PEA-15 C-Terminal Tail Allosterically Modulates Death-Effector Domain Conformation and Facilitates Protein–Protein Interactions


{kind=link}
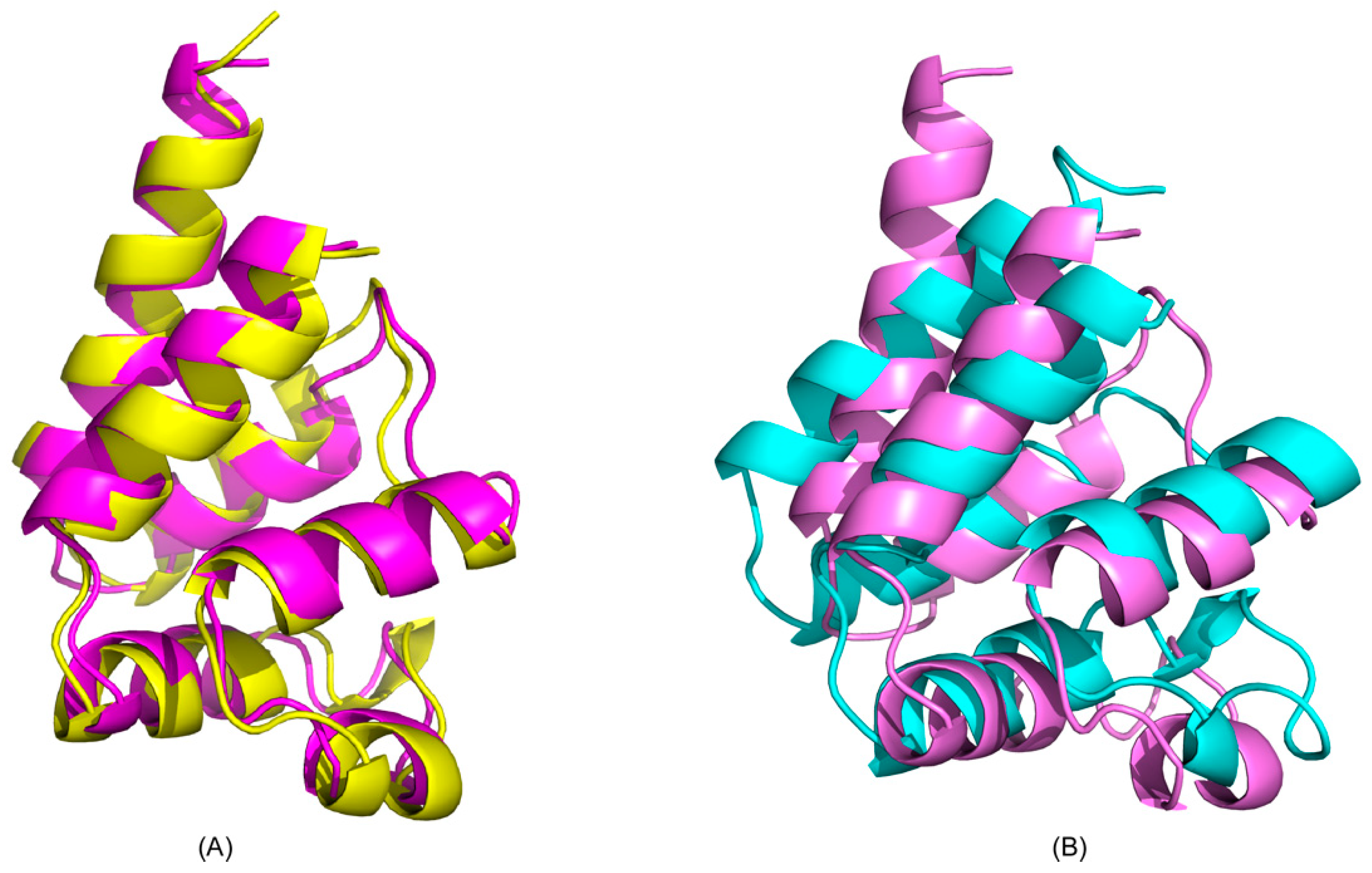{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
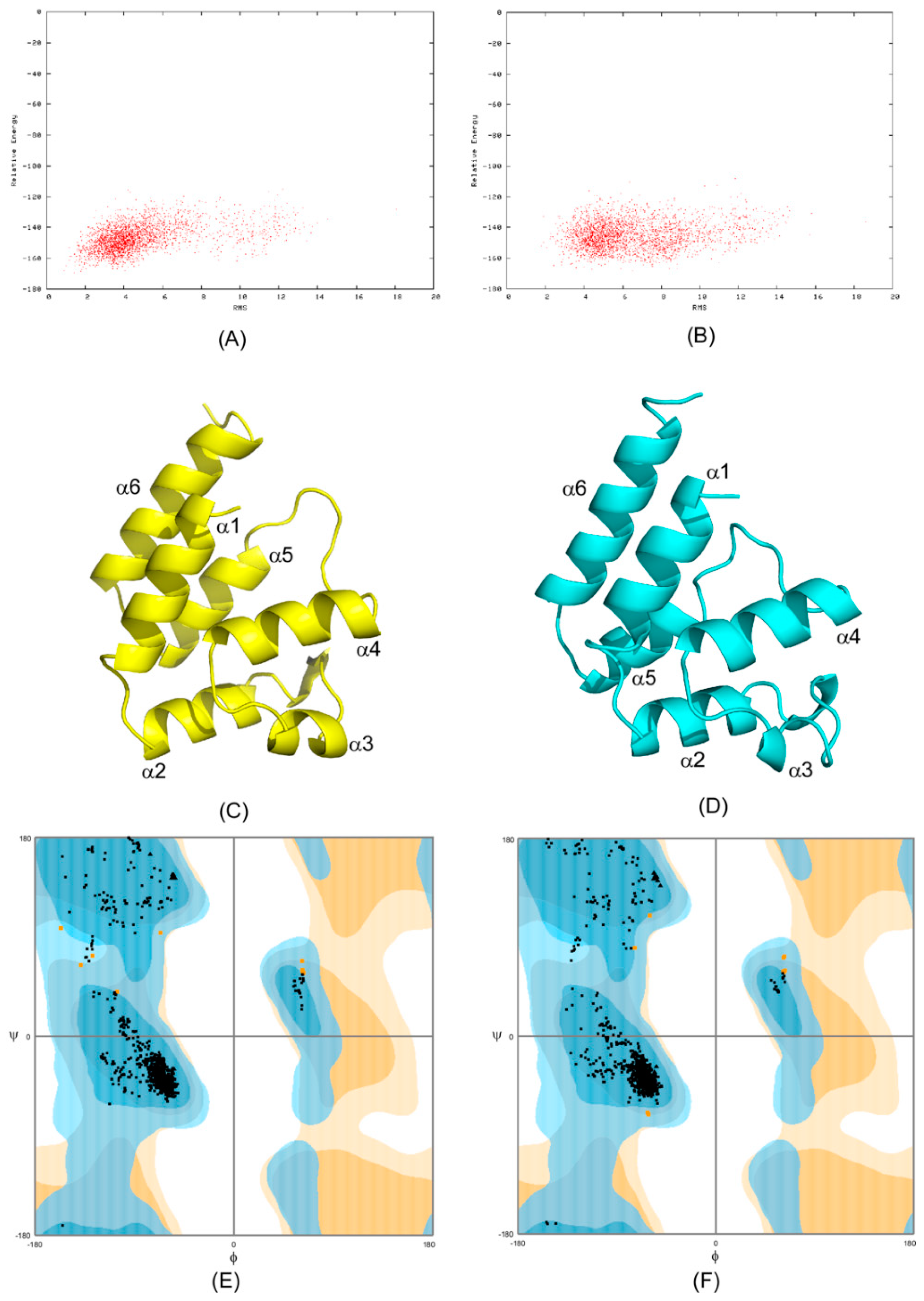
2.1. CS-Rosetta Models of PEA-15 in Free and ERK2-Bound Forms
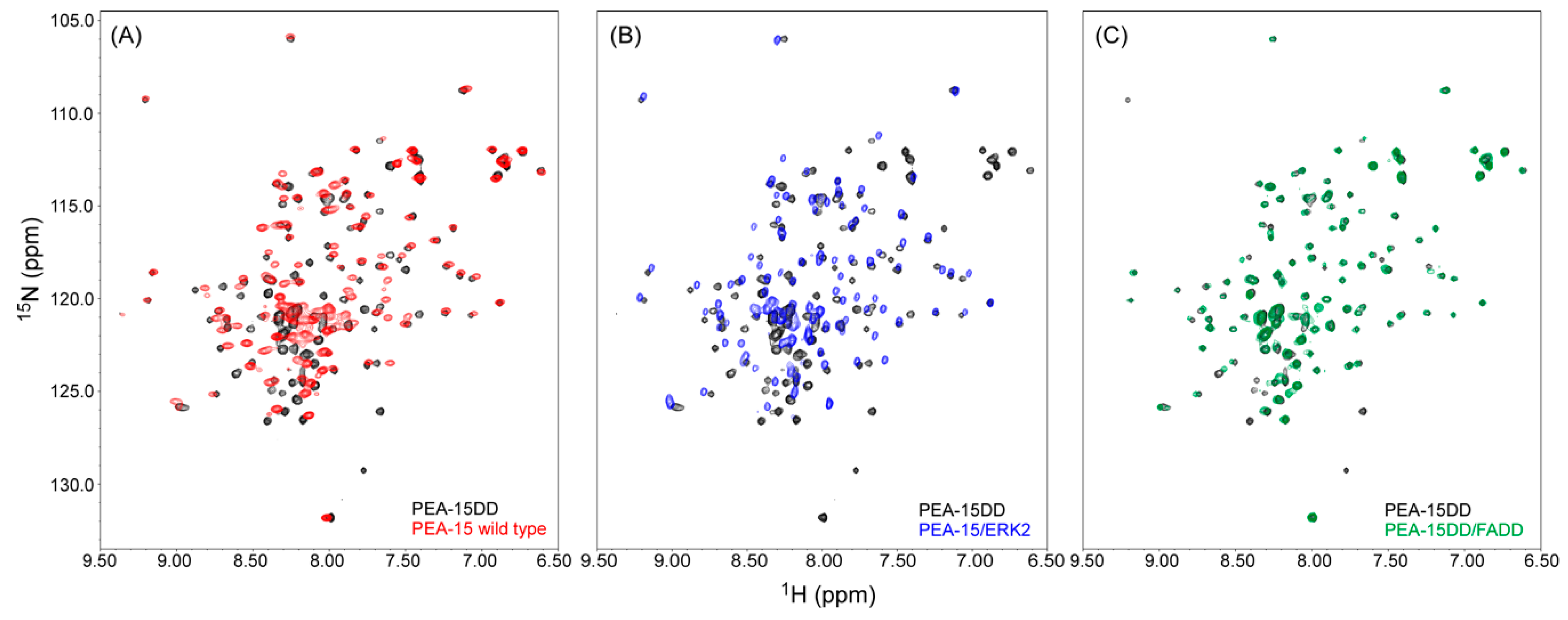
2.2. Recognition of ERK2 by PEA-15 C-Terminal Tail Allosterically Induces Conformational Change at the DED
2.3. Charge-Triad Residues Putatively Modulate Conformational Change of the DED
2.4. PEA-15 C-Terminal Tail Phosphorylation Allosterically Modulates DED Conformation to Accommodate FADD Binding
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Mutagenesis
4.3. NMR Spectroscopy
4.4. De Novo Structure Generation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PEA-15 | Phosphoprotein enriched in astrocytes, 15 kDa |
| ERK | Extracellular-signal regulated kinase |
| FADD | Fas-associated death domain |
| DED | Death-effector domain |
| NMR | Nuclear magnetic resonance |
| DISC | Death-inducing signaling complex |
| PDB | Protein Data Bank |
| BMRB | Biological Magnetic Resonance Data Bank |
| RMSD | Root-mean-square deviation |
References
- Greig, F.H.; Nixon, G.F. Phosphoprotein Enriched in Astrocytes (Pea)-15: A Potential Therapeutic Target in Multiple Disease States. Pharmacol. Therap. 2014, 143, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Fiory, F.; Spinelli, R.; Raciti, G.A.; Parrillo, L.; D’Esposito, V.; Formisano, P.; Miele, C.; Beguinot, F. Targetting Ped/Pea-15 for Diabetes Treatment. Expert Opin. Targets 2017, 21, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Redina, O.; Altshuller, Y.M.; Yamazaki, M.; Ramos, J.; Chneiweiss, H.; Kanaho, Y.; Frohman, M.A. Regulation of Expression of Phospholipase D1 and D2 by PEA-15, a Novel Protein That Interacts with Them. J. Biol. Chem. 2000, 275, 