Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Epithelial–Mesenchymal Transition
Role of Epithelial–Mesenchymal Transition in Cancer
3. Extracellular-Signal Regulated Kinase: Structure and Function
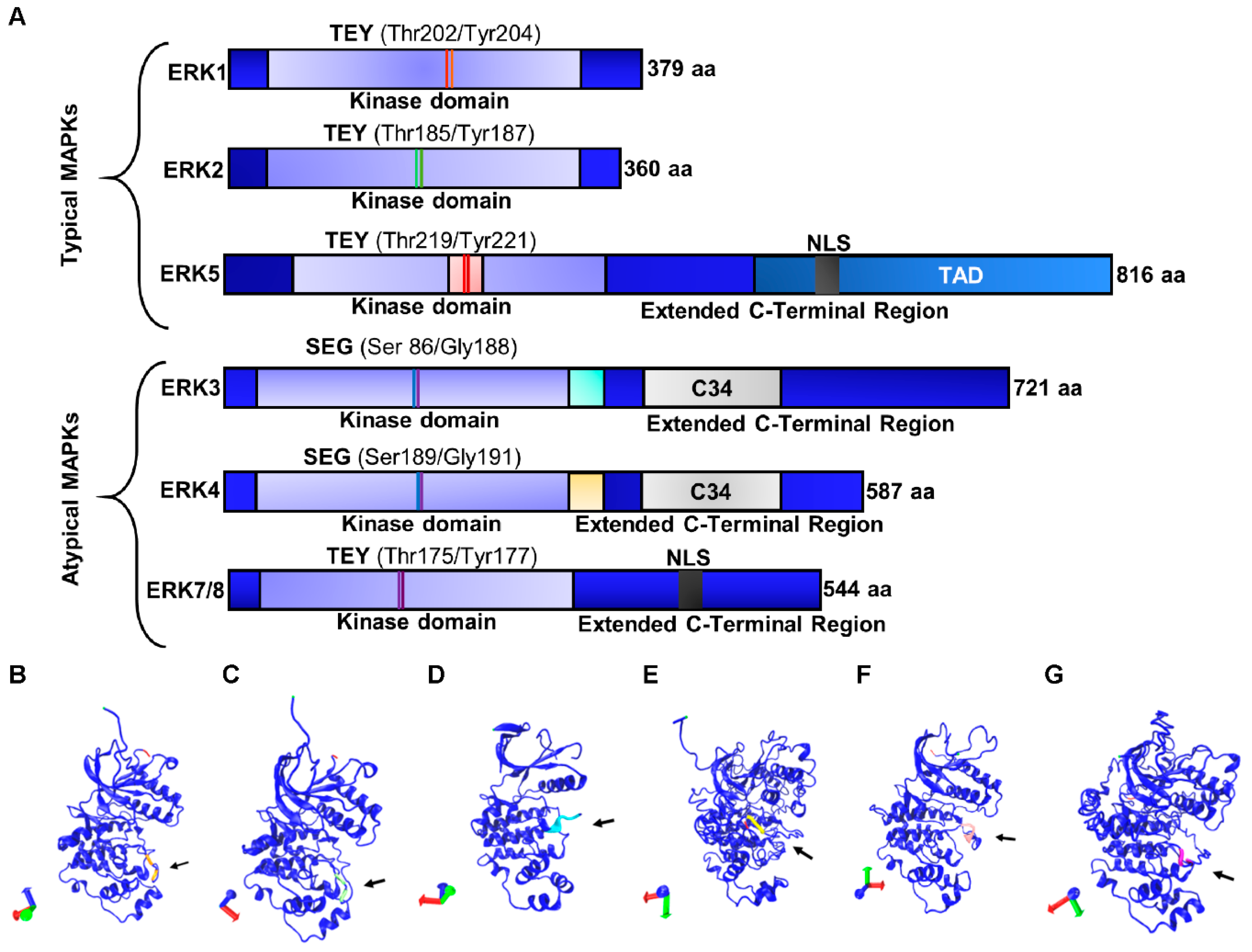
3.1. Typical Members of the ERK Subfamily
3.1.1. ERK1/2
3.1.2. ERK5
3.2. Atypical Members of the ERK Subfamily
3.2.1. ERK3/4
3.2.2. ERK7/8
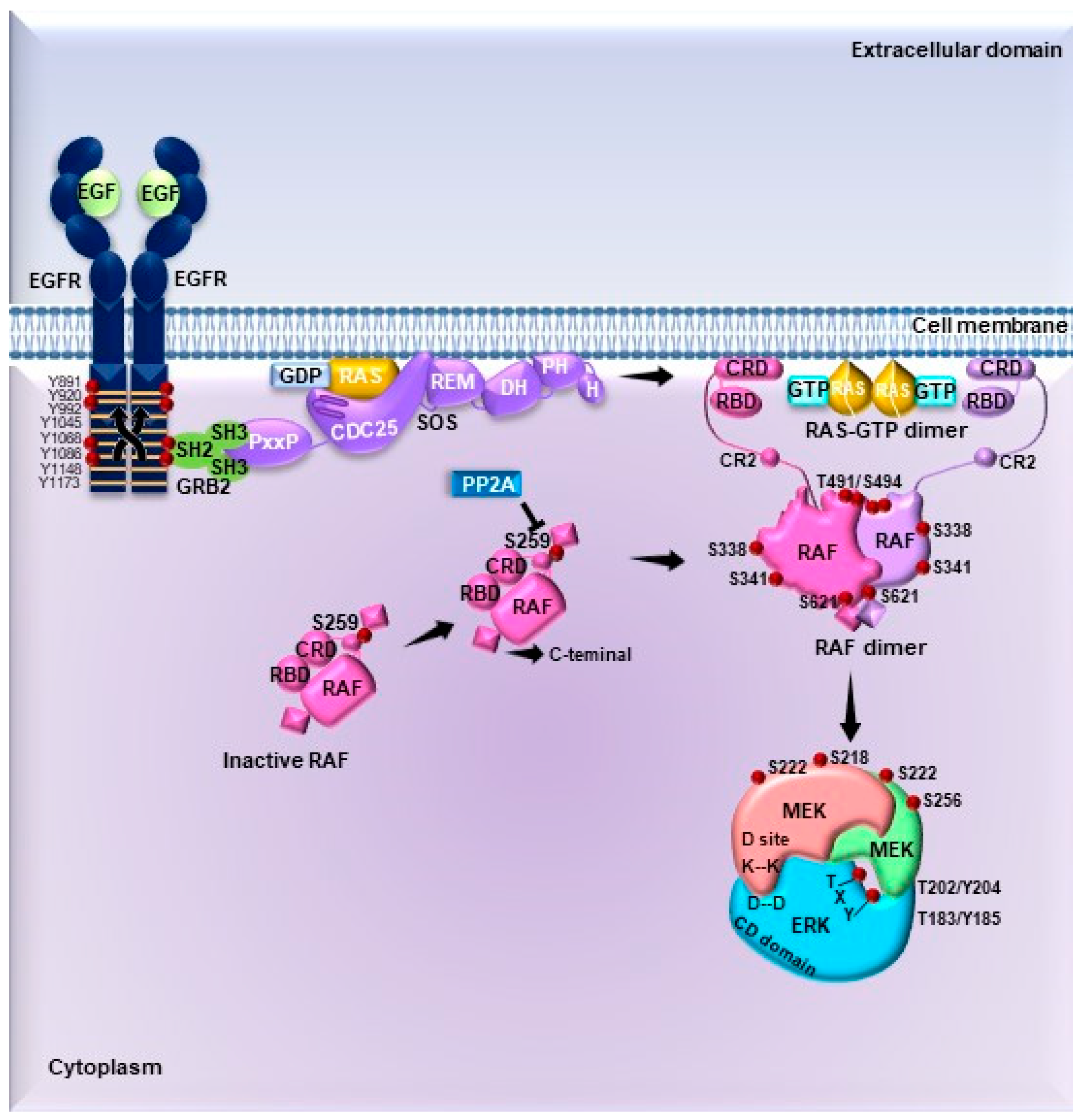
4. Signaling Pathways Driving ERK Activation: From Membrane Receptors to Substrates
4.1. Signal Transduction by MAPKs
4.2. Substrates of ERK1/2
4.2.1. Cytosolic Substrates
4.2.2. Nuclear Substrates
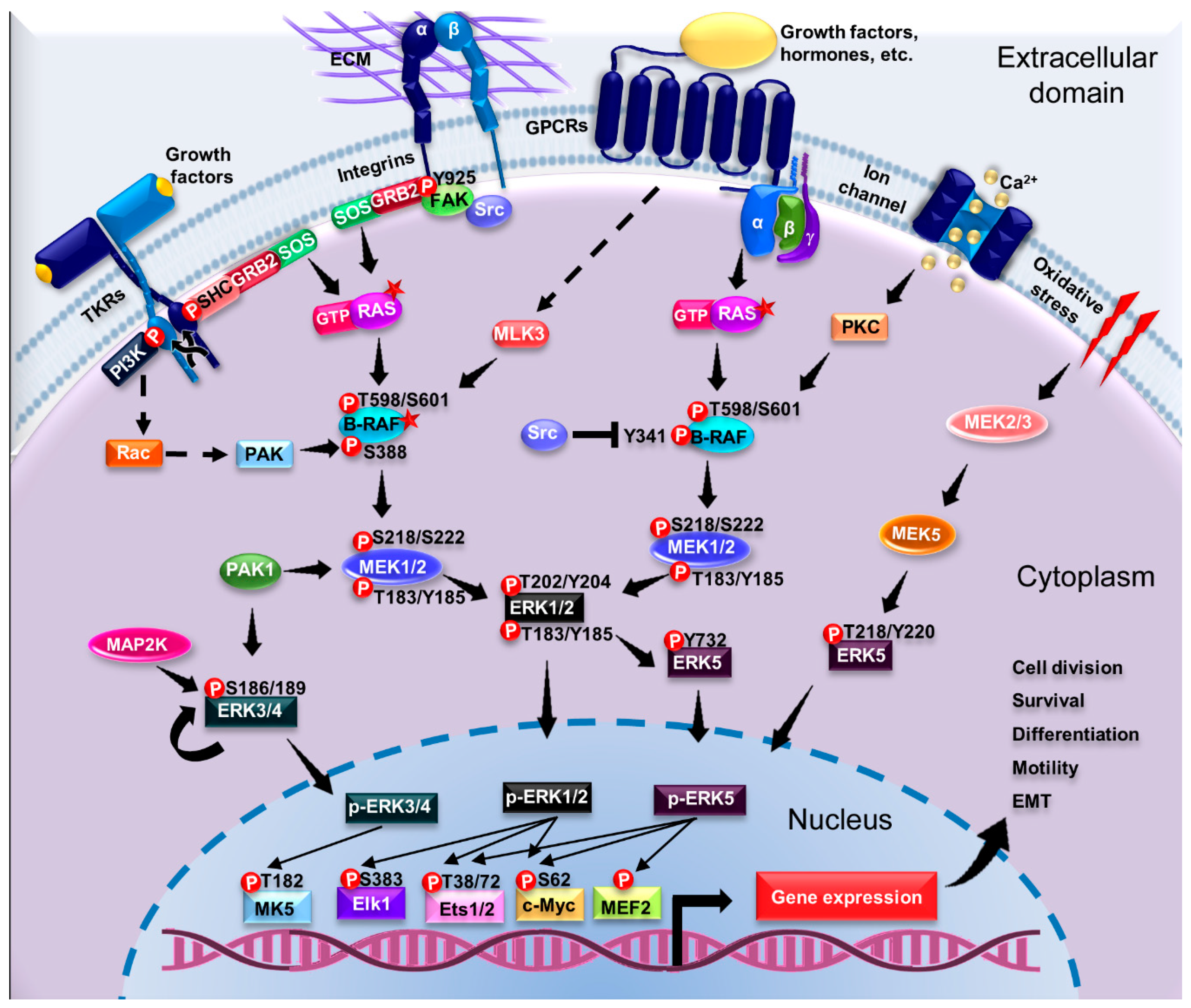
4.3. Activation of ERK1/2 Pathway by G Protein-Coupled Receptors
4.4. Regulation of the ERK1/2 Pathway
4.5. Activation of Pathways Dependent on Atypical ERK Members
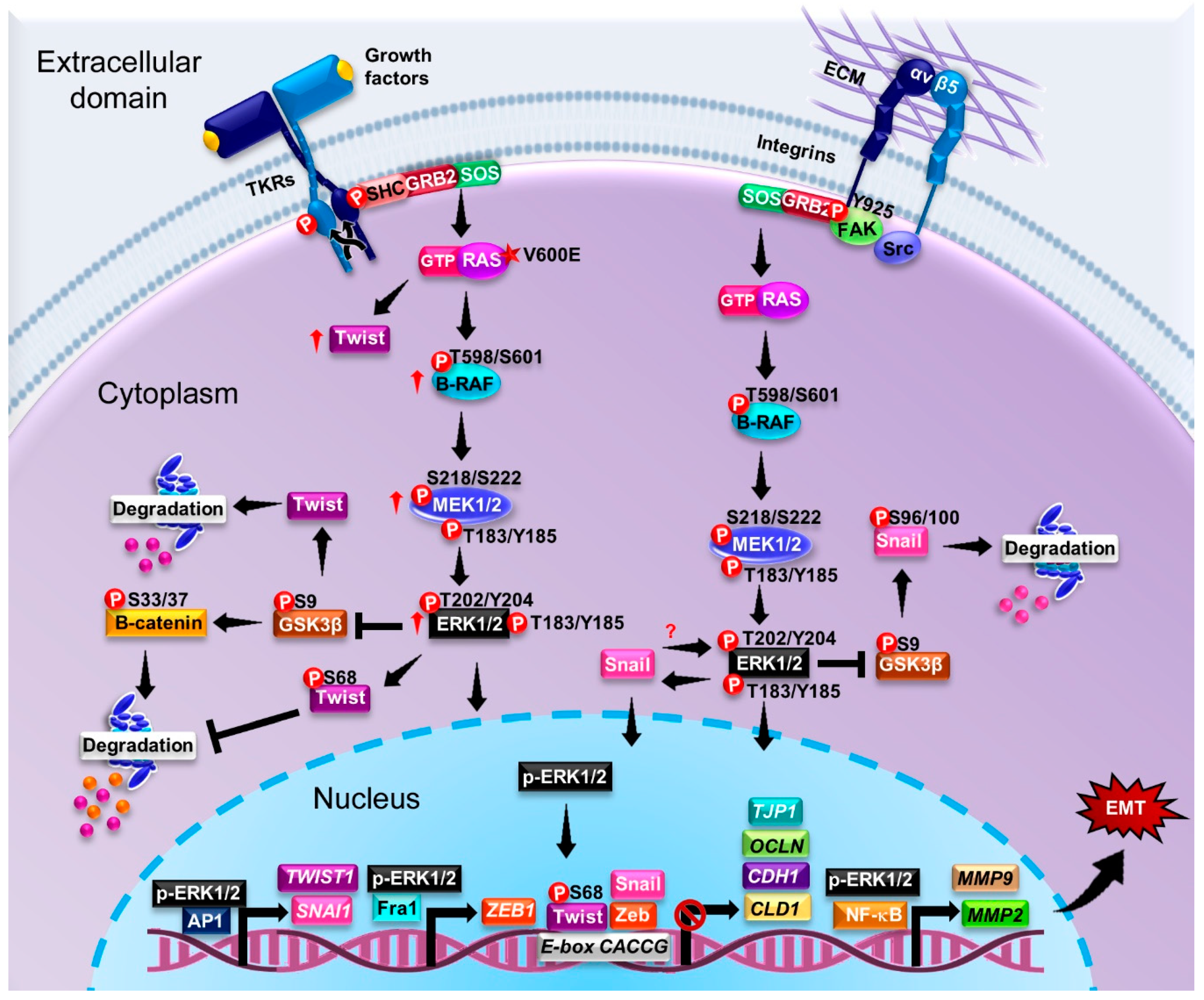
5. ERK-Activating Mutations in the MAPK Signaling Pathway and Their Contributions to EMT and Cancer Progression
p-ERK as a Marker of Cancer
6. Relationship between ERK and Epithelial–Mesenchymal Transition in Various Cancer Types
6.1. Lung Cancer
6.2. Breast Cancer
6.3. Colorectal Cancer
6.4. Cervical and Ovarian Cancer
6.5. Prostate Cancer
6.6. Other Types of Cancer
7. ERK as a Therapeutic Target in Cancer
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial—Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Gloushankova, N.; Zhitnyak, I.; Rubtsova, S. Role of Epithelial-Mesenchymal Transition in Tumor Progression. Biochemistry (Moscow) 2018, 83, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Das, V.; Bhattacharya, S.; Chikkaputtaiah, C.; Hazra, S.; Pal, M. The basics of epithelial–mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-mesenChymal Transition in Cancer; Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2018. [Google Scholar]
- Kim, D.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.-H. Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: A comprehensive overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Smith, B.; Bhowmick, N. Role of EMT in metastasis and therapy resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juárez-Cruz, J.; Mendoza-Catalán, M.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling Pathways Induced by Leptin during Epithelial–Mesenchymal Transition in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 3493. [Google Scholar] [CrossRef] [PubMed]
- Vyas-Read, S.; Wang, W.; Kato, S.; Colvocoresses-Dodds, J.; Fifadara, N.H.; Gauthier, T.W.; Helms, M.N.; Carlton, D.P.; Brown, L.A.S. Hyperoxia induces alveolar epithelial-to-mesenchymal cell transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 306, L326–L340. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.T.; Xu, Y.M. Regulation of human mitogen-activated protein kinase 15 (extracellular signal-regulated kinase 7/8) and its functions: A recent update. J. Cell. Physiol. 2019, 234, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xing, X.; Li, X.; Guo, Q.; Xu, T.; Xu, K. ZNF259 promotes breast cancer cells invasion and migration via ERK/GSK3β/snail signaling. Cancer Manag. Res. 2018, 10, 3159. [Google Scholar] [CrossRef]
- Yasuda, T. MAP kinase cascades in antigen receptor signaling and physiology. In B Cell Receptor Signaling; Springer: New York, NY, USA, 2015; pp. 211–231. [Google Scholar]
- Kahnamouyi, S.; Nouri, M.; Farzadi, L.; Darabi, M.; Hosseini, V.; Mehdizadeh, A. The role of mitogen-activated protein kinase–extracellular receptor kinase pathway in female fertility outcomes: A focus on pituitary gonadotropins regulation. Ther. Adv. Endocrinol. Metab. 2018, 9, 209–215. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Ro, E.J.; Choi, K.-Y. Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. Npj Precis. Oncol. 2018, 2, 5. [Google Scholar] [CrossRef]
- Mehdizadeh, A.; Somi, M.H.; Darabi, M.; Jabbarpour-Bonyadi, M. Extracellular signal-regulated kinase 1 and 2 in cancer therapy: A focus on hepatocellular carcinoma. Mol. Biol. Rep. 2016, 43, 107–116. [Google Scholar] [CrossRef]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1161–1176. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. In MAP Kinase Signaling Protocols; Springer: New York, NY, USA, 2010; pp. 3–38. [Google Scholar]
- Pouysségur, J.; Lenormand, P. ERK1 and ERK2 map kinases: Specific roles or functional redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar]
- Saba-El-Leil, M.K.; Vella, F.D.; Vernay, B.; Voisin, L.; Chen, L.; Labrecque, N.; Ang, S.L.; Meloche, S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Christen, R.; Lovern, M.; Clifford, A.M.; Yue, J.-X.; Goss, G.G.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 present functional redundancy in tetrapods despite higher evolution rate of ERK1. BMC Evol. Biol. 2015, 15, 179. [Google Scholar]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Lehmann, K.; Jefferies, H.B.; McMahon, M.; Downward, J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001, 15, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Lidke, D.S.; Huang, F.; Post, J.N.; Rieger, B.; Wilsbacher, J.; Thomas, J.L.; Pouyssegur, J.; Jovin, T.M.; Lenormand, P. ERK nuclear translocation is dimerization-independent but controlled by the rate of phosphorylation. J. Biol. Chem. 2010, 285, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017, 284, 4177–4195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radu, M.; Lyle, K.; Hoeflich, K.P.; Villamar-Cruz, O.; Koeppen, H.; Chernoff, J. p21-activated kinase 2 regulates endothelial development and function through the Bmk1/Erk5 pathway. Mol. Cell. Biol. 2015, 35, 3990–4005. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and cell proliferation: Nuclear localization is what matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-N.; Sun, L.-H.; Wang, M.; Yan, M. Research progress of the role and mechanism of extracellular signal-regulated protein kinase 5 (ERK5) pathway in pathological pain. J. Zhejiang Univ. Sci. B 2016, 17, 733–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Johnson, G.L. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J. Biol. Chem. 2003, 278, 36989–36992. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, J.; Wang, X.; Kharebava, G.; Tournier, C. A novel MAPK docking site in the N-terminus of MEK5a organizes the components of the ERK5 signaling pathway. Mol. Cell. Biol. 2005, 25, 9820–9828. [Google Scholar] [CrossRef] [PubMed]
