Engaging Cytotoxic T and NK Cells for Immunotherapy in Chronic Lymphocytic Leukemia

Abstract
:1. Introduction
2. Immunomodulation of Effector Cells in CLL
2.1. CD4+ and CD8+ T Cells
2.2. NK Cells
3. Responses to Immunotherapy in CLL
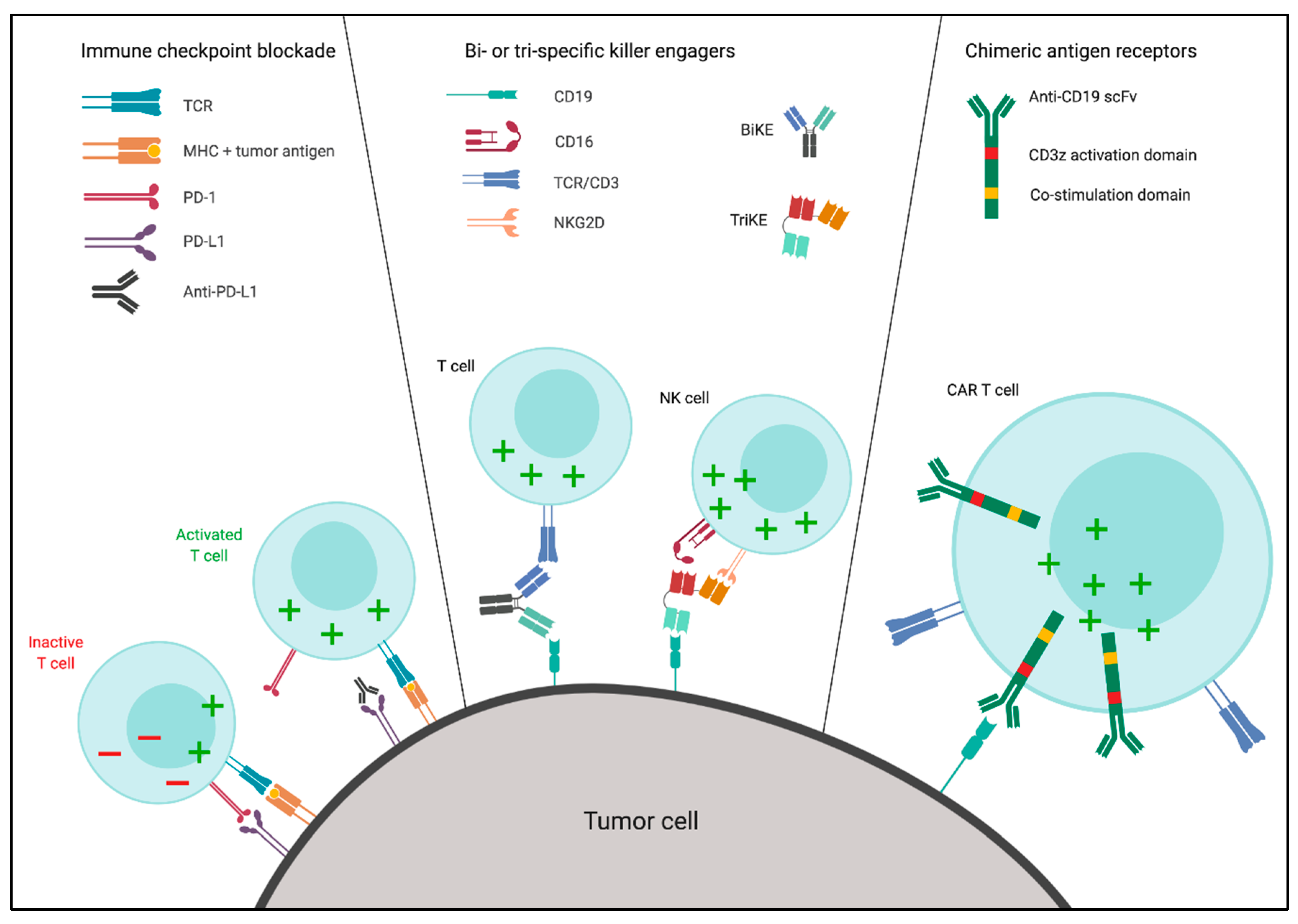
3.1. Immune Checkpoint Blockade (ICB)
3.2. Bi- and Tri-Specific Killer Engagers
3.3. Chimeric Antigen Receptors
3.4. Combination Strategies and the Role of Small-Molecule Inhibitors
4. Concluding Remarks
Funding
Conflicts of Interest
References
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. New Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. New Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. New Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef]
- Dreger, P.; Schetelig, J.; Andersen, N.; Corradini, P.; van Gelder, M.; Gribben, J.; Kimby, E.; Michallet, M.; Moreno, C.; Stilgenbauer, S.; et al. Managing high-risk CLL during transition to a new treatment era: Stem cell transplantation or novel agents? Blood 2014, 124, 3841–3849. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. New Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Gao, W.J.; You, J.; Wu, L.H.; Liu, J.L.; Wang, Z.X. The efficacy of anti-CD19 chimeric antigen receptor T cells for B-cell malignancies. Cytotherapy 2019, 21, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourgheysari, B.; Bruton, R.; Parry, H.; Billingham, L.; Fegan, C.; Murray, J.; Moss, P. The number of cytomegalovirus-specific CD4+ T cells is markedly expanded in patients with B-cell chronic lymphocytic leukemia and determines the total CD4+ T-cell repertoire. Blood 2010, 116, 2968–2974. [Google Scholar] [CrossRef] [Green Version]
- Mackus, W.J.; Frakking, F.N.; Grummels, A.; Gamadia, L.E.; De Bree, G.J.; Hamann, D.; Van Lier, R.A.; Van Oers, M.H. Expansion of CMV-specific CD8+CD45RA+CD27- T cells in B-cell chronic lymphocytic leukemia. Blood 2003, 102, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Te Raa, G.D.; Pascutti, M.F.; Garcia-Vallejo, J.J.; Reinen, E.; Remmerswaal, E.B.; ten Berge, I.J.; van Lier, R.A.; Eldering, E.; van Oers, M.H.; Tonino, S.H.; et al. CMV-specific CD8+ T-cell function is not impaired in chronic lymphocytic leukemia. Blood 2014, 123, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Palma, M.; Gentilcore, G.; Heimersson, K.; Mozaffari, F.; Nasman-Glaser, B.; Young, E.; Rosenquist, R.; Hansson, L.; Osterborg, A.; Mellstedt, H. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 2017, 102, 562–572. [Google Scholar] [CrossRef]
- Tinhofer, I.; Weiss, L.; Gassner, F.; Rubenzer, G.; Holler, C.; Greil, R. Difference in the relative distribution of CD4+ T-cell subsets in B-CLL with mutated and unmutated immunoglobulin (Ig) VH genes: Implication for the course of disease. J. Immunother. 2009, 32, 302–309. [Google Scholar] [CrossRef]
