FKRR-MVSF: A Fuzzy Kernel Ridge Regression Model for Identifying DNA-Binding Proteins by Multi-View Sequence Features via Chou’s Five-Step Rule


Abstract
:1. Introduction
2. Results
2.1. Data Sets
2.2. Measurements
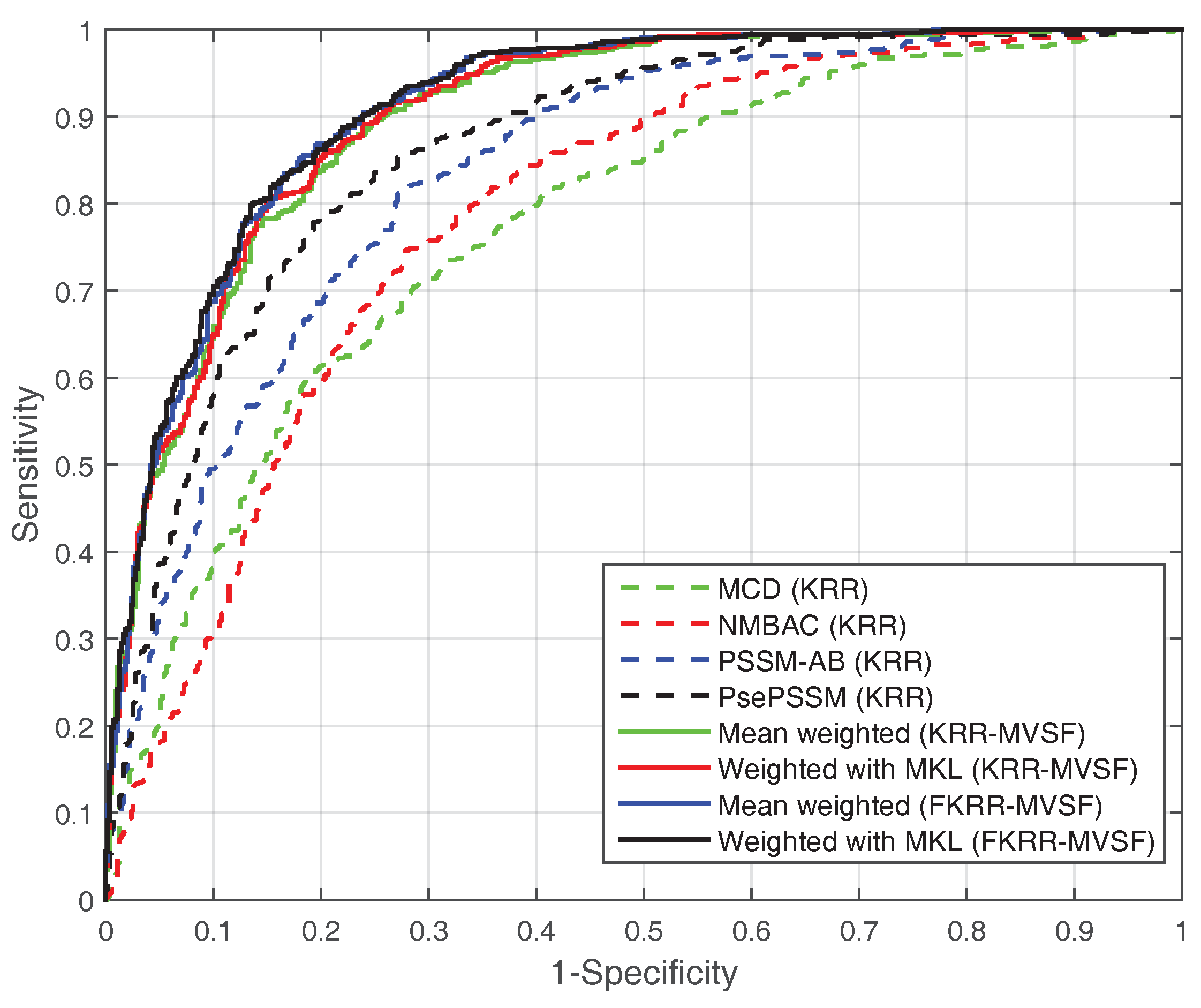
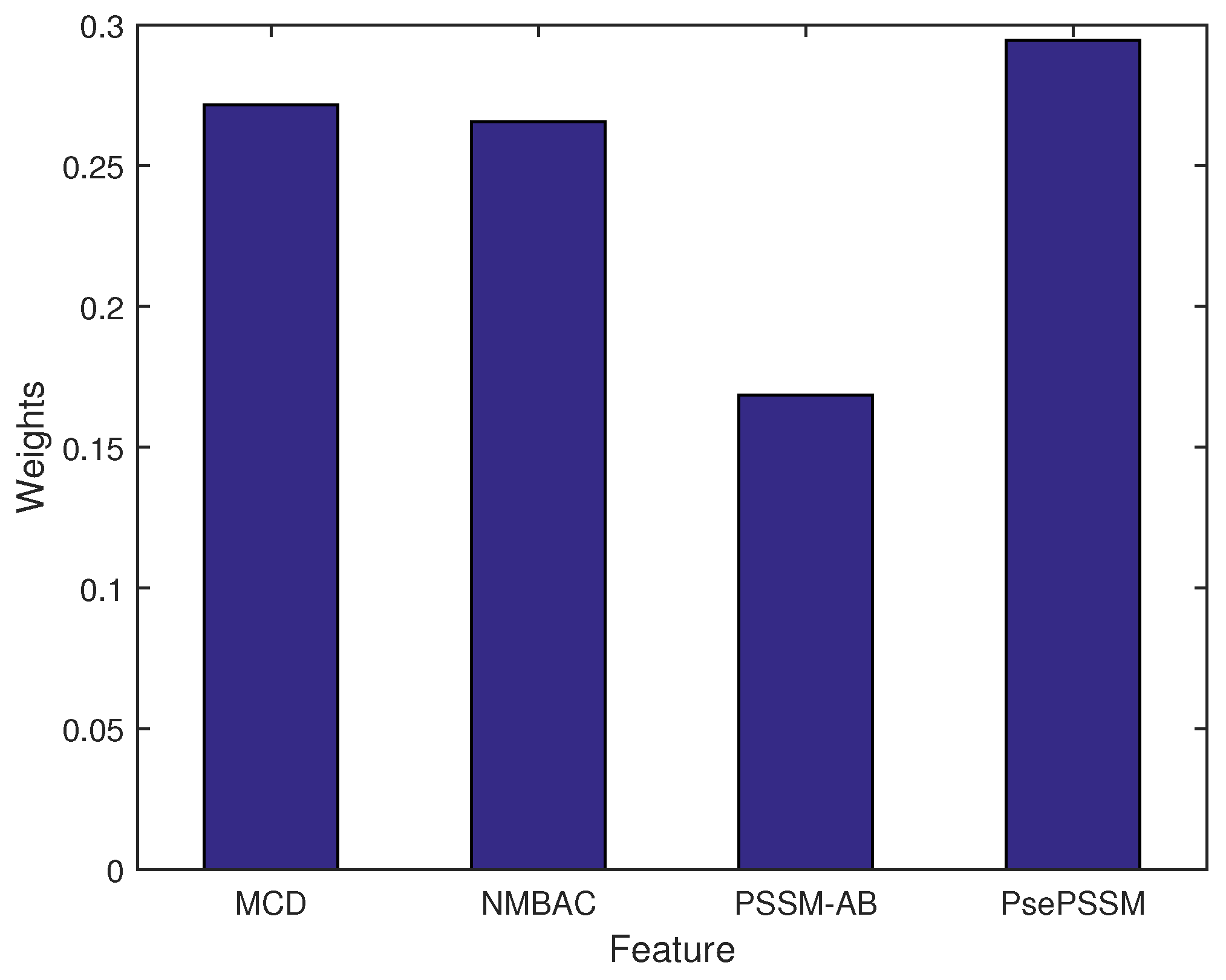
2.3. Performance Analysis of Different Features on the PDB1075 Data Set
2.4. Performance on an Independent DataSet of PDB186
3. Discussion
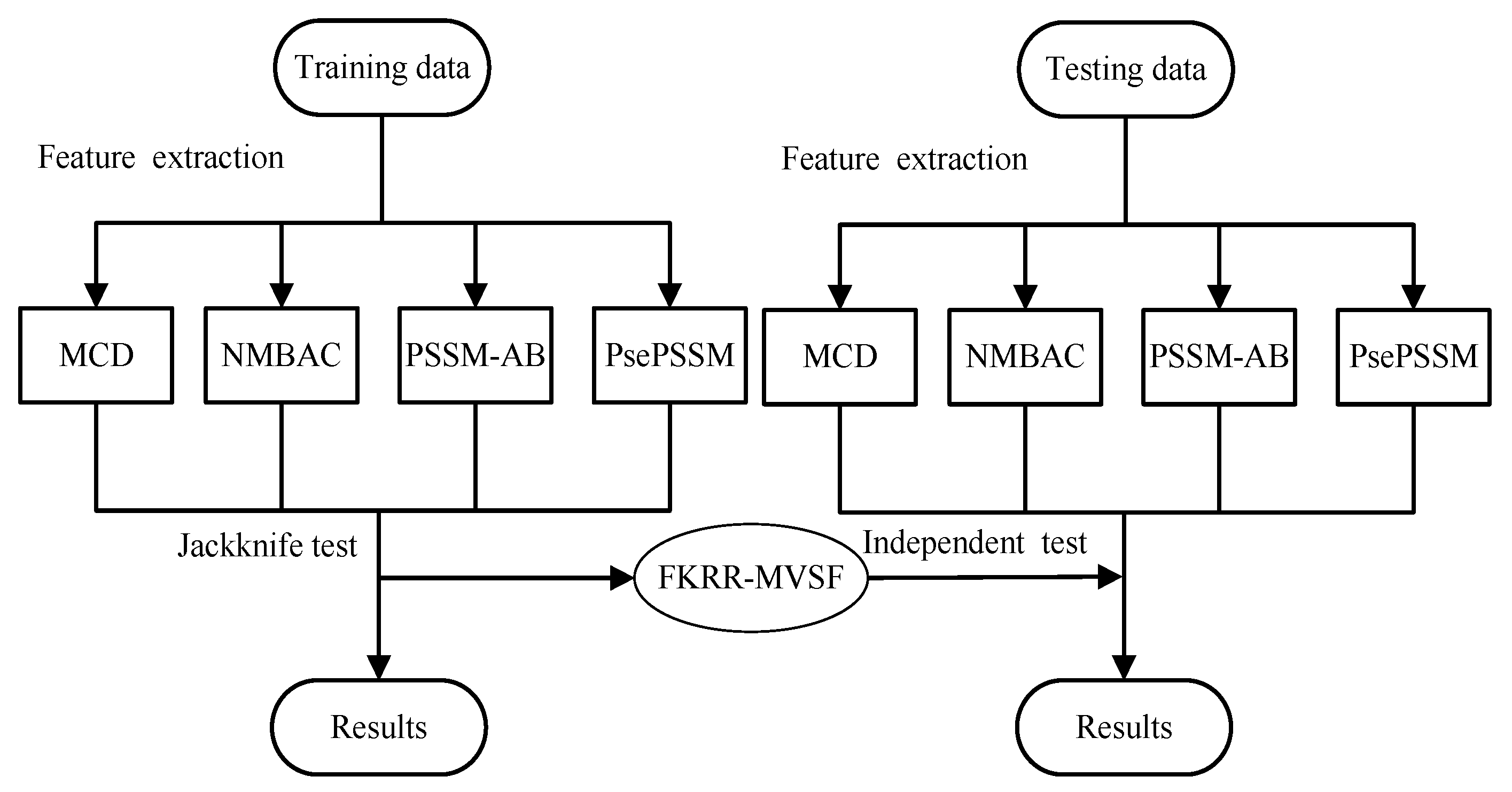
4. Materials and Methods
4.1. Feature Extraction
4.1.1. MCD Feature
4.1.2. NMBAC Feature
4.1.3. PSSM-AB Feature
4.1.4. PsePSSM Feature
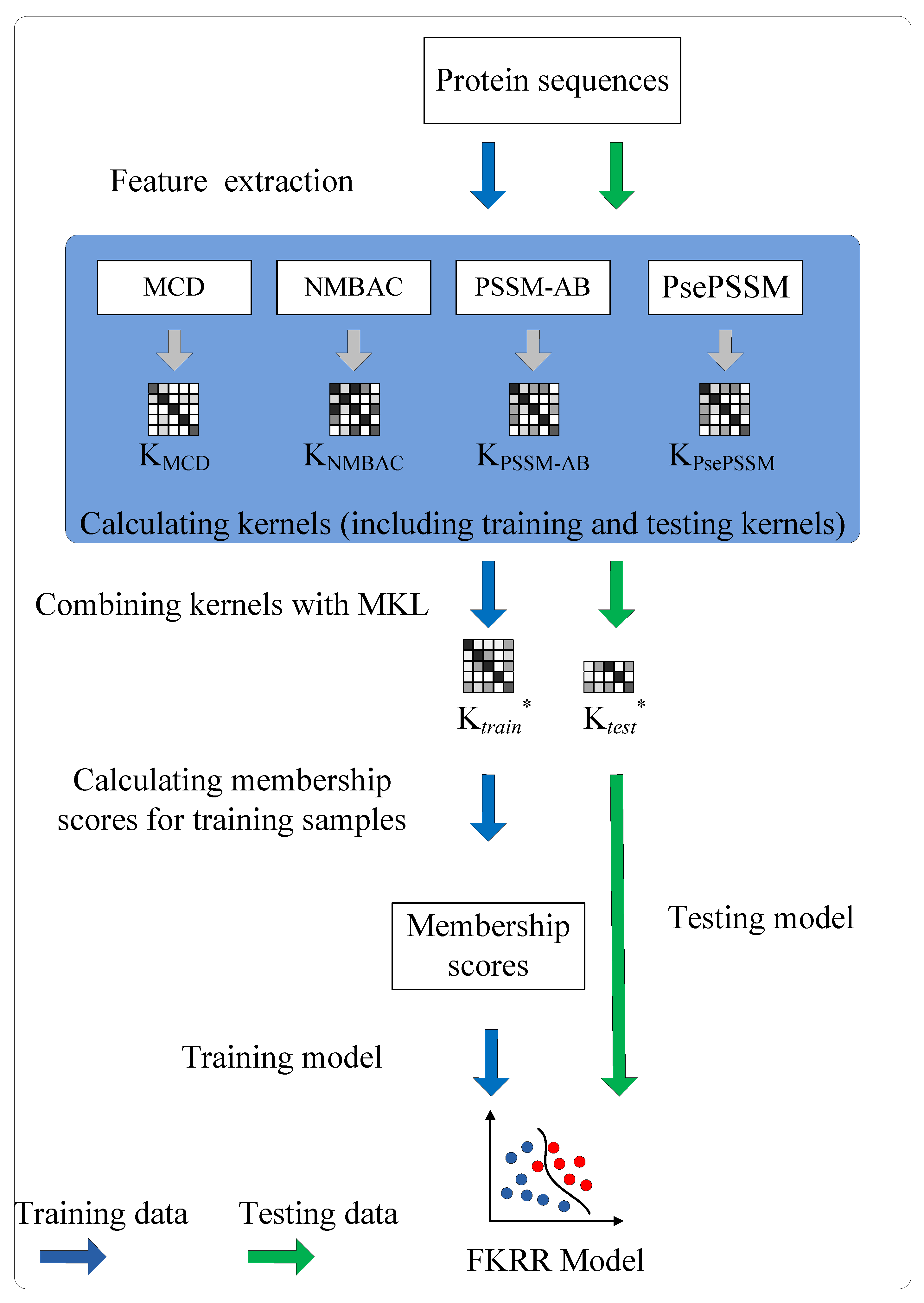
4.2. Multiple Kernel Learning
4.3. Fuzzy Kernel Ridge Regression
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chou, K.C.; Tomasselli, A.G.; Heinrikson, R.L. Prediction of the Tertiary Structure of a Caspase-9/Inhibitor Complex. FEBS Lett. 2000, 470, 249–256. [Google Scholar] [CrossRef]
- Chou, K.C.; Jones, D.; Heinrikson, R.L. Prediction of the tertiary structure and substrate binding site of caspase-8. FEBS Lett. 1997, 419, 49–54. [Google Scholar] [CrossRef]
- Chou, K.C. Insights from modelling the 3D structure of the extracellular domain of α7 nicotinic acetylcholine receptor. Biochem. Biophys. Res. Commun. 2004, 319, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.L.; Fu, L.; Nie, X. Using ensemble SVM to identify human GPCRs N-linked glycosylation sites based on the general form of Chou’s PseAAC. Protein Eng. Des. Sel. 2013, 26, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, J.; Wu, L. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS ONE 2013, 8, e55844. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Ding, H.; Lin, H. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef]