- Kalous, J.; Tetkova, A.; Kubelka, M.; Susor, A. Importance of ERK1/2 in regulation of protein translation during oocyte meiosis. Int. J. Mol. Sci. 2018, 19, 698. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.-D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Lee, J.-D. Role of the BMK1/ERK5 signaling pathway: Lessons from knockout mice. J. Mol. Med. 2004, 82, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, R.; Salinas, S.; Philips, A.; Hipskind, R.A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003, 22, 5387. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, S.; Dumitriu, G.; Moens, U. Tumour promoting and suppressing roles of the atypical MAP kinase signalling pathway ERK3/4-MK5. J. Mol. Signal. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Déléris, P.; Rousseau, J.; Coulombe, P.; Rodier, G.; Tanguay, P.L.; Meloche, S. Activation loop phosphorylation of the atypical MAP kinases ERK3 and ERK4 is required for binding, activation and cytoplasmic relocalization of MK5. J. Cell. Physiol. 2008, 217, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.K.; Kuo, W.-L.; Hershenson, M.B.; Rosner, M.R. Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol. Cell. Biol. 1999, 19, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Henrich, L.M.; Smith, J.A.; Kitt, D.; Errington, T.M.; Nguyen, B.; Traish, A.M.; Lannigan, D.A. Extracellular signal-regulated kinase 7, a regulator of hormone-dependent estrogen receptor destruction. Mol. Cell. Biol. 2003, 23, 5979–5988. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.-H.; Lee, J.; Kim, K.M.; Kim, S.; Kim, D.-H.; Park, J. Overexpression of MAPK15 in gastric cancer is associated with copy number gain and contributes to the stability of c-Jun. Oncotarget 2015, 6, 20190. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-M.; Zhu, F.; Cho, Y.-Y.; Carper, A.; Peng, C.; Zheng, D.; Yao, K.; Lau, A.T.; Zykova, T.A.; Kim, H.-G. Extracellular signal-regulated kinase 8–mediated c-Jun phosphorylation increases tumorigenesis of human colon cancer. Cancer Res. 2010, 70, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Watson, U.; Vasudevan, L.; Saini, D.K. ERK activation pathways downstream of GPCRs. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 338, pp. 79–109. [Google Scholar]
- Ramos, J.W. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, re13. [Google Scholar] [CrossRef]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, Š. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar]
- Eblen, S.T.; Slack-Davis, J.K.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J.; Catling, A.D. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol. Cell. Biol. 2004, 24, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.; Liao, J.; Cathcart, M.K.; Wolfman, A. Constitutive association of c-N-Ras with c-Raf-1 and protein kinase C epsilon in latent signaling modules. J. Biol. Chem. 2001, 276, 29079–29090. [Google Scholar] [CrossRef] [PubMed]
- Chadee, D.N.; Kyriakis, J.M. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat. Cell Biol. 2004, 6, 770. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Yang, Y.; Ward, R.; Liu, Y.; Guo, X.-X.; Xu, T.-R. A-Raf: A new star of the family of raf kinases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Saito, Y.; Campbell, D.; Cohen, P.; Sithanandam, G.; Rapp, U.; Ashworth, A.; Marshall, C.; Cowley, S. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994, 13, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 2003, 162, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Gotoh, I.; Adachi, M.; Gotoh, Y.; Nishida, E. A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade role of nuclear export signal of MAP kinase kinase. J. Biol. Chem. 1997, 272, 32642–32648. [Google Scholar] [CrossRef]
- Berti, D.A.; Seger, R. The nuclear translocation of ERK. In ERK Signaling; Springer: New York, NY, USA, 2017; pp. 175–194. [Google Scholar]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Payne, D.; Rossomando, A.; Martino, P.; Erickson, A.; Her, J.; Shabanowitz, J.; Hunt, D.; Weber, M.; Sturgill, T. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991, 10, 885–892. [Google Scholar] [CrossRef]
- Zehorai, E.; Yao, Z.; Plotnikov, A.; Seger, R. The subcellular localization of MEK and ERK—A novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol. Cell. Endocrinol. 2010, 314, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, A.J.; Frankson, E.; Bardwell, L. Selectivity of docking sites in MAPK kinases. J. Biol. Chem. 2009, 284, 13165–13173. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, K.A.; Shapiro, P. Quantitative Analysis of ERK2 Interactions with Substrate Proteins; Federation of American Societies for Experimental Biology: Bethesda, MD, USA, 2011. [Google Scholar]
