Cdh4 Down-Regulation Impairs in Vivo Infiltration and Malignancy in Patients Derived Glioblastoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
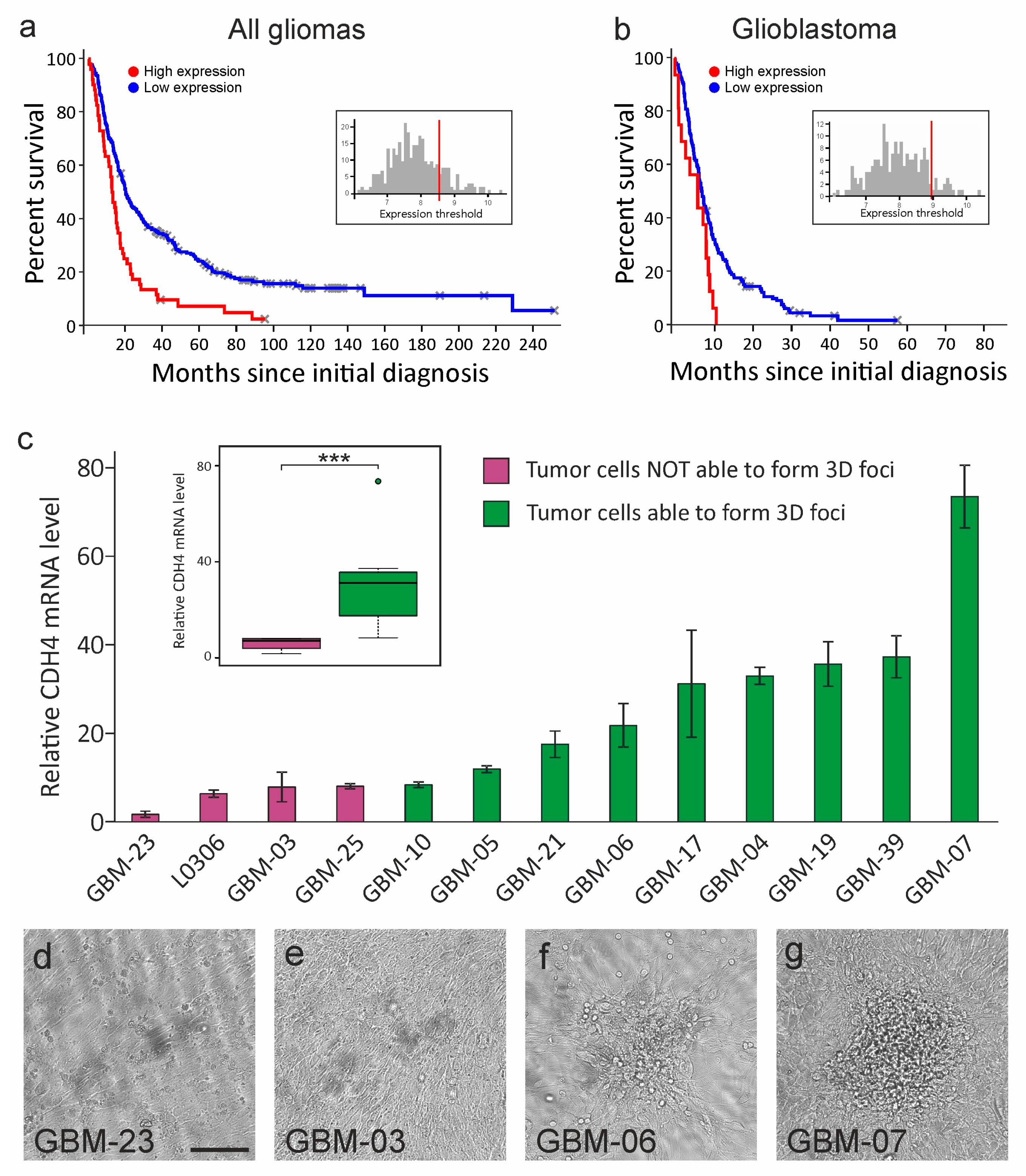
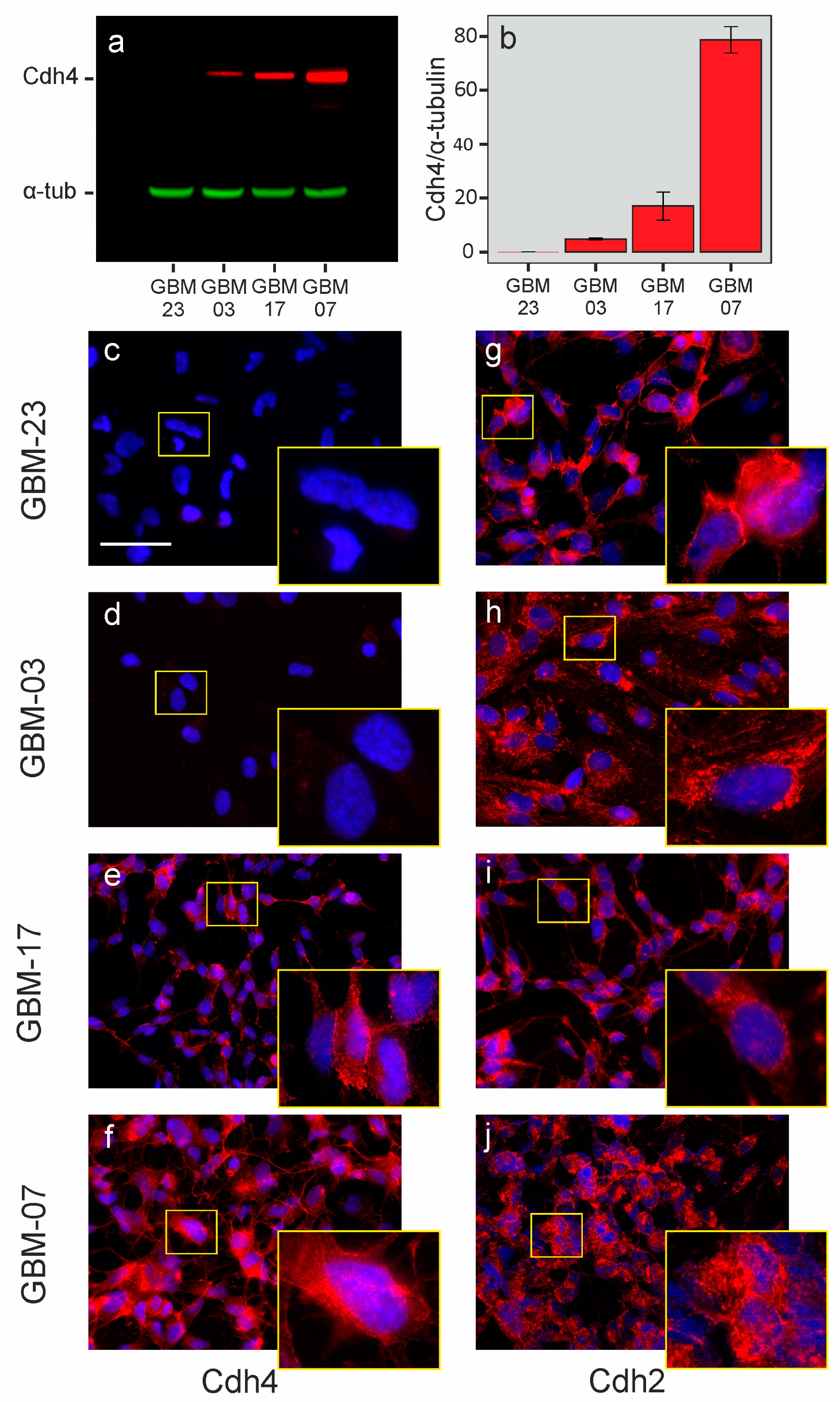
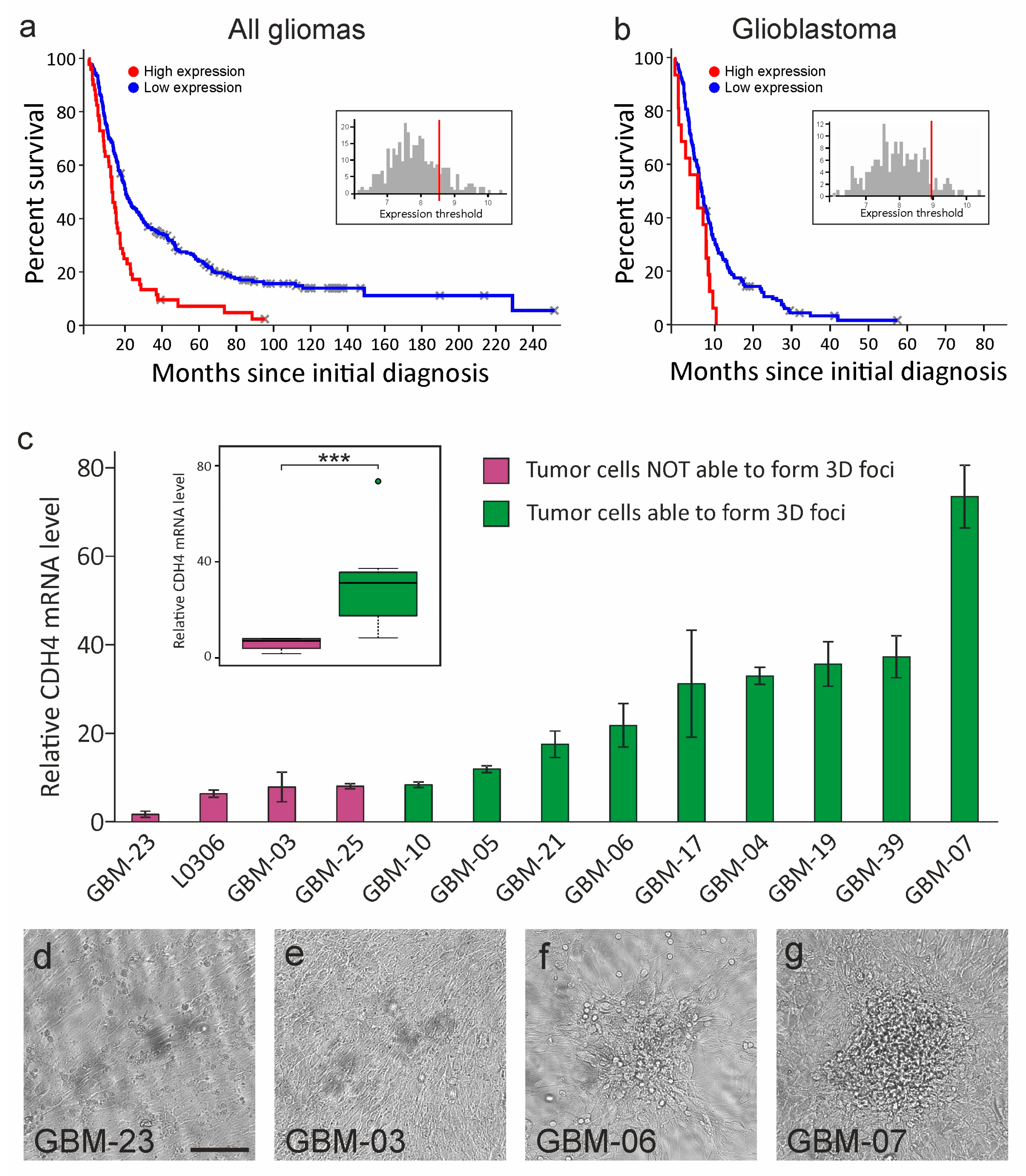
2.1. Cdh4 Is Heterogeneously Expressed by Human Gliomas and Highlights Tumors Ability to Bypass the Cell–Cell Contact Inhibition of Proliferation
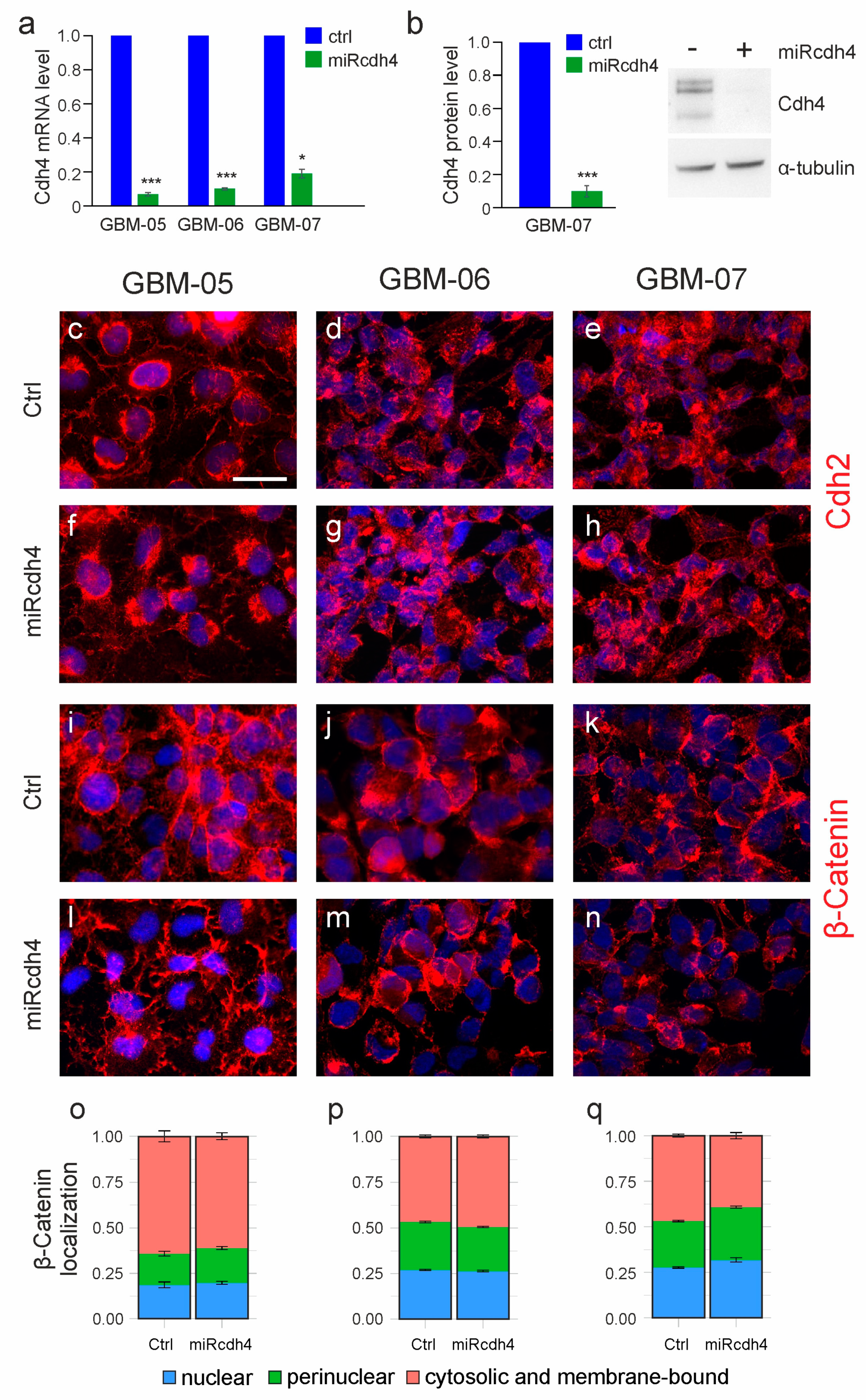
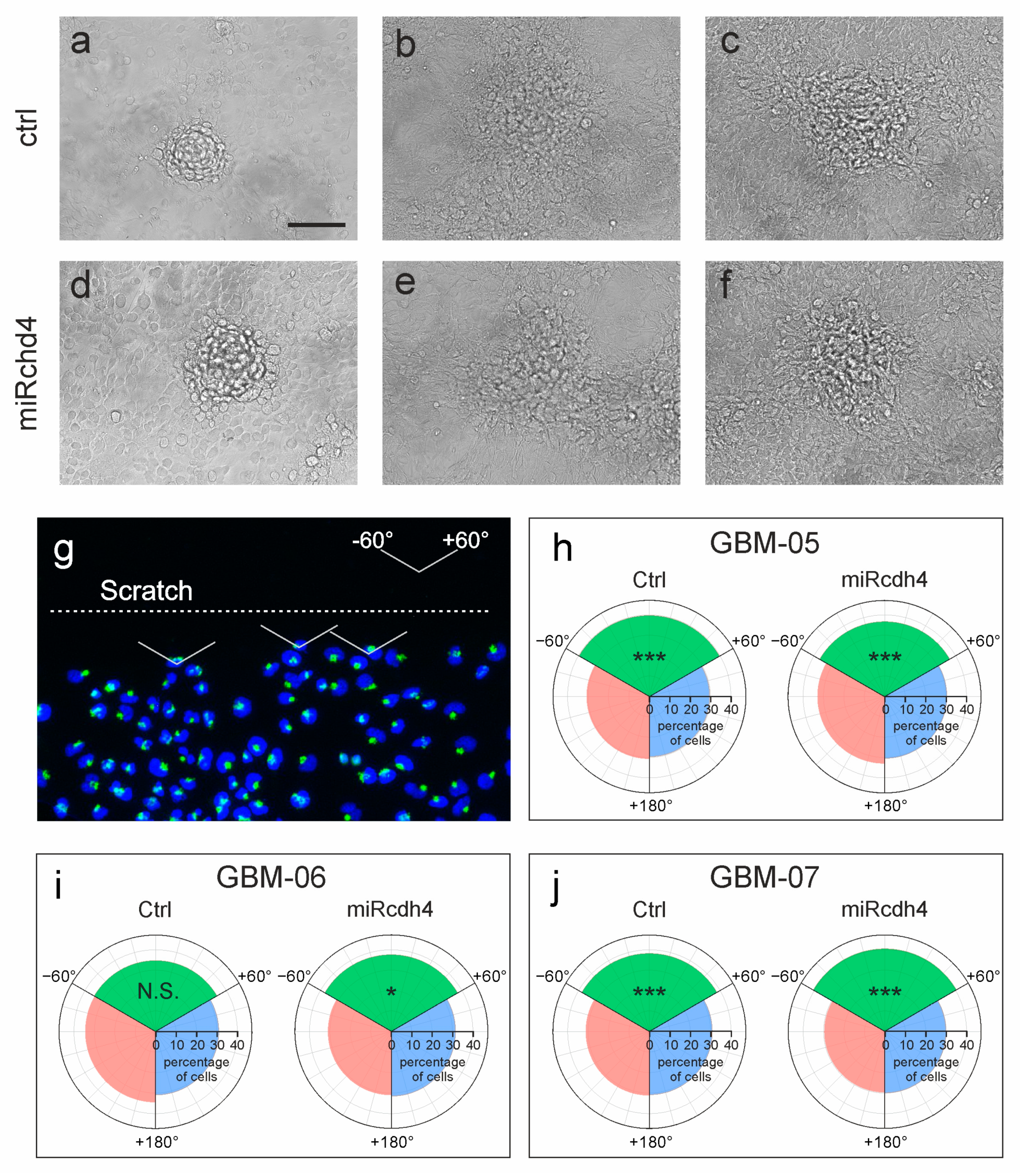
2.2. The Silencing of Cdh4 Is Not Sufficient to Restore Cell–Cell Contact Inhibition of Proliferation
2.3. The Silencing of Cdh4 Reduces Tumor Infiltration and Proliferation
3. Discussion
4. Materials and Methods
4.1. Vectors
4.2. Glioma Initiating Cells Culture
4.3. Animal Procedures
4.4. Immunostaining
4.5. Quantitative PCR and Western Blot
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EMT | epithelial to mesenchymal transition |
| Cdh1 | E-Cadherin |
| Cdh2 | N-Cadherin |
| CIP | cell-cell inhibition of proliferation |
| CIM | cell-cell inhibition of migration |
| GIC | glioblastoma initiating cell |
| OPBA | Animal Ethics Committee |
| NOD/SCID | NOD.CB17-Prkdcscid/J |
References
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and molecular pathology of glioma. Nat. Rev. Neurol. 2006, 2, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Pal, I.; Banik, P.; Chakraborty, S.; Borkar, S.A.; Dey, G.; Mukherjee, A.; Mandal, M. Insights into molecular therapy of glioma: Current challenges and next generation blueprint. Acta. Pharm. Sin. 2017, 38, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell Mol. Life Sci. 2018, 75, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef] [PubMed]