- Chou, K.C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015, 11, 218–234. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J. Theor. Biol. 2016, 394, 223–230. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B. iCar-PseCp: Identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget 2016, 7, 34558–34570. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Qiu, W.R. iDNA-Methyl: Identifying DNA methylation sites via pseudo trinucleotide composition. Anal. Biochem. 2015, 474, 69–77. [Google Scholar] [CrossRef]
- Xiao, X.; Min, J.L.; Lin, W.Z.; Liu, Z.; Cheng, X. iDrug-Target: Predicting the interactions between drug compounds and target proteins in cellular networking via the benchmark dataset optimization approach. J. Biomol. Struct. Dyn. 2015, 33, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X. iPPI-Esml: An ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J. Theor. Biol. 2015, 377, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Lin, H. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS Struct. Funct. Genet. 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lei, T.; Jin, D.; Lin, H. PseKNC: A flexible web-server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 2014, 456, 53–60. [Google Scholar] [CrossRef]
- Wei, L.; Luan, S.; Nagai, L.; Su, R.; Zou, Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics 2019, 35, 1326–1333. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene Subsequence Embedding for Prediction of Mammalian N6-Methyladenosine Sites from mRNA. RNA 2019, 25, 205–218. [Google Scholar] [CrossRef]
- Jia, C.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018, 34, 2029–2036. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, L.; Lu, L.; Zou, Q. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 2018, 34, 2425–2432. [Google Scholar] [CrossRef] [Green Version]
