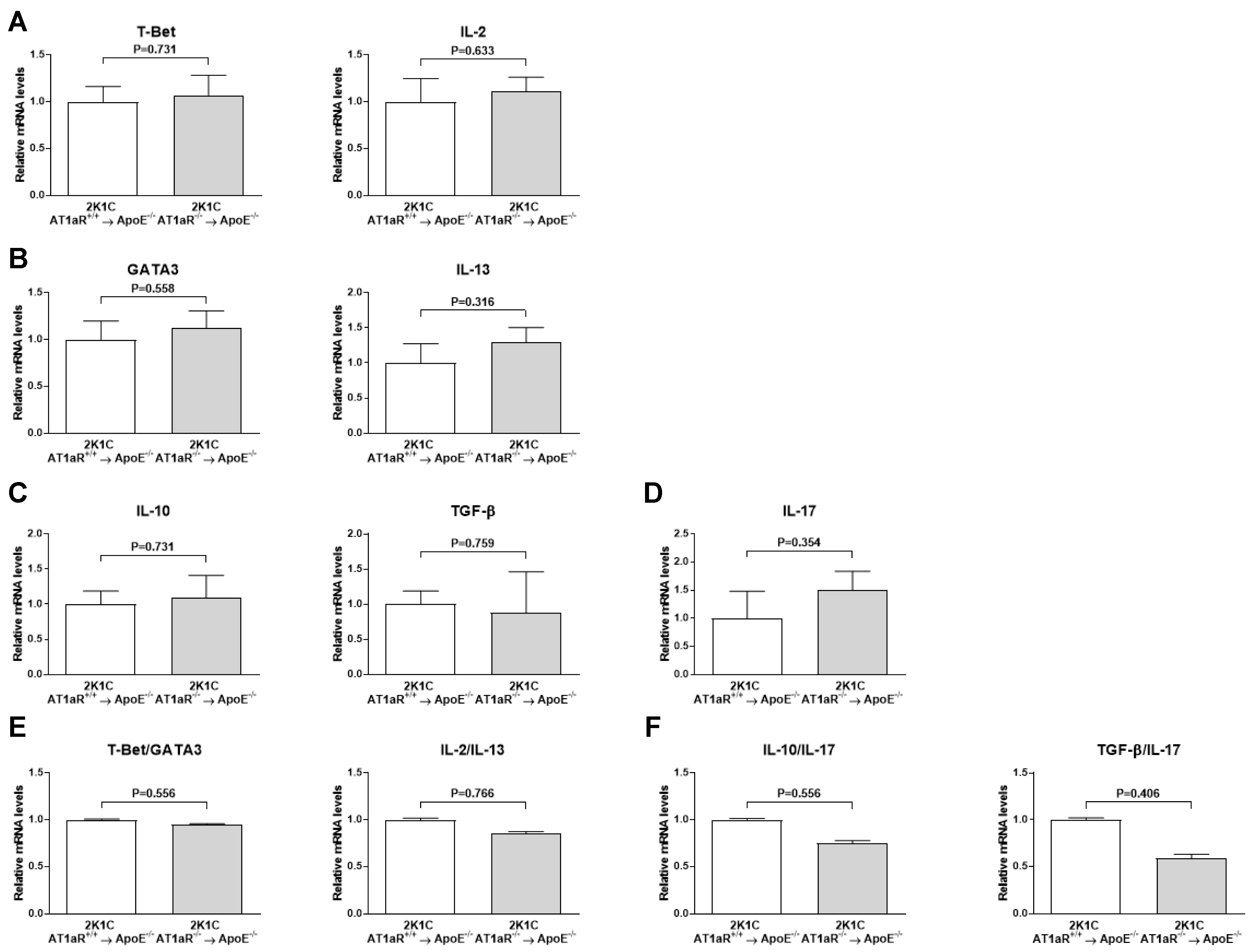
2.7. AT1aR Deficiency in BM-Derived Cells Does Not Affect Systemic T Helper Cells Subtypes
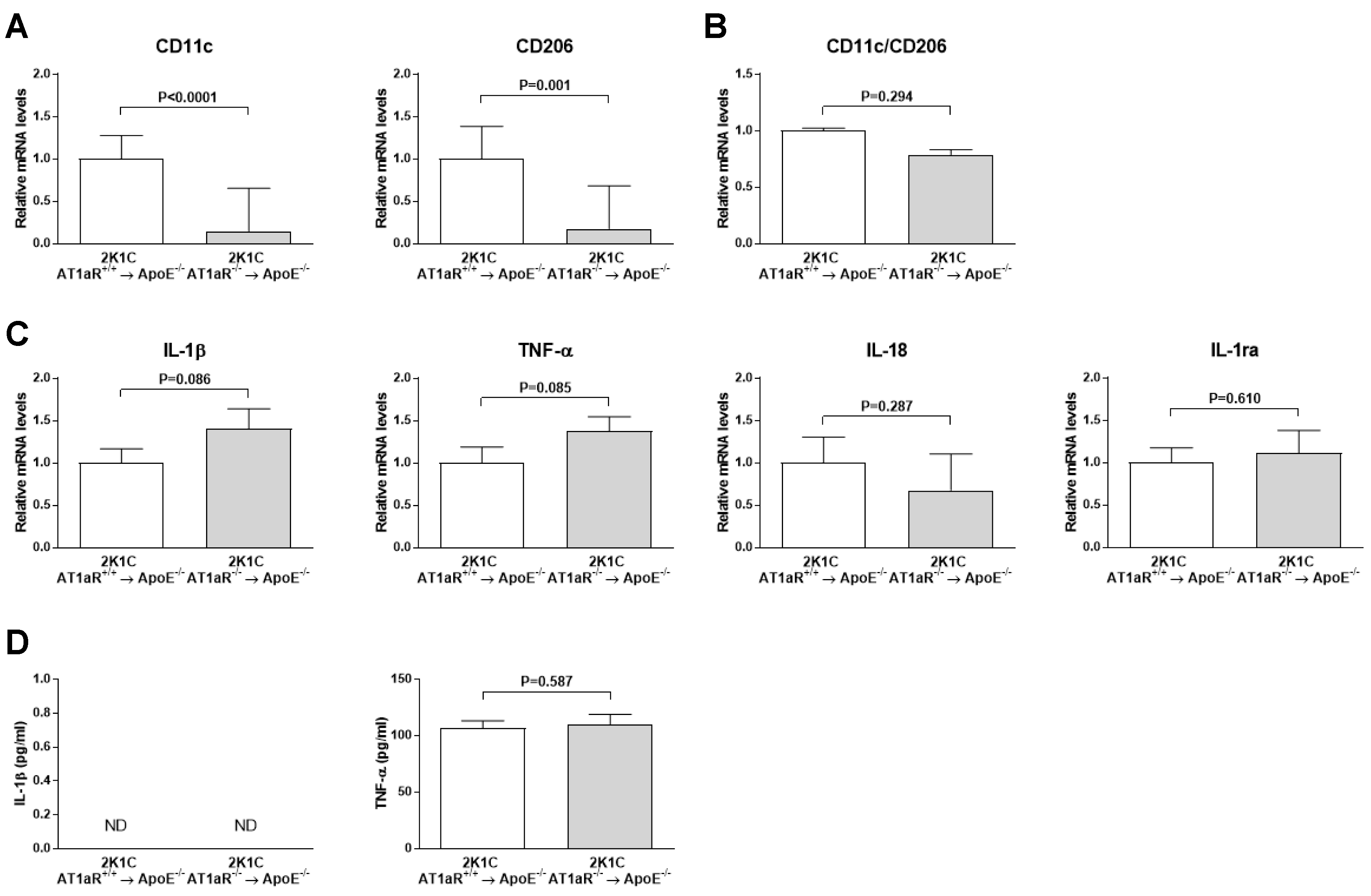
2.8. AT1aR Deficiency in BM-Derived Cells Does Not Modulate Systemic Macrophages Phenotype
2.9. AT1aR Deficiency in BM-Derived Cells Does Not Impact Circulating Leukocyte Profile
3. Discussion
4. Materials and Methods
4.1. Animals and Bone Marrow Transplantation
4.2. Mouse Model of Ang II-Mediated Vulnerable Atherosclerotic Plaques
4.3. Determination of Atherosclerotic Plaque Size by Histology
4.4. Evaluation of Atherosclerotic Plaque Staging and Phenotype by Histology and Immunohistochemical Analyses
4.5. Quantification of Gene Expression by Real-Time RT-PCR
4.6. Plasma Cholesterol and Cytokines Determination
4.7. Analysis of Blood Leukocytes by Flow Cytometry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Chrysochou, C.; Kalra, P.A. Epidemiology and natural history of atherosclerotic renovascular disease. Prog. Cardiovasc. Dis. 2009, 52, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Mazzolai, L.; Duchosal, M.A.; Korber, M.; Bouzourene, K.; Aubert, J.F.; Hao, H.; Vallet, V.; Brunner, H.R.; Nussberger, J.; Gabbiani, G.; et al. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE−/− mice 3. Hypertension 2004, 44, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, M.; Mazzolai, L. Angiotensin II as an Inducer of Atherosclerosis: Evidence from Mouse Studies. Clin. Exp. Cardiol. 2013, S1. [Google Scholar] [CrossRef]
- Vijayaraghavan, K.; Deedwania, P. Renin-angiotensin-aldosterone blockade for cardiovascular disease prevention. Cardiol. Clin. 2011, 29, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Blessing, E.; Preusch, M.; Kranzhofer, R.; Kinscherf, R.; Marx, N.; Rosenfeld, M.E.; Isermann, B.; Weber, C.M.; Kreuzer, J.; Grafe, J.; et al. Anti-atherosclerotic properties of telmisartan in advanced atherosclerotic lesions in apolipoprotein E deficient mice. Atherosclerosis 2008, 199, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, D.; Enomoto, S.; Hirata, Y.; Nagai, R.; Sata, M. The angiotensin receptor blocker, telmisartan, reduces and stabilizes atherosclerosis in ApoE and AT1aR double deficient mice. Biomed. Pharmacother. 