- Sheridan, D.L.; Kong, Y.; Parker, S.A.; Dalby, K.N.; Turk, B.E. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J. Biol. Chem. 2008, 283, 19511–19520. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Dimitri, C.A.; Yoon, S.-O.; Dowdle, W.; Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 2010, 38, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, K.; Pim, D.; Massimi, P.; Thomas, M.; Tomaić, V.; Subbaiah, V.; Kranjec, C.; Nakagawa, S.; Yano, T.; Taketani, Y. The cell polarity regulator hScrib controls ERK activation through a KIM site-dependent interaction. Oncogene 2010, 29, 5311. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuderland, D.; Seger, R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, S.M.; Chouinard, C.R.; Labadorf, A.; Lam, C.J.; Schmelzle, K.; Fraenkel, E.; White, F.M. Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci. Signal. 2011, 4, rs11. [Google Scholar] [CrossRef] [PubMed]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Deak, M.; Clifton, A.D.; Lucocq, J.M.; Alessi, D.R. Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.M.; Nebreda, A.R. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem. J. 2003, 372, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Richards, S.A.; Dreisbach, V.C.; Murphy, L.O.; Blenis, J. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol. Cell. Biol. 2001, 21, 7470–7480. [Google Scholar] [CrossRef] [PubMed]
- Frödin, M.; Jensen, C.J.; Merienne, K.; Gammeltoft, S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000, 19, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Warn-Cramer, B.J.; Lampe, P.D.; Kurata, W.E.; Kanemitsu, M.Y.; Loo, L.W.; Eckhart, W.; Lau, A.F. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J. Biol. Chem. 1996, 271, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef]
- Pulido, R.; Zúñiga, Á.; Ullrich, A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998, 17, 7337–7350. [Google Scholar] [CrossRef] [Green Version]
- Perlson, E.; Hanz, S.; Ben-Yaakov, K.; Segal-Ruder, Y.; Seger, R.; Fainzilber, M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 2005, 45, 715–726. [Google Scholar] [CrossRef]
- Lewis, T.S.; Hunt, J.B.; Aveline, L.D.; Jonscher, K.R.; Louie, D.F.; Yeh, J.M.; Nahreini, T.S.; Resing, K.A.; Ahn, N.G. Identification of novel MAP kinase pathway signaling targets by functional proteomics and mass spectrometry. Mol. Cell 2000, 6, 1343–1354. [Google Scholar] [CrossRef]
- Pol, A.; Lu, A.; Pons, M.; Peiró, S.; Enrich, C. Epidermal Growth Factor-mediated Caveolin Recruitment to Early Endosomes and MAPK Activation role of cholesterol and actin cytoskeleton. J. Biol. Chem. 2000, 275, 30566–30572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Ishibe, S.; Joly, D.; Zhu, X.; Cantley, L.G. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell 2003, 12, 1275–1285. [Google Scholar] [CrossRef]
- Ishibe, S.; Joly, D.; Liu, Z.-X.; Cantley, L.G. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 2004, 16, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zeng, W.; Su, J.; Kuang, Y.; He, Y.; Zhao, S.; Zhang, J.; Ma, W.; Bode, A.M.; Dong, Z. Cyclin-dependent kinase 2 (CDK2) is a key mediator for EGF-induced cell transformation mediated through the ELK4/c-Fos signaling pathway. Oncogene 2016, 35, 1170. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.O.; Smith, S.; Chen, R.-H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556. [Google Scholar] [CrossRef] [PubMed]
- Lowes, V.L.; Ip, N.Y.; Wong, Y.H. Integration of signals from receptor tyrosine kinases and G protein-coupled receptors. Neurosignals 2002, 11, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Luttrell, L.M.; Lefkowitz, R.J. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene 2001, 20, 1532. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Signalling: Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639. [Google Scholar] [CrossRef]
- Chung, K.Y. Structural aspects of GPCR-G protein coupling. Toxicol. Res. 2013, 29, 149. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qiao, Y.; Li, Z. New insights into modes of GPCR activation. Trends Pharmacol. Sci. 2018, 39, 367–386. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827. [Google Scholar] [CrossRef]
- Ebisuya, M.; Kondoh, K.; Nishida, E. The duration, magnitude and compartmentalization of ERK MAP kinase activity: Mechanisms for providing signaling specificity. J. Cell Sci. 2005, 118, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Vetterkind, S.; Poythress, R.H.; Lin, Q.Q.; Morgan, K.G. Hierarchical scaffolding of an ERK1/2 activation pathway. Cell Commun. Signal. 2013, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.-G.; Li, Z.; Sacks, D.B. IQGAP1 modulates activation of B-Raf. Proc. Natl. Acad. Sci. USA 2007, 104, 10465–10469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, M.; Mathers, J.; Dickinson, R.J.; Mandl, M.; Keyse, S.M. Both nuclear-cytoplasmic shuttling of the dual specificity phosphatase MKP-3 and its ability to anchor MAP kinase in the cytoplasm are mediated by a conserved nuclear export signal. J. Biol. Chem. 2004, 279, 41882–41891. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Roux, D.; Lenormand, P.; Dowd, S.; Keyse, S.; Pouysségur, J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999, 18, 664–674. [Google Scholar] [CrossRef] [Green Version]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci. 2008, 13, 4258–4275. [Google Scholar] [CrossRef]
- Mace, P.D.; Wallez, Y.; Egger, M.F.; Dobaczewska, M.K.; Robinson, H.; Pasquale, E.B.; Riedl, S.J. Structure of ERK2 bound to PEA-15 reveals a mechanism for rapid release of activated MAPK. Nat. Commun. 2013, 4, 1681. [Google Scholar] [CrossRef] [PubMed]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.-C.; Canton, B.; Nguyen, X.-T.; Barnier, J.-V.; Camonis, J.; Ginsberg, M.H. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Adachi, M.; Fukuda, M.; Nishida, E. Nuclear export of map kinase (ERK) involves a map kinase kinase (Mek-dependent) active transport mechanism. J. Cell Biol. 2000, 148, 849–856. [Google Scholar] [CrossRef]