- Xuan, P.; Han, K.; Guo, Y.; Li, J.; Li, X.; Zhong, Y.; Zhang, Z.; Ding, J. Prediction of potential disease-associated microRNAs by using neural network. Mol. Ther. -Nucleic Acids 2019, 16, 566–575. [Google Scholar]
- Liu, B.; Jiang, S.; Zou, Q. HITS-PR-HHblits: Protein remote homology detection by combining pagerank and hyperlink-induced topic search. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ding, Y.; Su, L.; Tang, J.; Zou, Q. Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 2018, 117, 212–217. [Google Scholar] [CrossRef]
- Ru, X.; Li, L.; Zou, Q. Incorporating Distance-based Top-n-gram and Random Forest to Identify Electron Transport Proteins. J. Proteome Res. 2019, 18, 2931–2939. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Guo, F.; Liu, X.; Zou, Q. Application of Machine Learning in Microbiology. Front. Microbiol. 2019, 10, 827. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou’s PseAAC and Physicochemical Distance Transformation. Mol. Inform. 2015, 34, 8–17. [Google Scholar] [CrossRef]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinform. 2007, 8, 463. [Google Scholar] [CrossRef]
- Lipman, D.J.; Zhang, J.; Madden, T.L. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Liu, B.; Xu, J.; Lan, X.; Xu, R.; Zhou, J.; Wang, X.; Chou, K.C. iDNA-Prot|dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. PLoS ONE 2014, 9, e106691. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2016, 384, 135–144. [Google Scholar] [CrossRef]
- Nimrod, G.; Schushan, M.; Szilágyi, A.; Leslie, C.; Ben-Tal, N. iDBPs: A web server for the identification of DNA binding proteins. Bioinformatics 2010, 26, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Hussain, W.; Khan, S.D.; Rasool, N.; Khan, S.A. SPalmitoylC-PseAAC: A sequence-based model developed via Chou’s five-step rule and general PseAAC for identifying S-palmitoylation sites in proteins. Anal. Biochem. 