- Kubelt, C.; Hattermann, K.; Sebens, S.; Mehdorn, H.M.; Held-Feindt, J. Epithelial-to-mesenchymal transition in paired human primary and recurrent glioblastomas. Int. J. Oncol. 2015, 46, 2515–2525. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro. Oncol. 2014, 16, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Zarkoob, H.; Taube, J.H.; Singh, S.K.; Mani, S.A.; Kohandel, M. Investigating the link between molecular subtypes of glioblastoma, epithelial-mesenchymal transition, and CD133 cell surface protein. PLoS ONE 2013, 8, e64169. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, M.; Nakamura, S.; Machon, O.; Krauss, S.; Radice, G.L.; Takeichi, M. N-cadherin mediates cortical organization in the mouse brain. Dev. Biol. 2007, 304, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Inuzuka, H.; Miyatani, S.; Takeichi, M. R-cadherin: A novel Ca(2+)-dependent cell-cell adhesion molecule expressed in the retina. Neuron 1991, 7, 69–79. [Google Scholar] [CrossRef]
- Asano, K.; Kubo, O.; Tajika, Y.; Huang, M.C.; Takakura, K.; Ebina, K.; Suzuki, S. Expression and role of cadherins in astrocytic tumors. Brain Tumor Pathol. 1997, 14, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Cai, J.; Jiang, C. CDH2 expression is of prognostic significance in glioma and predicts the efficacy of temozolomide therapy in patients with glioblastoma. Oncol. Lett. 2018, 15, 7415–7422. [Google Scholar] [PubMed]
- Utsuki, S.; Sato, Y.; Oka, H.; Tsuchiya, B.; Suzuki, S.; Fujii, K. Relationship between the expression of E-, N-cadherins and beta-catenin and tumor grade in astrocytomas. J. Neurooncol 2002, 57, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Camand, E.; Peglion, F.; Osmani, N.; Sanson, M.; Etienne-Manneville, S. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J. Cell Sci. 2012, 125, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, G.; Magro, G.; Cardile, V.; Coco, M.; Marzagalli, R.; Castrogiovanni, P.; Imbesi, R.; Graziano, A.C.; Barone, F.; Di Rosa, M.; et al. Characterization of matrix metalloproteinase-2 and -9, ADAM-10 and N-cadherin expression in human glioblastoma multiforme. Cell Tissue Res. 2015, 362, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chang, R.; Xu, F.; Gao, Y.; Yang, F.; Wang, C.; Xiao, J.; Su, Z.; Bi, Y.; Wang, L.; et al. N-Glycosylation at Asn 402 Stabilizes N-Cadherin and Promotes Cell-Cell Adhesion of Glioma Cells. J. Cell Biochem. 2017, 118, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Appolloni, I.; Barilari, M.; Caviglia, S.; Gambini, E.; Reisoli, E.; Malatesta, P. A cadherin switch underlies malignancy in high-grade gliomas. Oncogene 2015, 34, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Krishna, K.K.; Hertel, N.; Redies, C. Cadherin expression in the somatosensory cortex: Evidence for a combinatorial molecular code at the single-cell level. Neuroscience 2011, 175, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Babb, S.G.; Kotradi, S.M.; Shah, B.; Chiappini-Williamson, C.; Bell, L.N.; Schmeiser, G.; Chen, E.; Liu, Q.; Marrs, J.A. Zebrafish R-cadherin (Cdh4) controls visual system development and differentiation. Dev. Dyn. 2005, 233, 930–945. [Google Scholar] [CrossRef] [PubMed]
- Akins, M.R.; Benson, D.L.; Greer, C.A. Cadherin expression in the developing mouse olfactory system. J. Comp. Neurol. 2007, 501, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Redies, C.; Engelhart, K.; Takeichi, M. Differential expression of N- and R-cadherin in functional neuronal systems and other structures of the developing chicken brain. J. Comp. Neurol. 1993, 333, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Redies, C.; Inuzuka, H.; Takeichi, M. Restricted expression of N- and R-cadherin on neurites of the developing chicken CNS. J. Neurosci 1992, 12, 3525–3534. [Google Scholar] [CrossRef] [Green Version]