- Gorgun, G.; Holderried, T.A.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgun, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Gassner, F.J.; Ratswohl, C.; Zaborsky, N.; Rebhandl, S.; Schubert, M.; Steiner, M.; Gutjahr, J.C.; Pleyer, L.; Egle, A.; et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology 2017, 7, e1371399. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhou, Y.; Yu, Q.; Zheng, S.; Wang, Z.; Huang, Q. Elevated levels of follicular T helper cells and their association with therapeutic effects in patients with chronic lymphocytic leukaemia. Immunol. Lett. 2018, 197, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Jak, M.; Tromp, J.M.; Derks, I.A.; Remmerswaal, E.B.; Thijssen, R.; van Attekum, M.H.; van Bochove, G.G.; Luijks, D.M.; Pals, S.T.; et al. IL-21 and CD40L signals from autologous T cells can induce antigen-independent proliferation of CLL cells. Blood 2013, 122, 3010–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, M.; Kochanek, M.; Darabi, K.; Popov, A.; Jensen, M.; Endl, E.; Knolle, P.A.; Thomas, R.K.; von Bergwelt-Baildon, M.; Debey, S.; et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005, 106, 2018–2025. [Google Scholar] [CrossRef] [Green Version]
- D’Arena, G.; Laurenti, L.; Minervini, M.M.; Deaglio, S.; Bonello, L.; De Martino, L.; De Padua, L.; Savino, L.; Tarnani, M.; De Feo, V.; et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk. Res. 2011, 35, 363–368. [Google Scholar] [CrossRef]
- De Matteis, S.; Molinari, C.; Abbati, G.; Rossi, T.; Napolitano, R.; Ghetti, M.; Di Rora, A.G.L.; Musuraca, G.; Lucchesi, A.; Rigolin, G.M.; et al. Immunosuppressive Treg cells acquire the phenotype of effector-T cells in chronic lymphocytic leukemia patients. J. Transl. Med. 2018, 16, 172. [Google Scholar] [CrossRef]
- Piper, K.P.; Karanth, M.; McLarnon, A.; Kalk, E.; Khan, N.; Murray, J.; Pratt, G.; Moss, P.A. Chronic lymphocytic leukaemia cells drive the global CD4+ T cell repertoire towards a regulatory phenotype and leads to the accumulation of CD4+ forkhead box P3+ T cells. Clin. Exp. Immunol. 2011, 166, 154–163. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Vlachonikola, E.; Karypidou, M.; Stalika, E.; Bikos, V.; Gemenetzi, K.; Maramis, C.; Siorenta, A.; Anagnostopoulos, A.; Pospisilova, S.; et al. Restrictions in the T-cell repertoire of chronic lymphocytic leukemia: High-throughput immunoprofiling supports selection by shared antigenic elements. Leukemia 2017, 31, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Vardi, A.; Puiggros, A.; Gomez-Llonin, A.; Muro, M.; Rodriguez-Rivera, M.; Stalika, E.; Abella, E.; Gimeno, E.; Lopez-Sanchez, M.; et al. Restricted T cell receptor repertoire in CLL-like monoclonal B cell lymphocytosis and early stage CLL. Oncoimmunology 2018, 7, e1432328. [Google Scholar] [CrossRef] [PubMed]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Anandappa, A.J.; Sun, J.; Kim, J.; Leet, D.E.; Bozym, D.J.; Chen, C.; Williams, L.; Shukla, S.A.; Zhang, W.; et al. A cloning and expression system to probe T cell receptor specificity and assess functional avidity to neoantigens. Blood 2018, 132, 1911–1921. [Google Scholar] [CrossRef]
- Orange, J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525, quiz 526. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, D.; Lanuza, P.M.; Gomez, N.; Muntasell, A.; Cisneros, E.; Moraru, M.; Azaceta, G.; Anel, A.; Martinez-Lostao, L.; Villalba, M.; et al. Activated Allogeneic NK Cells Preferentially Kill Poor Prognosis B-Cell Chronic Lymphocytic Leukemia Cells. Front. Immunol. 2016, 7, 454. [Google Scholar] [CrossRef]