2019, 568, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Progresses in predicting post-translational modification. Int. J. Pept. Res. Ther. 2019. [Google Scholar] [CrossRef]
- Awais, M.; Hussain, W.; Khan, Y.D.; Rasool, N.; Khan, S.A. iPhosH-PseAAC: Identify phosphohistidine sites in proteins by blending statistical moments and position relative features according to the Chou’s 5-step rule and general pseudo amino acid composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Ma, Z.; Zhao, X. dForml(KNN)-PseAAC: Detecting formylation sites from protein sequences using K-nearest neighbor algorithm via Chou’s 5-step rule and pseudo components. J. Theor. Biol. 2019, 470, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review, five-step rule). J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Advance in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Curr. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef]
- Liu, B.; Wu, H. Pse-in-One 2.0: An improved package of web servers for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nat. Sci. 2017, 9, 67–91. [Google Scholar] [CrossRef]
- Rose, P.W.; Prli, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB Protein Data Bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, 345–356. [Google Scholar] [CrossRef]
- Lou, W.; Wang, X.; Chen, F.; Chen, Y.; Jiang, B.; Zhang, H. Sequence Based Prediction of DNA-Binding Proteins Based on Hybrid Feature Selection Using Random Forest and Gaussian Naïve Bayes. PLoS ONE 2014, 9, e86703. [Google Scholar] [CrossRef]
- Lin, W.; Fang, J.; Xiao, X. iDNA-Prot: Identification of DNA Binding Proteins Using Random Forest with Grey Model. PLoS ONE 2011, 6, e24756. [Google Scholar] [CrossRef]