2010, 64, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Sasamura, H.; Azegami, T.; Itoh, H. Regression of atherosclerosis in apolipoprotein E-deficient mice is feasible using high-dose angiotensin receptor blocker, candesartan. J. Atheroscler. Thromb. 2012, 19, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Murayama, T.; Yokode, M.; Kita, T.; Fujita, M.; Kishimoto, C. Olmesartan, a novel angiotensin II type 1 receptor antagonist, reduces severity of atherosclerosis in apolipoprotein E deficient mice associated with reducing superoxide production. Nutr. Metab. Cardiovasc. Dis. 2010, 21, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Czech, T.; van Eickels, M.; Fleming, I.; Bohm, M.; Nickenig, G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation 2004, 110, 3062–3067. [Google Scholar] [CrossRef] [PubMed]
- Nussberger, J.; Aubert, J.F.; Bouzourene, K.; Pellegrin, M.; Hayoz, D.; Mazzolai, L. Renin inhibition by aliskiren prevents atherosclerosis progression: Comparison with irbesartan, atenolol, and amlodipine 1. Hypertension 2008, 51, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, M.; Szostak, J.; Bouzourene, K.; Aubert, J.F.; Berthelot, A.; Nussberger, J.; Laurant, P.; Mazzolai, L. Running Exercise and Angiotensin II Type I Receptor Blocker Telmisartan Are Equally Effective in Preventing Angiotensin II-Mediated Vulnerable Atherosclerotic Lesions. J. Cardiovasc. Pharmacol. Therapeutics 2016. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S. Inflammation in atherosclerosis. Arch. Cardiovasc. Dis. 2016, 109, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Cassis, L.A.; Rateri, D.L.; Lu, H.; Daugherty, A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, D.; Sata, M.; Ishizaka, N.; Nagai, R. Critical role of bone marrow angiotensin II type 1 receptor in the pathogenesis of atherosclerosis in apolipoprotein E deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Koga, J.; Egashira, K.; Matoba, T.; Kubo, M.; Ihara, Y.; Iwai, M.; Horiuchi, M.; Sunagawa, K. Essential role of angiotensin II type 1a receptors in the host vascular wall, but not the bone marrow, in the pathogenesis of angiotensin II-induced atherosclerosis. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2008, 31, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Ishida, J.; Nagano, K.; Honjo, K.; Sugaya, T.; Takeda, N.; Sugiyama, F.; Yagami, K.; Fujita, T.; Nangaku, M.; et al. Deterioration of atherosclerosis in mice lacking angiotensin II type 1A receptor in bone marrow-derived cells. Lab. Investig. J. Tech. Methods Pathol. 