35224–35232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerolama, C.; Vigliotta, G.; Trencia, A.; Maitan, M.A.; Caruso, M.; Miele, C.; Oriente, F.; Santopietro, S.; Formisano, P.; Beguinot, F. Protein Kinase C (Pkc)-A Activation Inhibits Pkc-Ζ and Mediates the Action of Ped/Pea-15 on Glucose Transport in the L6 Skeletal Muscle Cells. Diabetes 2001, 50, 1244–1252. [Google Scholar]
- Valentino, R.; Lupoli, G.A.; Raciti, G.A.; Oriente, F.; Farinaro, E.; Valle, E.D.; Salomone, M.; Riccardi, G.; Vaccaro, O.; Donnarumma, G.; et al. The PEA15 Gene Is Overexpressed and Related to Insulin Resistance in Healthy First-Degree Relatives of Patients with Type 2 Diabetes. Diabetologia 2006, 49, 3058–3066. [Google Scholar] [CrossRef]
- Condorelli, G.; Vigliotta, G.; Cafieri, A.; Trencia, A.; Andalo, P.; Oriente, F.; Miele, C.; Caruso, M.; Formisano, P.; Beguinot, F. PED/PEA-15: An Anti-Apoptotic Molecule That Regulates Fas/Tnfr1-Induced Apoptosis. Oncogene 1999, 18, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Kitsberg, D.; Formstecher, E.; Fauquet, M.; Kubes, M.; Cordier, J.; Canton, B.; Pan, G.H.; Rolli, M.; Glowinski, J.; Chneiweiss, H. Knock-out of the Neural Death Effector Domain Protein Pea-15 Demonstrates That Its Expression Protects Astrocytes from Tnfα-Induced Apoptosis. J. Neurol. 1999, 19, 8244–8251. [Google Scholar] [CrossRef]
- Hao, C.; Beguinot, F.; Condorelli, G.; Trencia, A.; van Meir, E.G.; Yong, V.W.; Parney, I.F.; Roa, W.H.; Petruk, K.C. Induction and Intracellular Regulation of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (Trail) Mediated Apotosis in Human Malignant Glioma Cells. Cancer Res. 2001, 61, 1162–1170. [Google Scholar]
- Ramos, J.W.; Hughes, P.E.; Renshaw, M.W.; Schwartz, M.A.; Formstecher, E.; Chneiweiss, H.; Ginsberg, M.H. Death Effector Domain Protein PEA-15 Potentiates Ras Activation of Extracellular Signal Receptor-Activated Kinase by an Adhesion-Independent Mechanism. Mol. Biol. Cell 2000, 11, 2863–2872. [Google Scholar] [CrossRef]