- Kumar, K.K.; Pugalenthi, G.; Suganthan, P.N. DNA-Prot: Identification of DNA Binding Proteins from Protein Sequence Information using Random Forest. J. Biomol. Struct. Dyn. 2009, 26, 679–686. [Google Scholar] [CrossRef]
- Liu, B.; Wang, S.; Wang, X. DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation. Sci. Rep. 2015, 5, 15479. [Google Scholar] [CrossRef] [Green Version]
- Adilina, S.; Farid, D.; Shatabda, S. Effective DNA binding protein prediction by using key features via Chou’s general PseAAC. J. Theor. Biol. 2019, 460, 64–78. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, J.; Wang, H. Identifying DNA-binding proteins by combining support vector machine and PSSM distance transformation. BMC Syst. Biol. 2014, 9, e86703. [Google Scholar] [CrossRef]
- Liu, X.; Gong, X.; Yu, H.; Xu, J. A Model Stacking Framework for Identifying DNA Binding Proteins by Orchestrating Multi-View Features and Classifiers. Genes 2018, 9, 394. [Google Scholar] [CrossRef]
- Feng, Z.P.; Zhang, C.T. Prediction of membrane protein types based on the hydrophobic index of amino acids. J. Protein Chem. 2000, 19, 269–275. [Google Scholar] [CrossRef]
- Ding, Y.J.; Tang, J.J.; Guo, F. Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinform. 2016, 17, 398–410. [Google Scholar] [CrossRef]
- Jeong, J.C.; Lin, X.; Chen, X.W. On position-specific scoring matrix for protein function prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef]
- You, Z.H.; Zhu, L.; Zheng, C.H.; Yu, H.J.; Deng, S.P.; Ji, Z. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinform. 2014, 15, S9. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. MemType-2L: A Web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem. Biophys. Res. Commun. 2007, 360, 339–345. [Google Scholar] [CrossRef]