- Maillet, M.; Purcell, N.H.; Sargent, M.A.; York, A.J.; Bueno, O.F.; Molkentin, J.D. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J. Biol. Chem. 2008, 283, 31246–31255. [Google Scholar] [CrossRef] [PubMed]
- Kucharska, A.; Rushworth, L.K.; Staples, C.; Morrice, N.A.; Keyse, S.M. Regulation of the inducible nuclear dual-specificity phosphatase DUSP5 by ERK MAPK. Cell. Signal. 2009, 21, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.; Slack, D.N.; Keyse, S.M. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Mol. Cell. Biol. 2005, 25, 1830–1845. [Google Scholar] [CrossRef]
- Volmat, V.; Camps, M.; Arkinstall, S.; Pouysségur, J.; Lenormand, P. The nucleus, a site for signal termination by sequestration and inactivation of p42/p44 MAP kinases. J. Cell Sci. 2001, 114, 3433–3443. [Google Scholar]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef]
- Kamakura, S.; Moriguchi, T.; Nishida, E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 1999, 274, 26563–26571. [Google Scholar] [CrossRef]
- Abe, J.-I.; Takahashi, M.; Ishida, M.; Lee, J.-D.; Berk, B.C. C-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1 (BMK1). J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef]
- Xu, B.-E.; Stippec, S.; Lenertz, L.; Lee, B.-H.; Zhang, W.; Lee, Y.-K.; Cobb, M.H. WNK1 activates ERK5 by an MEKK2/3-dependent mechanism. J. Biol. Chem. 2004, 279, 7826–7831. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wei, X.; Kesavan, K.; Garrington, T.P.; Fan, R.; Mei, J.; Anderson, S.M.; Gelfand, E.W.; Johnson, G.L. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol. Cell. Biol. 2003, 23, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-M.; Yang, F.; Zhang, Y.-X. Preconditioning-induced activation of ERK5 is dependent on moderate Ca2+ influx via NMDA receptors and contributes to ischemic tolerance in the hippocampal CA1 region of rats. Life Sci. 2006, 79, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.-H.; Hayashi, M.; Tapping, R.I.; Kato, Y.; Lee, J.-D. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J. Biol. Chem. 1999, 274, 36035–36038. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef] [PubMed]
- Watson, F.L.; Heerssen, H.M.; Bhattacharyya, A.; Klesse, L.; Lin, M.Z.; Segal, R.A. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 2001, 4, 981. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Boulton, T.G.; Cobb, M.H. ERK3 is a constitutively nuclear protein kinase. J. Biol. Chem. 1996, 271, 8951–8958. [Google Scholar] [CrossRef] [PubMed]
- Åberg, E.; Perander, M.; Johansen, B.; Julien, C.; Meloche, S.; Keyse, S.M.; Seternes, O.-M. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J. Biol. Chem. 2006, 281, 35499–35510. [Google Scholar] [CrossRef] [PubMed]
- Kant, S. Characterization of the Protein Kinases ERK3 and ERK4 and Their Interaction with MK5. Ph.D. Thesis, Hannover Medical School, Hannover, Germany, 2006. [Google Scholar]
- Schumacher, S.; Laaß, K.; Kant, S.; Shi, Y.; Visel, A.; Gruber, A.D.; Kotlyarov, A.; Gaestel, M. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 2004, 23, 4770–4779. [Google Scholar] [CrossRef]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef]
- Perander, M.; Al-Mahdi, R.; Jensen, T.C.; Nunn, J.A.; Kildalsen, H.; Johansen, B.; Gabrielsen, M.; Keyse, S.M.; Seternes, O.-M. Regulation of atypical MAP kinases ERK3 and ERK4 by the phosphatase DUSP2. Sci. Rep. 2017, 7, 43471. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Coulombe, P.; Meloche, S. Nuclear export of ERK3 by a CRM1-dependent mechanism regulates its inhibitory action on cell cycle progression. J. Biol. Chem. 2003, 278, 42615–42624. [Google Scholar] [CrossRef] [PubMed]
- Klinger, S.; Turgeon, B.; Lévesque, K.; Wood, G.A.; Aagaard-Tillery, K.M.; Meloche, S. Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc. Natl. Acad. Sci. USA 2009, 106, 16710–16715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saelzler, M.P.; Spackman, C.C.; Liu, Y.; Martinez, L.C.; Harris, J.P.; Abe, M.K. ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5. J. Biol. Chem. 2006, 281, 16821–16832. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.K.; Saelzler, M.P.; Espinosa, R.; Kahle, K.T.; Hershenson, M.B.; Le Beau, M.M.; Rosner, M.R. ERK8, a new member of the mitogen-activated protein kinase family. J. Biol. Chem. 2002, 277, 16733–16743. [Google Scholar] [CrossRef] [PubMed]
- Klevernic, I.V.; Stafford, M.J.; Morrice, N.; Peggie, M.; Morton, S.; Cohen, P. Characterization of the reversible phosphorylation and activation of ERK8. Biochem. J. 2006, 394, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small Gtpases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Lyons, J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr. Opin. Pharmacol. 2005, 5, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Lou, D.; McCormick, F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013, 3, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Stolze, B.; Reinhart, S.; Bulllinger, L.; Fröhling, S.; Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 2015, 5, 8535. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.; Wiese, M.; Rowland, A.; Kichenadasse, G.; McKinnon, R.; Karapetis, C. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2014, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Elkhadragy, L.; Alsaran, H.; Morel, M.; Long, W. Activation loop phosphorylation of ERK3 is important for its kinase activity and ability to promote lung cancer cell invasiveness. J. Biol. Chem. 2018, 293, 16193–16205. [Google Scholar] [CrossRef] [Green Version]
- Röring, M.; Herr, R.; Fiala, G.J.; Heilmann, K.; Braun, S.; Eisenhardt, A.E.; Halbach, S.; Capper, D.; Von Deimling, A.; Schamel, W.W. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012, 31, 2629–2647. [Google Scholar] [CrossRef] [Green Version]