- Formstecher, E.J.; Ramos, W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.-C.; Canton, B.; Nguyen, X.-T.; Barnier, J.-V.; Camonis, J.; Ginsberg, M.H.; et al. Pea-15 Mediates Cytoplasmic Sequestration of Erk Map Kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Glading, A.J.; Koziol, A.; Krueger, J.; Ginsberg, M.H. PEA-15 Inhibits Tumor Cell Invasion by Binding to Extracellular Signal-Regulated Kinase 1/2. Cancer Res. 2007, 67, 1536–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, F.H.; Kennedy, S.; Gibson, G.; Ramos, J.W.; Nixon, G.F. Pea-15 (Phosphoprotein Enriched in Astrocytes 15) Is a Protective Mediator in the Vasculature and Is Regulated During Neointimal Hyperplasia. J. Am. Heart Assoc. 2017, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.; Chou, F.-L.; Glading, A.; Schaefer, E.; Ginsberg, M.H. Phosphorylation of Phosphoprotein Enriched in Astrocytes (Pea-15) Regulates Extracellular Signal-Regulated Kinase-Dependent Transcription and Cell Proliferation. Mol. Biol. Cell 2005, 16, 3552–3561. [Google Scholar] [CrossRef] [PubMed]
- Renganathan, H.; Vaidyanathan, H.; Knapinska, A.; Ramos, J.W. Phosphorylation of Pea-15 Switches Its Binding Specificity from Erk/Mapk to Fadd. Biochem. J. 2005, 390, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.; Opoku-Ansah, J.; Ramos, J.W. Phosphorylation Is the Switch That Turns Pea-15 from Tumor Suppressor to Tumor Promoter. Small Gtpases 2012, 3, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, K.; Aten, S.; Queiroz, L.S.; Sullivan, K.; Oberdick, J.; Hoyt, K.R.; Obrietan, K. Circadian Expression and Functional Characterization of Pea-15 within the Mouse Suprachiasmatic Nucleus. Eur. J. Neurosci. 2018, 47, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Kaoud, T.; Ashwini, S.; Devkota, K.; Mitra, R.H.; Rana, S.; Abramczyk, O.; Warthaka, M.; Mark, S.L.; Girvin, E.; Riggs, A.F.; et al. Activated Erk2 Is a Monomer in Vitro with or without Divalent Cations and When Complexed to the Cytoplasmic Scaffold Pea-15. Biochemistry 2011, 50, 4568–4578. [Google Scholar] [CrossRef]