- Wang, W.T.; Zhu, H.Y.; Wu, Y.J.; Xia, Y.; Wu, J.Z.; Wu, W.; Liang, J.H.; Wang, L.; Fan, L.; Li, J.Y.; et al. Elevated absolute NK cell counts in peripheral blood predict good prognosis in chronic lymphocytic leukemia. J. Cancer Res. Clin. Oncol. 2018, 144, 449–457. [Google Scholar] [CrossRef]
- Veuillen, C.; Aurran-Schleinitz, T.; Castellano, R.; Rey, J.; Mallet, F.; Orlanducci, F.; Pouyet, L.; Just-Landi, S.; Coso, D.; Ivanov, V.; et al. Primary B-CLL resistance to NK cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J. Clin. Immunol. 2012, 32, 632–646. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Garff-Tavernier, M.; Decocq, J.; de Romeuf, C.; Parizot, C.; Dutertre, C.A.; Chapiro, E.; Davi, F.; Debre, P.; Prost, J.F.; Teillaud, J.L.; et al. Analysis of CD16+CD56dim NK cells from CLL patients: Evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia 2011, 25, 101–109. [Google Scholar] [CrossRef]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-Garcia, E.; Payer, A.R.; Villa-Alvarez, M.; Fernandez-Guizan, A.; Lopez-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.M.; Stevens, T.; Oldreive, C.; Zadran, B.; McSkeane, T.; Rudzki, Z.; Paneesha, S.; Chadwick, C.; Stankovic, T.; Pratt, G.; et al. NK cell function is markedly impaired in patients with chronic lymphocytic leukaemia but is preserved in patients with small lymphocytic lymphoma. Oncotarget 2016, 7, 68513–68526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, A.W.t.; Jillab, M.; Smith, M.R.; Alpaugh, R.K.; Cole, M.E.; Litwin, S.; Millenson, M.M.; Al-Saleem, T.; Cohen, A.D.; Campbell, K.S. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology 2017, 6, e1330235. [Google Scholar] [CrossRef] [PubMed]
- Hilpert, J.; Grosse-Hovest, L.; Grunebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Reiners, K.S.; Topolar, D.; Henke, A.; Simhadri, V.R.; Kessler, J.; Sauer, M.; Bessler, M.; Hansen, H.P.; Tawadros, S.; Herling, M.; et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013, 121, 3658–3665. [Google Scholar] [CrossRef]
- Rizzo, R.; Audrito, V.; Vacca, P.; Rossi, D.; Brusa, D.; Stignani, M.; Bortolotti, D.; D’Arena, G.; Coscia, M.; Laurenti, L.; et al. HLA-G is a component of the chronic lymphocytic leukemia escape repertoire to generate immune suppression: Impact of the HLA-G 14 base pair (rs66554220) polymorphism. Haematologica 2014, 99, 888–896. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1226720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, B.; da Silva Nardi, F.; Schramm, S.; Kraemer, T.; Celik, A.A.; Durig, J.; Horn, P.A.; Duhrsen, U.; Nuckel, H.; Rebmann, V. HLA-E allelic genotype correlates with HLA-E plasma levels and predicts early progression in chronic lymphocytic leukemia. Cancer 2017, 123, 814–823. [Google Scholar] [CrossRef]