- Thevakumaran, N.; Lavoie, H.; Critton, D.A.; Tebben, A.; Marinier, A.; Sicheri, F.; Therrien, M. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol. 2015, 22, 37. [Google Scholar] [CrossRef]
- Wimmer, R.; Baccarini, M. Partner exchange: Protein–protein interactions in the Raf pathway. Trends Biochem. Sci. 2010, 35, 660–668. [Google Scholar] [CrossRef]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 2013, 49, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Samuels, Y. Analysis of the genome to personalize therapy for melanoma. Oncogene 2010, 29, 5545–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcila, M.E.; Drilon, A.; Sylvester, B.E.; Lovly, C.M.; Borsu, L.; Reva, B.; Kris, M.G.; Solit, D.B.; Ladanyi, M. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin. Cancer Res. 2015, 21, 1935–1943. [Google Scholar] [CrossRef]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Hanafiah, N.M.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P. Molecular signaling in multiple myeloma: Association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; Martin, P.; Herzog, S.; Misawa, Y.; Daly, R.; Reth, M. Functional analysis of the regulatory requirements of B-Raf and the B-Raf V600E oncoprotein. Oncogene 2006, 25, 6262. [Google Scholar] [CrossRef] [PubMed]
- Köhler, M.; Ehrenfeld, S.; Halbach, S.; Lauinger, M.; Burk, U.; Reischmann, N.; Cheng, S.; Spohr, C.; Uhl, F.M.; Köhler, N. B-Raf deficiency impairs tumor initiation and progression in a murine breast cancer model. Oncogene 2019, 38, 1324. [Google Scholar] [CrossRef]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461, 542. [Google Scholar] [CrossRef]
- Ritt, D.A.; Monson, D.M.; Specht, S.I.; Morrison, D.K. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol. Cell. Biol. 2010, 30, 806–819. [Google Scholar] [CrossRef]
- Eisenhardt, A.E.; Olbrich, H.; Roring, M.; Janzarik, W.; Anh, T.N.; Cin, H.; Remke, M.; Witt, H.; Korshunov, A.; Pfister, S.M.; et al. Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int. J. Cancer 2011, 129, 2297–2303. [Google Scholar] [CrossRef]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK mutations and amplification confer resistance to ERK-inhibitor therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Zhang, P.; You, J. Post-translational modifications of EMT transcriptional factors in cancer metastasis. Open Life Sci. 2016, 11, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Jin, H.; Gao, D.; Lieftink, C.; Evers, B.; Jin, G.; Xue, Z.; Wang, L.; Beijersbergen, R.L.; Qin, W. Phospho-ERK is a biomarker of response to a synthetic lethal drug combination of sorafenib and MEK inhibition in liver cancer. J. Hepatol. 2018, 69, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Han, T.; Ma, R.-H.; Zhu, Y.-L.; Jia, Y.-N.; Du, J.-J.; Chen, Y.; Jiang, X.-J.; Xie, X.-D.; Guo, X. SHP2 promotes laryngeal cancer growth through the Ras/Raf/Mek/Erk pathway and serves as a prognostic indicator for laryngeal cancer. Int. J. Oncol. 2014, 44, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Mizushima, T.; Ide, H.; Jiang, G.; Goto, T.; Nagata, Y.; Netto, G.J.; Miyamoto, H. ATF2 promotes urothelial cancer outgrowth via cooperation with androgen receptor signaling. Endocr. Connect. 2018, 7, 1397–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Zhao, P.; Li, S.-Q.; Xiao, W.-K.; Yin, X.-Y.; Peng, B.-G.; Liang, L.-J. Prognostic impact of pERK in advanced hepatocellular carcinoma patients treated with sorafenib. Eur. J. Surg. Oncol. (EJSO) 2013, 39, 974–980. [Google Scholar] [CrossRef]
- Lok, G.T.; Chan, D.W.; Liu, V.W.; Hui, W.W.; Leung, T.H.; Yao, K.; Ngan, H.Y. Aberrant activation of ERK/FOXM1 signaling cascade triggers the cell migration/invasion in ovarian cancer cells. PLoS ONE 2011, 6, e23790. [Google Scholar] [CrossRef]
- Hong, S.-K.; Park, J.-R.; Kwon, O.-S.; Kim, K.-T.; Bae, G.-Y.; Cha, H.-J. Induction of integrin β3 by sustained ERK activity promotes the invasiveness of TGFβ-induced mesenchymal tumor cells. Cancer Lett. 2016, 376, 339–346. [Google Scholar] [CrossRef]
- Chiu, L.; Hsin, I.; Yang, T.; Sung, W.; Chi, J.; Chang, J.; Ko, J.; Sheu, G. The ERK–ZEB1 pathway mediates epithelial–mesenchymal transition in pemetrexed resistant lung cancer cells with suppression by vinca alkaloids. Oncogene 2017, 36, 242. [Google Scholar] [CrossRef]
- Kwon, O.-S.; Hong, S.-K.; Kwon, S.-J.; Go, Y.-H.; Oh, E.; Cha, H.-J. BCL2 induced by LAMTOR3/MAPK is a druggable target of chemoradioresistance in mesenchymal lung cancer. Cancer Lett. 2017, 403, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Foulds, C.E.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Tsai, S.Y.; Tsai, M.-J. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Investig. 2012, 122, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Cao, F.-L.; Li, N.; Gao, X.; Su, X.; Jiang, X. Leptin induces epithelial-to-mesenchymal transition via activation of the ERK signaling pathway in lung cancer cells. Oncol. Lett. 2018, 16, 4782–4788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Wu, R.; Xie, W.; Zhu, M.; Xie, C.; Li, X.; Zhu, J.; Zhu, W.; Wu, J.; Geng, S. Curcumin reverses tobacco smoke-induced epithelial-mesenchymal transition by suppressing the MAPK pathway in the lungs of mice. Mol. Med. Rep. 2018, 17, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Javaid, S.; Zhang, J.; Smolen, G.A.; Yu, M.; Wittner, B.S.; Singh, A.; Arora, K.S.; Madden, M.W.; Desai, R.; Zubrowski, M.J. MAPK7 regulates EMT features and modulates the generation of CTCs. Mol. Cancer Res. 2015, 13, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Mali, A.V.; Joshi, A.A.; Hegde, M.V.; Kadam, S.S. Enterolactone modulates the ERK/NF-κB/Snail signaling pathway in triple-negative breast cancer cell line MDA-MB-231 to revert the TGF-β-induced epithelial–mesenchymal transition. Cancer Biol. Med. 2018, 15, 137. [Google Scholar] [PubMed]