2008, 88, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Rateri, D.L.; Feldman, D.L.; Jr, R.J.; Fukamizu, A.; Ishida, J.; Oesterling, E.G.; Cassis, L.A.; Daugherty, A. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J. Clin. Investig. 2008, 118, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Yancey, P.G.; Zuo, Y.; Ma, L.J.; Kaseda, R.; Fogo, A.B.; Ichikawa, I.; Linton, M.F.; Fazio, S.; Kon, V. Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Peshkova, I.O.; Schaefer, G.; Koltsova, E.K. Atherosclerosis and aortic aneurysm—Is inflammation a common denominator? FEBS J. 2016, 283, 1636–1652. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Weber, C.; Lutgens, E. Regulation of atherosclerotic plaque inflammation. J. Intern. Med. 2015, 278, 462–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gragnano, F.; Calabro, P. Role of dual lipid-lowering therapy in coronary atherosclerosis regression: Evidence from recent studies. Atherosclerosis 2018, 269, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Yamaguchi, S.; Nishizawa, T. Cholesterol-lowering pattern affects the progression of atherosclerosis in apolipoprotein E deficient mice. J. Pharmacol. Sci. 2016, 132, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Parolini, C.; Bjorndal, B.; Busnelli, M.; Manzini, S.; Ganzetti, G.S.; Dellera, F.; Ramsvik, M.; Bruheim, I.; Berge, R.K.; Chiesa, G. Effect of Dietary Components from Antarctic Krill on Atherosclerosis in apoE-Deficient Mice. Mol. Nutr. Food. Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; Corsa, B.A.; Zhou, J.; Yang, H.; Li, H.; Tang, Y.W.; Babaev, V.R.; Major, A.S.; Linton, M.F.; Fazio, S.; et al. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am. J. Physiol. Renal. Physiol. 2011, 300, F1203–F1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaka, S.; Usui, I.; Kanatani, Y.; Ikutani, M.; Takasaki, I.; Tsuneyama, K.; Tabuchi, Y.; Bukhari, A.; Yamazaki, Y.; Suzuki, H.; et al. Telmisartan improves insulin resistance and modulates adipose tissue macrophage polarization in high-fat-fed mice. Endocrinology 2011, 152, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Zotes, T.M.; Arias, C.F.; Fuster, J.J.; Spada, R.; Perez-Yague, S.; Hirsch, E.; Wymann, M.; Carrera, A.C.; Andres, V.; Barber, D.F. PI3K p110gamma deletion attenuates murine atherosclerosis by reducing macrophage proliferation but not polarization or apoptosis in lesions. PLoS ONE 2013, 8, e72674. [Google Scholar] [CrossRef] [PubMed]