- He, J.; Chang, S.F.; Xie, L. Fast Kernel learning for Spatial Pyramid Matching. In Proceedings of the 2008 IEEE Conference on Computer Vision and Pattern Recognition, Anchorage, AK, USA, 23–28 June 2008; pp. 1–7. [Google Scholar]
- Chou, K.C. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr. Proteom. 2009, 6, 262–274. [Google Scholar] [CrossRef]
- Chen, W.; Lin, H. Pseudo nucleotide composition or PseKNC: An effective formulation for analyzing genomic sequences. Mol. Biosyst. 2015, 11, 2620–2634. [Google Scholar] [CrossRef]
- Liu, B.; Yang, F.; Huang, D.S. iPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2018, 34, 33–40. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Zhou, X.; Lin, H. iRNA(m6A)-PseDNC: Identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018, 561, 59–65. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H. iRNA-3typeA: Identifying 3-types of modification at RNA’s adenosine sites. Mol. Ther.-Nucleic Acid 2018, 11, 468–474. [Google Scholar] [CrossRef]
- Lin, H.; Deng, E.Z.; Ding, H.; Chen, W. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961–12972. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Sets | PDB1075 | PDB186 |
|---|---|---|
| Positive | 525 | 93 |
| Negative | 550 | 93 |
| Total | 1075 | 186 |
| Feature Type | Model | ACC | SN | Spec | MCC | AUC |
|---|---|---|---|---|---|---|
| MCD | KRR | 0.7070 | 0.7086 | 0.7088 | 0.4139 | 0.7751 |
| NMBAC | KRR | 0.7284 | 0.7181 | 0.7382 | 0.4564 | 0.7857 |
| PSSM-AB | KRR | 0.7553 | 0.7695 | 0.7418 | 0.5113 | 0.8352 |
| PsePSSM | KRR | 0.7944 | 0.7905 | 0.7982 | 0.5886 | 0.8637 |
| MW a | KRR | 0.8195 | 0.8362 | 0.8036 | 0.6398 | 0.8998 |
| MKL | KRR | 0.8214 | 0.8438 | 0.8000 | 0.6439 | 0.9032 |
| MCD | SVM | 0.7088 | 0.7345 | 0.6819 | 0.4171 | 0.7611 |
| NMBAC | SVM | 0.7116 | 0.6909 | 0.7333 | 0.4244 | 0.7706 |