- Stoykova, A.; Gotz, M.; Gruss, P.; Price, J. Pax6-dependent regulation of adhesive patterning, R-cadherin expression and boundary formation in developing forebrain. Development 1997, 124, 3765–3777. [Google Scholar] [PubMed]
- Adhikary, S.; Chakravarti, D.; Terranova, C.; Sengupta, I.; Maitituoheti, M.; Dasgupta, A.; Srivastava, D.K.; Ma, J.; Raman, A.T.; Tarco, E.; et al. Atypical plant homeodomain of UBR7 functions as an H2BK120Ub ligase and breast tumor suppressor. Nat. Commun. 2019, 10, 1398. [Google Scholar] [CrossRef]
- Agiostratidou, G.; Li, M.; Suyama, K.; Badano, I.; Keren, R.; Chung, S.; Anzovino, A.; Hulit, J.; Qian, B.; Bouzahzah, B.; et al. Loss of retinal cadherin facilitates mammary tumor progression and metastasis. Cancer Res. 2009, 69, 5030–5038. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, X.; Cui, N.; Liang, Y. Cadherins Associate with Distinct Stem Cell-Related Transcription Factors to Coordinate the Maintenance of Stemness in Triple-Negative Breast Cancer. Stem Cells Int. 2017, 2017, 5091541. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, G.; Zhang, C.; Lin, M.; Liu, X.; Zeng, Y.; Liu, J. Long non-coding RNA linc-cdh4-2 inhibits the migration and invasion of HCC cells by targeting R-cadherin pathway. Biochem. Biophys. Res. Commun. 2016, 480, 348–354. [Google Scholar] [CrossRef]
- Li, Z.; Su, D.; Ying, L.; Yu, G.; Mao, W. Study on expression of CDH4 in lung cancer. World J. Surg. Oncol. 2017, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Lu, J.; Zou, C.; Shao, Y.; Chen, Y.; Narala, S.; Fang, H.; Xu, H.; Wang, J.; Shen, J.; et al. CDH4 is a novel determinant of osteosarcoma tumorigenesis and metastasis. Oncogene 2018, 37, 3617–3630. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.; Mukouyama, Y.S.; Gilbert, M.R.; Jackson, S. Targeting WNT Signaling for Multifaceted Glioblastoma Therapy. Front. Cell Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Iorio, P.D.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, A.; Louvard, D.; Singer, S.J. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc. Natl. Acad. Sci. USA 1982, 79, 2603–2607. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Shen, Y.C.; Babb-Clendenon, S.G.; Rostedt, J.; Liu, B.; Barald, K.F.; Marrs, J.A.; Liu, Q. Cadherin-4 plays a role in the development of zebrafish cranial ganglia and lateral line system. Dev. Dyn. 2007, 236, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Andrews, G.L.; Mastick, G.S. R-cadherin is a Pax6-regulated, growth-promoting cue for pioneer axons. J. Neurosci. 2003, 23, 9873–9880. [Google Scholar] [CrossRef]
- Gliem, M.; Weisheit, G.; Mertz, K.D.; Endl, E.; Oberdick, J.; Schilling, K. Expression of classical cadherins in the cerebellar anlage: Quantitative and functional aspects. Mol. Cell Neurosci 2006, 33, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Hertel, N.; Redies, C. Absence of layer-specific cadherin expression profiles in the neocortex of the reeler mutant mouse. Cereb. Cortex 2011, 21, 1105–1117. [Google Scholar] [CrossRef]
- Martinez-Garay, I.; Gil-Sanz, C.; Franco, S.J.; Espinosa, A.; Molnar, Z.; Mueller, U. Cadherin 2/4 signaling via PTP1B and catenins is crucial for nucleokinesis during radial neuronal migration in the neocortex. Development 2016, 143, 2121–2134. [Google Scholar] [CrossRef]
- Hertel, N.; Redies, C.; Medina, L. Cadherin expression delineates the divisions of the postnatal and adult mouse amygdala. J. Comp. Neurol. 2012, 520, 3982–4012. [Google Scholar] [CrossRef]