- Lotz, M.; Ranheim, E.; Kipps, T.J. Transforming growth factor beta as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. J. Exp. Med. 1994, 179, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Hofland, T. XVII International Workshop on Chronic Lymphocytic Leukemia 2017 May 12--15, 2017, New York. Leuk. Lymphoma 2017, 58, 1–240, abstract 289. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.S.; Illmer, T.; et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. New Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet. Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef]
- McClanahan, F.; Hanna, B.; Miller, S.; Clear, A.J.; Lichter, P.; Gribben, J.G.; Seiffert, M. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood 2015, 126, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Motta, M.; Rassenti, L.; Shelvin, B.J.; Lerner, S.; Kipps, T.J.; Keating, M.J.; Wierda, W.G. Increased expression of CD152 (CTLA-4) by normal T lymphocytes in untreated patients with B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1788–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet. Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; Andre, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755.e1715. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.; Gruen, M.; Wuchter, C.; Schriever, F.; Kufer, P.; Dreier, T.; Hanakam, F.; Baeuerle, P.A.; Bommert, K.; Karawajew, L.; et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia 2003, 17, 900–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.; Pepper, C.; Brennan, P.; Nagorsen, D.; Man, S.; Fegan, C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica 2013, 98, 1930–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, H.R.; Qi, J.; Cook, E.M.; Nichols, C.; Dadashian, E.L.; Underbayev, C.; Herman, S.E.M.; Saba, N.S.; Keyvanfar, K.; Sun, C.; et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood 2018, 132, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lam, C.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clin. Cancer Res. 2017, 23, 1506–1518. [Google Scholar] [CrossRef]
- Circosta, P.; Elia, A.R.; Landra, I.; Machiorlatti, R.; Todaro, M.; Aliberti, S.; Brusa, D.; Deaglio, S.; Chiaretti, S.; Bruna, R.; et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. Oncoimmunology 2018, 7, e1341032. [Google Scholar] [CrossRef] [PubMed]
- Stanglmaier, M.; Faltin, M.; Ruf, P.; Bodenhausen, A.; Schroder, P.; Lindhofer, H. Bi20 (fBTA05), a novel trifunctional bispecific antibody (anti-CD20 x anti-CD3), mediates efficient killing of B-cell lymphoma cells even with very low CD20 expression levels. Int. J. Cancer 2008, 123, 1181–1189. [Google Scholar] [CrossRef]
- Gohil, S.H.; Evans, R.; Harasser, M.; El-Kholy, M.; Paredes-Moscosso, S.R.; Della Peruta, M.; Nathwani, A.C. Ibrutinib enhances the efficacy of ROR1 bispecific T cell engager mediated cytotoxicity in chronic lymphocytic leukaemia. Br. J. Haematol. 2019, 186, 380–382. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Li, X.; Peng, H.; Cook, E.M.; Dadashian, E.L.; Wiestner, A.; Park, H.; Rader, C. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc. Natl. Acad. Sci. USA 2018, 115, E5467–E5476. [Google Scholar] [CrossRef]