- Callaway, K.; Abramczyk, O.; Martin, L.; Dalby, K.N. The Anti-Apoptotic Protein PEA-15 Is a Tight Binding Inhibitor of Erk1 and Erk2, Which Blocks Docking Interactions at the d-Recruitment Site. Biochemistry 2007, 46, 9187–9198. [Google Scholar] [CrossRef]
- Hill, J.M.; Vaidyanathan, H.; Ramos, J.W.; Ginsberg, M.H.; Werner, M.H. Recognition of Erk Map Kinase by Pea-15 Reveals a Common Docking Site within the Death Domain and Death Effector Domain. Embo J. 2002, 21, 6494–6504. [Google Scholar] [CrossRef]
- Twomey, E.C.; Wei, Y. High-Definition Nmr Structure of Ped/Pea-15 Death Effector Domain Reveals Details of Key Polar Side Chain Interactions. Biophys. Res. Commun. 2012, 424, 141–146. [Google Scholar] [CrossRef]
- Twomey, E.; Dana, C.; Cordasco, F.; Wei, Y. Profound Conformational Changes of Ped/Pea-15 in Erk2 Complex Revealed by Nmr Backbone Dynamics. BBA Proteins Proteom. 2012, 1824, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.; Dana, C.; Cordasco, F.; Kozuch, S.D.; Wei, Y. Substantial Conformational Change Mediated by Charge-Triad Residues of the Death Effector Domain in Protein-Protein Interactions. PLoS ONE 2013, 8, e83421. [Google Scholar] [CrossRef] [PubMed]
- Mace, P.D.; Wallez, Y.; Egger, M.F.; Dobaczewska, M.K.; Robinson, H.; Pasquale, B.; Riedl, S.J. Structure of Erk2 Bound to PEA-15 Reveals a Mechanism for Rapid Release of Activated Mapk. Nat. Commun. 2013, 4, 1681. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent Blind Protein Structure Generation from Nmr Chemical Shift Data. Proc. Natl. Acad. Sci. USA 2008, 105, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Vernon, R.; Baker, D.; Bax, A. De Novo Protein Structure Generation from Incomplete