| PSSM-AB | SVM | 0.7693 | 0.6981 | 0.8438 | 0.5467 | 0.8391 |
| PsePSSM | SVM | 0.7851 | 0.7472 | 0.8247 | 0.5731 | 0.8566 |
| MW a | SVM | 0.8201 | 0.8232 | 0.8170 | 0.6421 | 0.9011 |
| MKL | SVM | 0.8299 | 0.8541 | 0.8057 | 0.6568 | 0.9101 |
| MW a | FKRR | 0.8270 | 0.8533 | 0.8018 | 0.6554 | 0.9094 |
| MKL | FKRR | 0.8326 | 0.8571 | 0.8091 | 0.6664 | 0.9115 |
| Methods | ACC (%) | MCC | SN (%) | Spec (%) |
|---|---|---|---|---|
| IDNA-Prot | 75.40 | 0.50 | 83.81 | 64.73 |
| DNAbinder | 73.95 | 0.48 | 68.57 | 79.09 |
| DNA-Prot | 72.55 | 0.44 | 82.67 | 59.76 |
| iDNAPro-PseAAC | 76.56 | 0.53 | 75.62 | 77.45 |
| IDNA-Prot|dis | 77.30 | 0.54 | 79.40 | 75.27 |
| Kmer1+ACC | 75.23 | 0.50 | 76.76 | 73.76 |
| Local-DPP | 79.10 | 0.59 | 84.80 | 73.60 |
| PseDNA-Pro | 76.55 | 0.53 | 79.61 | 73.63 |
| Adilina’s work | 70.21 | 0.41 | 61.00 | 79.70 |
| Our method (FKRR-MVSF) | 83.26 | 0.67 | 85.71 | 80.91 |
| Methods | ACC (%) | MCC | SN (%) | Spec (%) |
|---|---|---|---|---|
| IDNA-Prot | 67.2 | 0.344 | 67.7 | 66.7 |
| DNA-Prot | 61.8 | 0.240 | 69.9 | 53.8 |
| IDNA-Prot|dis | 72.0 | 0.445 | 79.5 | 64.5 |
| DNAbinder | 60.8 | 0.216 | 57.0 | 64.5 |
| DBPPred | 76.9 | 0.538 | 79.6 | 74.2 |
| Kmer1+ACC | 71.0 | 0.431 | 82.8 | 59.1 |
| iDNAPro-PseAAC | 71.5 | 0.442 | 82.8 | 60.2 |
| Local-DPP | 79.0 | 0.625 | 92.5 | 65.6 |
| Adilina’s work | 82.3 | 0.670 | 95.0 | 69.9 |
| MSFBinder (SVM) | 81.7 | 0.640 | 89.3 | 74.2 |
| Our method (FKRR-MVSF) | 81.7 | 0.676 | 98.9 | 64.5 |
| Amino Acid | H | VSC | P1 | P2 | SASA | NCISC |
|---|---|---|---|---|---|---|
| A | 0.62 | 27.5 | 8.1 | 0.046 | 1.181 | 0.007187 |
| C | 0.29 | 44.6 | 5.5 | 0.128 | 1.461 | −0.03661 |
| D | −0.9 | 40 | 13 | 0.105 | 1.587 | −0.02382 |
| E | −0.74 | 62 | 12.3 | 0.151 | 1.862 | 0.006802 |