- Zhang, Y.-Q.; Wei, X.-L.; Liang, Y.-K.; Chen, W.-L.; Zhang, F.; Bai, J.-W.; Qiu, S.-Q.; Du, C.-W.; Huang, W.-H.; Zhang, G.-J. Over-expressed twist associates with markers of epithelial mesenchymal transition and predicts poor prognosis in breast cancers via ERK and Akt activation. PLoS ONE 2015, 10, e0135851. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Elsum, I.A.; Martin, C.; Humbert, P.O. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J. Cell Sci. 2013, 126, 3990–3999. [Google Scholar] [CrossRef]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [PubMed]
- Navandar, M.; Garding, A.; Sahu, S.K.; Pataskar, A.; Schick, S.; Tiwari, V.K. ERK signalling modulates epigenome to drive epithelial to mesenchymal transition. Oncotarget 2017, 8, 29269. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Duque, A.; Zuniga-Eulogio, M.D.; Dena-Beltran, J.; Castaneda-Saucedo, E.; Calixto-Galvez, M.; Mendoza-Catalán, M.A.; Ortuno-Pineda, C.; Navarro-Tito, N. Leptin induces partial epithelial-mesenchymal transition in a FAK-ERK dependent pathway in MCF10A mammary non-tumorigenic cells. Int. J. Clin. Exp. Pathol. 2017, 10, 10334–10342. [Google Scholar]
- Li, J.; Guo, Y.; Duan, L.; Hu, X.; Zhang, X.; Hu, J.; Huang, L.; He, R.; Hu, Z.; Luo, W. AKR1B10 promotes breast cancer cell migration and invasion via activation of ERK signaling. Oncotarget 2017, 8, 33694. [Google Scholar] [CrossRef] [PubMed]
- Sankpal, N.V.; Fleming, T.P.; Sharma, P.K.; Wiedner, H.J.; Gillanders, W.E. A double-negative feedback loop between EpCAM and ERK contributes to the regulation of epithelial–mesenchymal transition in cancer. Oncogene 2017, 36, 3706. [Google Scholar] [CrossRef]
- Al-Mahdi, R.; Babteen, N.; Thillai, K.; Holt, M.; Johansen, B.; Wetting, H.L.; Seternes, O.-M.; Wells, C.M. A novel role for atypical MAPK kinase ERK3 in regulating breast cancer cell morphology and migration. Cell Adhes. Migr. 2015, 9, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Ma, C.; Li, W.; Yang, S.; Liu, Z. miR-143 suppresses epithelial–mesenchymal transition and inhibits tumor growth of breast cancer through down-regulation of ERK5. Mol. Carcinog. 2016, 55, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, T.; Osada, S.; Yamada, A.; Kato, J.; Yawata, K.; Mori, R.; Imai, H.; Sasaki, Y.; Saito, S.; Tanaka, Y. Extracellular signal-regulated kinase and Akt activation play a critical role in the process of hepatocyte growth factor-induced epithelial-mesenchymal transition. Int. J. Oncol. 2013, 42, 556–564. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Xu, C.; Zhou, Z.; Zhu, Z.; You, T. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014, 355, 130–140. [Google Scholar] [CrossRef]
- Blaj, C.; Schmidt, E.M.; Lamprecht, S.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Oncogenic effects of high MAPK activity in colorectal cancer mark progenitor cells and persist irrespective of RAS mutations. Cancer Res. 2017, 77, 1763–1774. [Google Scholar] [CrossRef]
- Wu, L.; Han, L.; Zhou, C.; Wei, W.; Chen, X.; Yi, H.; Wu, X.; Bai, X.; Guo, S.; Yu, Y. TGF-β1-induced CK 17 enhances cancer stem cell-like properties rather than EMT in promoting cervical cancer metastasis via the ERK 1/2-MZF 1 signaling pathway. FEBS J. 2017, 284, 3000–3017. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xie, Y.; Cui, D.; Ma, Y.; Sui, L.; Zhu, C.; Kong, H.; Kong, Y. Osteopontin promotes invasion, migration and epithelial-mesenchymal transition of human endometrial carcinoma cell HEC-1A through AKT and ERK1/2 signaling. Cell. Physiol. Biochem. 2015, 37, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Chen, H.; Zhu, J.Y.; Wang, W.; Teng, Y.; Ding, H.-F.; Jing, Q.; Su, S.-B.; Huang, S. Epithelial–mesenchymal transition of ovarian cancer cells is sustained by Rac1 through simultaneous activation of MEK1/2 and Src signaling pathways. Oncogene 2017, 36, 1546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hu, Y.; Dai, L.; Wang, Y.; Zhou, J.; Wang, W.; Di, W.; Qiu, L. MicroRNA-7 inhibits tumor metastasis and reverses epithelial-mesenchymal transition through AKT/ERK1/2 inactivation by targeting EGFR in epithelial ovarian cancer. PLoS ONE 2014, 9, e96718. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Chang, H.; Liang, Q.; Guo, Z.; Wu, J. ZEB1 promotes prostate cancer proliferation and invasion through ERK1/2 signaling pathway. Eur. Rev. Med. Pharm. Sci. 2017, 21, 4032–4038. [Google Scholar]
- Wu, J.; Ivanov, A.I.; Fisher, P.B.; Fu, Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016, 5, e10734. [Google Scholar] [CrossRef]
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.-X.; He, H.-W.; Cai, X.-Y.; Zhou, J.; Cheng, Y.-F.; Fan, J. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3. [Google Scholar] [CrossRef]
- Xie, Y.-X.; Liao, R.; Pan, L.; Du, C.-Y. ERK pathway activation contributes to the tumor-promoting effects of hepatic stellate cells in hepatocellular carcinoma. Immunol. Lett. 2017, 188, 116–123. [Google Scholar] [CrossRef]
- Dong, F.; Liu, T.; Jin, H.; Wang, W. Chimaphilin inhibits human osteosarcoma cell invasion and metastasis through suppressing the TGF-β1-induced epithelial-to-mesenchymal transition markers via PI-3K/Akt, ERK1/2, and Smad signaling pathways. Can. J. Physiol. Pharmacol. 2017, 96, 1–7. [Google Scholar] [CrossRef]
- Hou, C.-H.; Lin, F.-L.; Hou, S.-M.; Liu, J.-F. Cyr61 promotes epithelial-mesenchymal transition and tumor metastasis of osteosarcoma by Raf-1/MEK/ERK/Elk-1/TWIST-1 signaling pathway. Mol. Cancer 2014, 13, 236. [Google Scholar] [CrossRef]