Chemical Shift Assignments. J. Biomol. NMR 2009, 43, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bryan, P.N.; He, Y.; Orban, J.; Baker, D.; Bax, A. De Novo Structure Generation Using Chemical Shifts for Proteins with High-Sequence Identity but Different Folds. Protein Sci. 2010, 19, 349–356. [Google Scholar] [CrossRef]
- Lange, O.F.; Rossi, P.; Sgourakis, N.G.; Song, Y.; Lee, H.-W.; Aramini, J.M.; Ertekin, A.; Xiao, R.; Acton, T.B.; Montelione, G.T.; et al. Determination of Solution Structures of Proteins up to 40 kda Using Cs-Rosetta with Sparse Nmr Data from Deuterated Samples. Proc. Natl. Acad. Sci. USA 2012, 109, 10873–10878. [Google Scholar] [CrossRef]
- Lee, J.; Bartholomeusz, C.; Krishnamurthy, S.; Liu, P.; Saso, H.; Lafortune, T.A.; Hortobagyi, G.N.; Ueno, N.T. PEA-15 Unphosphorylated at Both Serine 104 and Serine 116 Inhibits Ovarian Cancer Cell Tumorigenicity and Progression through Blocking Beta-Catenin. Oncogenesis 2012, 1, 22. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd.; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure Validation by Calpha Geometry: Phi,Psi and Cbeta Deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Callaway, K.; Rainey, M.A.; Dalby, K.N. Quantifying Erk2-Protein Interactions by Fluorescence Anisotropy: PEA-15 Inhibits Erk2 by Blocking the Binding of Dejl Domains. BBA Proteins Proteom. 2005, 1754, 316–323. [Google Scholar] [CrossRef]
- Valmiki, M.; Ramos, J. Death Effector Domain-Containing Proteins. Cell. Mol. Life Sci. 2009, 66, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Valmiki, M.K.; Nelson, D.A.; Caliva, M.J.; Geerts, D.; White, M.E.P.; Ramos, J.W. PEA-15 Potentiates H-Ras-Mediated Epithelial Cell Transformation through Phospholipase D. Oncogene 2012, 31, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Robbins, D.J.; Cobb, M.H.; Goldsmith, E.J. Crystallization and Preliminary X-Ray Studies of Extracellular Signal-Regulated