- Matsunaga, E.; Nambu, S.; Oka, M.; Iriki, A. Differential cadherin expression in the developing postnatal telencephalon of a New World monkey. J. Comp. Neurol. 2013, 521, 4027–4060. [Google Scholar] [CrossRef]
- Campbell, K.; Casanova, J. A role for E-cadherin in ensuring cohesive migration of a heterogeneous population of non-epithelial cells. Nat. Commun. 2015, 6, 7998. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Martinez-Rico, C.; Pincet, F.; Thiery, J.P.; Dufour, S. Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J. Cell Sci. 2010, 123, 712–722. [Google Scholar] [CrossRef] [Green Version]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar] [CrossRef]
- Shamir, E.R.; Ewald, A.J. Adhesion in mammary development: Novel roles for E-cadherin in individual and collective cell migration. Curr. Top. Dev. Biol. 2015, 112, 353–382. [Google Scholar]
- Kowalski, P.J.; Rubin, M.A.; Kleer, C.G. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003, 5, R217–R222. [Google Scholar] [CrossRef]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Delva, E.; Kowalczyk, A.P. Regulation of cadherin trafficking. Traffic 2009, 10, 259–267. [Google Scholar] [CrossRef]
- Adam, A.P. Regulation of Endothelial Adherens Junctions by Tyrosine Phosphorylation. Mediat. Inflamm. 2015, 2015, 272858. [Google Scholar] [CrossRef]
- Kowalczyk, A.P.; Nanes, B.A. Adherens junction turnover: Regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell. Biochem. 2012, 60, 197–222. [Google Scholar]
- McEwen, A.E.; Maher, M.T.; Mo, R.; Gottardi, C.J. E-cadherin phosphorylation occurs during its biosynthesis to promote its cell surface stability and adhesion. Mol. Biol. Cell 2014, 25, 2365–2374. [Google Scholar] [CrossRef]
- Chu, Y.S.; Eder, O.; Thomas, W.A.; Simcha, I.; Pincet, F.; Ben-Ze’ev, A.; Perez, E.; Thiery, J.P.; Dufour, S. Prototypical type I E-cadherin and type II cadherin-7 mediate very distinct adhesiveness through their extracellular domains. J. Biol. Chem. 2006, 281, 2901–2910. [Google Scholar] [CrossRef]