- Franca, C.N.; Izar, M.C.O.; Hortencio, M.N.S.; do Amaral, J.B.; Ferreira, C.E.S.; Tuleta, I.D.; Fonseca, F.A.H. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin. Sci. 2017, 131, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.; Zeng, Q.; Lu, Q.; Lin, Y.; Wu, B.; Yu, K.; Dong, Z.; Zhang, J.; Chai, M.; Liu, Y.; et al. Valsartan Attenuates Atherosclerosis via Upregulating the Th2 Immune Response in Prolonged Angiotensin II-Treated ApoE(−/−) Mice. Mol. Med. 2015, 21, 143–153. [Google Scholar] [PubMed]
- Momiyama, Y.; Adachi, H.; Fairweather, D.; Ishizaka, N.; Saita, E. Inflammation, Atherosclerosis and Coronary Artery Disease. Clin. Med. Insights Cardiol. 2014, 8 (Suppl. 3), 67–70. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Liu, Y.; Zhao, Y.; Aono, J.; Jones, K.L.; Heywood, E.B.; Howatt, D.; Binkley, C.M.; Daugherty, A.; Liang, Y.; et al. Deficiency of the NR4A orphan nuclear receptor NOR1 in hematopoietic stem cells accelerates atherosclerosis. Stem Cells 2014, 32, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, M.; Alonso, F.; Aubert, J.F.; Bouzourene, K.; Braunersreuther, V.; Mach, F.; Haefliger, J.A.; Hayoz, D.; Berthelot, A.; Nussberger, J.; et al. Swimming prevents vulnerable atherosclerotic plaque development in hypertensive 2-kidney, 1-clip mice by modulating angiotensin II type 1 receptor expression independently from hemodynamic changes. Hypertension 2009, 53, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, M.; Aubert, J.F.; Bouzourene, K.; Amstutz, C.; Mazzolai, L. Voluntary Exercise Stabilizes Established Angiotensin II-Dependent Atherosclerosis in Mice through Systemic Anti-Inflammatory Effects. PLoS ONE 2015, 10, e0143536. [Google Scholar] [CrossRef] [PubMed]
- Donners, M.M.; Wolfs, I.M.; Stoger, L.J.; van der Vorst, E.P.; Pottgens, C.C.; Heymans, S.; Schroen, B.; Gijbels, M.J.; de Winther, M. Hematopoietic miR155 deficiency enhances atherosclerosis and decreases plaque stability in hyperlipidemic mice. PLoS ONE 2012, 7, e35877. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | 2K1C AT1aR+/+ → ApoE−/− | 2K1C AT1aR−/− → ApoE−/− | p-Value |
|---|---|---|---|