- Ding, Q.; Miyazaki, Y.; Tsukasa, K.; Matsubara, S.; Yoshimitsu, M.; Takao, S. CD133 facilitates epithelial-mesenchymal transition through interaction with the ERK pathway in pancreatic cancer metastasis. Mol. Cancer 2014, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012, 72, 6382–6392. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, T.; Deng, Q.; Zhou, Q.; Sun, X.; Li, E.; Yu, D.; Zhong, C. Benzidine Induces Epithelial–Mesenchymal Transition of Human Bladder Cancer Cells through Activation of ERK5 Pathway. Mol. Cells 2018, 41, 188. [Google Scholar] [PubMed]
- Strippoli, R.; Loureiro, J.; Moreno, V.; Benedicto, I.; Lozano, M.L.P.; Barreiro, O.; Pellinen, T.; Minguet, S.; Foronda, M.; Osteso, M.T. Caveolin-1 deficiency induces a MEK-ERK1/2-Snail-1-dependent epithelial–mesenchymal transition and fibrosis during peritoneal dialysis. EMBO Mol. Med. 2015, 7, 102–123. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ge, Y.; Jiang, M.; Zhou, J.; Luo, D.; Fan, H.; Shi, L.; Lin, L.; Yang, L. MiR-592 promotes gastric Cancer proliferation, migration, and invasion through the PI3K/AKT and MAPK/ERK signaling pathways by targeting Spry2. Cell. Physiol. Biochem. 2018, 47, 1465–1481. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, X.; Feng, M.; Yao, L.; Deng, Z.; Zao, G.; Zhou, Y.; Chen, S.; Du, Z. Downregulation of RNF138 inhibits cellular proliferation, migration, invasion and EMT in glioma cells via suppression of the Erk signaling pathway. Oncol. Rep. 2018, 40, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Kubota, Y.; Nakamura, T.; Weng, J.S.; Tomida, T.; Saito, H.; Takekawa, M. MCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial-mesenchymal transition by regulating the co-repressor CtBP. Mol. Cell 2015, 58, 35–46. [Google Scholar] [CrossRef]
- Wu, J.; Cui, H.; Zhu, Z.; Wang, L. MicroRNA-200b-3p suppresses epithelial-mesenchymal transition and inhibits tumor growth of glioma through down-regulation of ERK5. Biochem. Biophys. Res. Commun. 2016, 478, 1158–1164. [Google Scholar] [CrossRef]
- Little, A.S.; Balmanno, K.; Sale, M.J.; Newman, S.; Dry, J.R.; Hampson, M.; Edwards, P.A.; Smith, P.D.; Cook, S.J. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci. Signal. 2011, 4, ra17. [Google Scholar] [CrossRef]
- Little, A.S.; Smith, P.D.; Cook, S.J. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2013, 32, 1207–1215. [Google Scholar] [CrossRef]
- Ohori, M.; Kinoshita, T.; Okubo, M.; Sato, K.; Yamazaki, A.; Arakawa, H.; Nishimura, S.; Inamura, N.; Nakajima, H.; Neya, M.; et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 2005, 336, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.K.; Muller, J.; Ritt, D.A.; Zhou, M.; Zhou, X.Z.; Copeland, T.D.; Conrads, T.P.; Veenstra, T.D.; Lu, K.P.; Morrison, D.K. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 2005, 17, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Kelley, E.H.; Methot, J.L.; Zhou, H.; Petrocchi, A.; Chen, H.; Hill, S.E.; Hinton, M.C.; Hruza, A.; Jung, J.O.; et al. Discovery of 1-(1H-Pyrazolo[4,3-c]pyridin-6-yl)urea Inhibitors of Extracellular Signal-Regulated Kinase (ERK) for the Treatment of Cancers. J. Med. Chem. 2016, 59, 6501–6511. [Google Scholar] [CrossRef] [PubMed]
- Mendzelevski, B.; Ferber, G.; Janku, F.; Li, B.T.; Sullivan, R.J.; Welsch, D.; Chi, W.; Jackson, J.; Weng, O.; Sager, P.T. Effect of ulixertinib, a novel ERK1/2 inhibitor, on the QT/QTc interval in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2018, 81, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagdanoff, J.T.; Jain, R.; Han, W.; Zhu, S.; Madiera, A.M.; Lee, P.S.; Ma, X.; Poon, D. Tetrahydropyrrolo-diazepenones as inhibitors of ERK2 kinase. Bioorg. Med. Chem. Lett. 2015, 25, 3788–3792. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Hancock, C.N.; Macias, A.T.; Joh, J.; Still, K.; Zhong, S.; MacKerell, A.D., Jr.; Shapiro, P. Characterization of ATP-independent ERK inhibitors identified through in silico analysis of the active ERK2 structure. Bioorg. Med. Chem. Lett. 2006, 16, 6281–6287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaikuad, A.; Tacconi, E.M.; Zimmer, J.; Liang, Y.; Gray, N.S.; Tarsounas, M.; Knapp, S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S., 3rd; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, 92352. [Google Scholar] [CrossRef]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef]
- Aronov, A.M.; Baker, C.; Bemis, G.W.; Cao, J.; Chen, G.; Ford, P.J.; Germann, U.A.; Green, J.; Hale, M.R.; Jacobs, M.; et al. Flipped out: Structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J. Med. Chem. 2007, 50, 1280–1287. [Google Scholar] [CrossRef]
- Aronov, A.M.; Tang, Q.; Martinez-Botella, G.; Bemis, G.W.; Cao, J.; Chen, G.; Ewing, N.P.; Ford, P.J.; Germann, U.A.; Green, J.; et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J. Med. Chem. 2009, 52, 6362–6368. [Google Scholar] [CrossRef] [PubMed]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagdanoff, J.T.; Jain, R.; Han, W.; Poon, D.; Lee, P.S.; Bellamacina, C.; Lindvall, M. Ligand efficient tetrahydro-pyrazolopyridines as inhibitors of ERK2 kinase. Bioorg. Med. Chem. Lett. 2015, 25, 3626–3629. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.A.; Bethel, P.; Cook, C.; Davies, E.; Debreczeni, J.E.; Fairley, G.; Feron, L.; Flemington, V.; Graham, M.A.; Greenwood, R.; et al. Structure-Guided Discovery of Potent and Selective Inhibitors of ERK1/2 from a Modestly Active and Promiscuous Chemical Start Point. J. Med. Chem. 2017, 60, 3438–3450. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, D.E.; Mayer, P.; Lindel, T. Chiroptical analysis of marine sponge alkaloids sharing the pyrrolopyrazinone core. Chemistry 2004, 10, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, J.H.; Zhao, B.X.; Zhao, J.; Su, L.; Dong, W.L.; Zhang, S.L.; Miao, J.Y. Microwave-assisted synthesis, crystal structure of pyrazolo[1,5-a]pyrazin-4(5H)-ones and their selective effects on lung cancer cells. Eur. J. Med. Chem. 2011, 46, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Wang, G.; Li, Y.; Hou, K.; Yuan, Y.; Zhang, L.-J.; Song, H.-R.; Shi, W. Synthesis and biological evaluation of new pyrrolopyrazinone compounds aspotential antitumor agents. Chin. Chem. Lett. 2013, 24, 619–621. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.W.; Liu, M.C.; Yee, D.; Yau, C.; van ‘t Veer, L.J.; Symmans, W.F.; Paoloni, M.; Perlmutter, J.; Hylton, N.M.; Hogarth, M.; et al. Adaptive Randomization of Neratinib in Early Breast Cancer. N. Engl. J. Med. 2016, 375, 11–22. [Google Scholar] [CrossRef]