- Thiery, J.P.; Engl, W.; Viasnoff, V.; Dufour, S. Biochemical and biophysical origins of cadherin selectivity and adhesion strength. Curr. Opin. Cell Biol. 2012, 24, 614–619. [Google Scholar] [CrossRef]
- Gotz, M.; Wizenmann, A.; Reinhardt, S.; Lumsden, A.; Price, J. Selective adhesion of cells from different telencephalic regions. Neuron 1996, 16, 551–564. [Google Scholar] [CrossRef]
- Shan, W.S.; Tanaka, H.; Phillips, G.R.; Arndt, K.; Yoshida, M.; Colman, D.R.; Shapiro, L. Functional cis-heterodimers of N- and R-cadherins. J. Cell Biol. 2000, 148, 579–590. [Google Scholar] [CrossRef]
- Kim, C.L.; Choi, S.H.; Mo, J.S. Role of the Hippo Pathway in Fibrosis and Cancer. Cells 2019, 8, 468. [Google Scholar] [CrossRef]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar]
- Carnero, A.; Paramio, J.M. The PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front. Oncol. 2014, 4, 252. [Google Scholar] [CrossRef]
- Dorard, C.; Vucak, G.; Baccarini, M. Deciphering the RAS/ERK pathway in vivo. Biochem. Soc. Trans. 2017, 45, 27–36. [Google Scholar] [CrossRef]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117 Pt 20, 4619–4628. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Hemmings, B.A. PKB/Akt-dependent regulation of cell motility. J. Natl. Cancer Inst. 2013, 105, 393–404. [Google Scholar] [CrossRef]
- Terrile, M.; Appolloni, I.; Calzolari, F.; Perris, R.; Tutucci, E.; Malatesta, P. PDGF-B-driven gliomagenesis can occur in the absence of the proteoglycan NG2. BMC Cancer 2010, 10, 550. [Google Scholar] [CrossRef]
- Appolloni, I.; Curreli, S.; Caviglia, S.; Barilari, M.; Gambini, E.; Pagano, A.; Malatesta, P. Role of Btg2 in the progression of a PDGF-induced oligodendroglioma model. Int. J. Mol. Sci. 2012, 13, 14667–14678. [Google Scholar] [CrossRef]