| BW gain (g) | 2.26 ± 0.32 | 1.76 ± 0.28 | 0.255 |
| MBP (mmHg) | 133 ± 3 | 132 ± 3 | 0.755 |
| HR (bpm) | 602 ± 15 | 626 ± 15 | 0.269 |
| PRA (ng/mL per h) | 13.1 ± 1.4 | 15.1 ± 1.2 | 0.289 |
| PRC (ng/mL per h) | 1307 ± 169 | 1282 ± 155 | 0.913 |
| PTC (g/L) | 6.5 ± 0.15 | 3.5 ± 0.10 | p < 0.0001 |
| Parameter | Markers | 2K1C AT1aR+/+ → ApoE−/− | 2K1C AT1aR−/− → ApoE−/− | p-Value |
|---|---|---|---|---|
| T cells (% of viable cells) | CD3+ B220− | 21.8 ± 1.3 | 16.2 ± 1.5 | 0.011 |
| T-helper cells (% of T cells) | CD4+ CD8− | 26.9 ± 1.0 | 26.6 ± 0.7 | 0.804 |
| Cytotoxic T cells (% of T cells) | CD4− CD8+ | 67.3 ± 1.9 | 70.6 ± 0.7 | 0.131 |
| B cells (% of viable cells) | CD3− B220+ | 15.3 ± 3.8 | 7.8 ± 1.1 | 0.082 |
| Granulocytes (% of viable cells) | CD11b+ Ly6G+ | 29.3 ± 5.0 | 35.4 ± 3.0 | 0.314 |
| Monocytes (% of viable cells) | CD11b+ Ly6G− | 12.3 ± 1.9 | 11.0 ± 1.8 | 0.536 |
| Classical monocytes (% of monocytes) | Ly6Chigh | 20.7 ± 3.2 | 22.0 ± 2.4 | 0.745 |
| Intermediate monocytes (% of monocytes) | Ly6Cmedium | 21.0 ± 1.1 | 19.7 ± 0.5 | 0.322 |
| Nonclassical monocytes (% of monocytes) | Ly6Clow | 58.3 ± 3.4 | 58.3 ± 2.5 | 0.987 |
| Classical/nonclassical monocytes | Ly6Chigh/Ly6Clow | 0.383 ± 0.076 | 0.393 ± 0.054 | 0.917 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrin, M.; Bouzourène, K.; Aubert, J.-F.; Nahimana, A.; Duchosal, M.A.; Mazzolai, L. Activation of Bone Marrow-Derived Cells Angiotensin (Ang) II Type 1 Receptor by Ang II Promotes Atherosclerotic Plaque Vulnerability. Int. J. Mol. Sci. 2018, 19, 2621. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092621
Pellegrin M, Bouzourène K, Aubert J-F, Nahimana A, Duchosal MA, Mazzolai L. Activation of Bone Marrow-Derived Cells Angiotensin (Ang) II Type 1 Receptor by Ang II Promotes Atherosclerotic Plaque Vulnerability. International Journal of Molecular Sciences. 2018; 19(9):2621. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092621
Chicago/Turabian StylePellegrin, Maxime, Karima Bouzourène, Jean-François Aubert, Aimable Nahimana, Michel A. Duchosal, and Lucia Mazzolai. 2018. "Activation of Bone Marrow-Derived Cells Angiotensin (Ang) II Type 1 Receptor by Ang II Promotes Atherosclerotic Plaque Vulnerability" International Journal of Molecular Sciences 19, no. 9: 2621. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092621