- De Claro, R.A.; McGinn, K.M.; Verdun, N.; Lee, S.L.; Chiu, H.J.; Saber, H.; Brower, M.E.; Chang, C.J.; Pfuma, E.; Habtemariam, B.; et al. FDA Approval: Ibrutinib for Patients with Previously Treated Mantle Cell Lymphoma and Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3586–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khozin, S.; Weinstock, C.; Blumenthal, G.M.; Cheng, J.; He, K.; Zhuang, L.; Zhao, H.; Charlab, R.; Fan, I.; Keegan, P.; et al. Osimertinib for the Treatment of Metastatic EGFR T790M Mutation-Positive Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Ohori, M.; Kinoshita, T.; Yoshimura, S.; Warizaya, M.; Nakajima, H.; Miyake, H. Role of a cysteine residue in the active site of ERK and the MAPKK family. Biochem. Biophys. Res. Commun. 2007, 353, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Worlikar, S.; MacKerell, A.D., Jr.; Shapiro, P.; Fletcher, S. Small-molecule inhibitors of the ERK signaling pathway: Towards novel anticancer therapeutics. ChemMedChem 2011, 6, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Hancock, C.N.; Macias, A.; Lee, E.K.; Yu, S.Y.; Mackerell, A.D., Jr.; Shapiro, P. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. J. Med. Chem. 2005, 48, 4586–4595. [Google Scholar] [CrossRef] [PubMed]
- Samadani, R.; Zhang, J.; Brophy, A.; Oashi, T.; Priyakumar, U.D.; Raman, E.P.; St John, F.J.; Jung, K.Y.; Fletcher, S.; Pozharski, E.; et al. Small-molecule inhibitors of ERK-mediated immediate early gene expression and proliferation of melanoma cells expressing mutated BRaf. Biochem. J. 2015, 467, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhard, K.A.; Chen, F.; Shapiro, P. Quantitative analysis of ERK2 interactions with substrate proteins: Roles for kinase docking domains and activity in determining binding affinity. J. Biol. Chem. 2011, 286, 2477–2485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, B.; Zheng, C.F.; Zhang, Z.Y. A bipartite mechanism for ERK2 recognition by its cognate regulators and substrates. J. Biol. Chem. 2003, 278, 29901–29912. [Google Scholar] [CrossRef] [PubMed]
- Robinson, F.L.; Whitehurst, A.W.; Raman, M.; Cobb, M.H. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. J. Biol. Chem. 2002, 277, 14844–14852. [Google Scholar] [CrossRef] [PubMed]
- Casar, B.; Pinto, A.; Crespo, P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaffold complexes. Mol. Cell 2008, 31, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.; Pinto, A.; Colon-Bolea, P.; Casar, B.; Jones, M.; Agudo-Ibanez, L.; Vidal, R.; Tenbaum, S.P.; Nuciforo, P.; Valdizan, E.M.; et al. Small Molecule Inhibition of ERK Dimerization Prevents Tumorigenesis by RAS-ERK Pathway Oncogenes. Cancer Cell 2015, 28, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | MAPK Family and Gene Name | UniProtKB Accession ID | Isoform | Length (Amino Acids) | Mass (Da) | Phosphorylation Sites |
|---|---|---|---|---|---|---|
| ERK1 | MAPK3 | P27361 | Isoform 1 Isoform 2 Isoform 3 | 379 335 357 | 43,136 38,275 40,088 | Thr202/Tyr204 |
| ERK2 | MAPK1 | P28482 | Isoform 1 Isoform 2 | 360 316 | 41,390 36,432 | Thr185/Tyr187 |
| ERK3 | MAPK6 | Q16659 | – | 721 | 82,681 | Ser186/Gly188 |
| ERK4 | MAPK4 | P31152 | – | 587 | 65,922 | Ser189/Gly191 |
| ERK5 | MAPK7 | Q13164 | Isoform 1 Isoform 2 Isoform 3 Isoform 4 | 816 677 533 451 | 88,386 73,218 59,328 50,152 | Thr219/Tyr221 |
| ERK7/8 | MAPK15 | Q8TD08 | Isoform 1 Isoform 2 Isoform 3 | 544 254 277 | 59,832 28,696 31,112 | Thr175 and Tyr177 |
| Inhibitor | Target | Mechanism | Reference |
|---|---|---|---|
| FR180204 | ERK1/2 | Interacts with the ATP-binding pocket and competes with the nucleotide avoiding the kinase activation | [207] |
| SCH772984 | ERK1/2, ERK4 | ATP competitive inhibitor of ERK1/2 preventing the activation of ERK by MEK | [208,209] |
| MK-8353 | ERK1/2 | Unknown mechanism | [209,210] |
| VTX-11e | ERK2 | Impair the phosphorylation of the kinase’s substrate p90RSK | [207] |
| BVD-523 | ERK1/2 | Unknown mechanism | [211] |
| “Compound 35” | ERK1/2 | Binds covalently to the ATP-binding pocket preventing the kinase activation | [212] |
| SF-3-030 | ERK2 | Inhibits ERK-dependent phosphorylation of ELK1 | [213] |
| DEL-22379 | ERK1/2 | Inhibits ERK dimerization without affecting phosphorylation or kinase activity | [207] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olea-Flores, M.; Zuñiga-Eulogio, M.D.; Mendoza-Catalán, M.A.; Rodríguez-Ruiz, H.A.; Castañeda-Saucedo, E.; Ortuño-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122885
Olea-Flores M, Zuñiga-Eulogio MD, Mendoza-Catalán MA, Rodríguez-Ruiz HA, Castañeda-Saucedo E, Ortuño-Pineda C, Padilla-Benavides T, Navarro-Tito N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer. International Journal of Molecular Sciences. 2019; 20(12):2885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122885
Chicago/Turabian StyleOlea-Flores, Monserrat, Miriam Daniela Zuñiga-Eulogio, Miguel Angel Mendoza-Catalán, Hugo Alberto Rodríguez-Ruiz, Eduardo Castañeda-Saucedo, Carlos Ortuño-Pineda, Teresita Padilla-Benavides, and Napoleón Navarro-Tito. 2019. "Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer" International Journal of Molecular Sciences 20, no. 12: 2885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122885