- Appolloni, I.; Alessandrini, F.; Ceresa, D.; Marubbi, D.; Gambini, E.; Reverberi, D.; Loiacono, F.; Malatesta, P. Progression from low- to high-grade in a glioblastoma model reveals the pivotal role of immunoediting. Cancer Lett. 2019, 442, 213–221. [Google Scholar] [CrossRef]
- Alessandrini, F.; Ceresa, D.; Appolloni, I.; Marubbi, D.; Malatesta, P. Noninvasive Monitoring of Glioma Growth in the Mouse. J. Cancer 2016, 7, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2008; Version 3.5.2; Available online: https://www.R-project.org (accessed on 23 July 2019).
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurman, N. Advanced methods of microscope control using muManager software. J. Biol. Methods 2014, 1, e10. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceresa, D.; Alessandrini, F.; Bosio, L.; Marubbi, D.; Reverberi, D.; Malatesta, P.; Appolloni, I. Cdh4 Down-Regulation Impairs in Vivo Infiltration and Malignancy in Patients Derived Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 4028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164028
Ceresa D, Alessandrini F, Bosio L, Marubbi D, Reverberi D, Malatesta P, Appolloni I. Cdh4 Down-Regulation Impairs in Vivo Infiltration and Malignancy in Patients Derived Glioblastoma Cells. International Journal of Molecular Sciences. 2019; 20(16):4028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164028
Chicago/Turabian StyleCeresa, Davide, Francesco Alessandrini, Lorenzo Bosio, Daniela Marubbi, Daniele Reverberi, Paolo Malatesta, and Irene Appolloni. 2019. "Cdh4 Down-Regulation Impairs in Vivo Infiltration and Malignancy in Patients Derived Glioblastoma Cells" International Journal of Molecular Sciences 20, no. 16: 4028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164028