| F | 1.19 | 115.5 | 5.2 | 0.29 | 2.228 | 0.037552 |
| G | 0.48 | 0 | 9 | 0 | 0.881 | 0.179052 |
| H | −0.4 | 79 | 10.4 | 0.23 | 2.025 | −0.01069 |
| I | 1.38 | 93.5 | 5.2 | 0.186 | 1.81 | 0.021631 |
| K | −1.5 | 100 | 11.3 | 0.219 | 2.258 | 0.017708 |
| L | 1.06 | 93.5 | 4.9 | 0.186 | 1.931 | 0.051672 |
| M | 0.64 | 94.1 | 5.7 | 0.221 | 2.034 | 0.002683 |
| N | −0.78 | 58.7 | 11.6 | 0.134 | 1.655 | 0.005392 |
| P | 0.12 | 41.9 | 8 | 0.131 | 1.468 | 0.239531 |
| Q | −0.85 | 80.7 | 10.5 | 0.18 | 1.932 | 0.049211 |
| R | −2.53 | 105 | 10.5 | 0.291 | 2.56 | 0.043587 |
| S | −0.18 | 29.3 | 9.2 | 0.062 | 1.298 | 0.004627 |
| T | −0.05 | 51.3 | 8.6 | 0.108 | 1.525 | 0.003352 |
| V | 1.08 | 71.5 | 5.9 | 0.14 | 1.645 | 0.057004 |
| W | 0.81 | 145.5 | 5.4 | 0.409 | 2.663 | 0.037977 |
| Y | 0.26 | 117.3 | 6.2 | 0.298 | 2.368 | 0.023599 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, Y.; Ding, Y.; Tang, J.; Guo, F.; Peng, L. FKRR-MVSF: A Fuzzy Kernel Ridge Regression Model for Identifying DNA-Binding Proteins by Multi-View Sequence Features via Chou’s Five-Step Rule. Int. J. Mol. Sci. 2019, 20, 4175. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174175
Zou Y, Ding Y, Tang J, Guo F, Peng L. FKRR-MVSF: A Fuzzy Kernel Ridge Regression Model for Identifying DNA-Binding Proteins by Multi-View Sequence Features via Chou’s Five-Step Rule. International Journal of Molecular Sciences. 2019; 20(17):4175. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174175
Chicago/Turabian StyleZou, Yi, Yijie Ding, Jijun Tang, Fei Guo, and Li Peng. 2019. "FKRR-MVSF: A Fuzzy Kernel Ridge Regression Model for Identifying DNA-Binding Proteins by Multi-View Sequence Features via Chou’s Five-Step Rule" International Journal of Molecular Sciences 20, no. 17: 4175. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174175