Kinase-2/Map Kinase with an Incorporated His-Tag. J. Mol. Biol. 1993, 233, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Yao, X. Trosy-Based Nmr Experiments for Nmr Studies of Large Biomolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2008, 52, 49–68. [Google Scholar] [CrossRef]
- Sattler, M.; Schleucher, J.; Griesinger, C. Heteronuclear Multidimensional Nmr Experiments for the Structure Determination of Proteins in Solution Employing Pulsed Field Gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 93–158. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. Nmrpipe: A Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Blevins, R.A. Nmr View: A Computer Program for the Visualization and Analysis of Nmr Data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef]
- Wei, Y. On the Quest of Cellular Functions of PEA-15 and the Therapeutic Opportunities. Pharmaceuticals 2015, 8, 455–473. [Google Scholar] [CrossRef] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crespo-Flores, S.L.; Cabezas, A.; Hassan, S.; Wei, Y. PEA-15 C-Terminal Tail Allosterically Modulates Death-Effector Domain Conformation and Facilitates Protein–Protein Interactions. Int. J. Mol. Sci. 2019, 20, 3335. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133335
Crespo-Flores SL, Cabezas A, Hassan S, Wei Y. PEA-15 C-Terminal Tail Allosterically Modulates Death-Effector Domain Conformation and Facilitates Protein–Protein Interactions. International Journal of Molecular Sciences. 2019; 20(13):3335. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133335
Chicago/Turabian StyleCrespo-Flores, Sergio L., Andres Cabezas, Sherouk Hassan, and Yufeng Wei. 2019. "PEA-15 C-Terminal Tail Allosterically Modulates Death-Effector Domain Conformation and Facilitates Protein–Protein Interactions" International Journal of Molecular Sciences 20, no. 13: 3335. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133335