- Vyas, M.; Schneider, A.C.; Shatnyeva, O.; Reiners, K.S.; Tawadros, S.; Kloess, S.; Kohl, U.; Hallek, M.; Hansen, H.P.; Pogge von Strandmann, E. Mono- and dual-targeting triplebodies activate natural killer cells and have anti-tumor activity in vitro and in vivo against chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1211220. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Kodal, B.; Hinderlie, P.; Kaminski, M.F.; Cooley, S.; Weisdorf, D.J.; Vallera, D.A.; Miller, J.S.; Bachanova, V. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. 2019, 3, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Geyer, M.B.; Riviere, I.; Senechal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Palomba, M.L.; Halton, E.; et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Geyer, M.B.; Riviere, I.; Senechal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Halton, E.; Lamanna, N.; et al. Autologous CD19-Targeted CAR T Cells in Patients with Residual CLL following Initial Purine Analog-Based Therapy. Mol. Ther. 2018, 26, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Lemal, R.; Tournilhac, O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J. Immunother. Cancer 2019, 7, 202. [Google Scholar] [CrossRef] [Green Version]
- Bair, S.M.; Porter, D.L. Accelerating chimeric antigen receptor therapy in chronic lymphocytic leukemia: The development and challenges of chimeric antigen receptor T-cell therapy for chronic lymphocytic leukemia. Am. J. Hematol. 2019, 94, S10–s17. [Google Scholar] [CrossRef]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Terakura, S.; Martens, A.C.; van Meerten, T.; Uchiyama, S.; Imai, M.; Sakemura, R.; Goto, T.; Hanajiri, R.; Imahashi, N.; et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015, 194, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Scarfo, I.; Ormhoj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Faitschuk, E.; Hombach, A.A.; Frenzel, L.P.; Wendtner, C.M.; Abken, H. Chimeric antigen receptor T cells targeting Fc mu receptor selectively eliminate CLL cells while sparing healthy B cells. Blood 2016, 128, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Huckelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in Expansion Potential of Naive Chimeric Antigen Receptor T Cells from Healthy Donors and Untreated Chronic Lymphocytic Leukemia Patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, I.; Kalland, I.; Kochenderfer, J.N.; Osterborg, A.; Uhlin, M.; Mattsson, J. CD19 Chimeric Antigen Receptor T Cells From Patients With Chronic Lymphocytic Leukemia Display an Elevated IFN-gamma Production Profile. J. Immunother. 2018, 41, 73–83. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8+ T cells and impede CAR T cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Palomba, M.L.; Batlevi, C.L.; Riviere, I.; Wang, X.; Senechal, B.; Furman, R.R.; Bernal, Y.; Hall, M.; Pineda, J.; et al. A Phase I First-in-Human Clinical Trial of CD19-Targeted 19-28z/4-1BBL “Armored” CAR T Cells in Patients with Relapsed or Refractory NHL and CLL Including Richter’s Transformation. Blood 2018, 132, 224. [Google Scholar] [CrossRef]
- Martyniszyn, A.; Krahl, A.C.; Andre, M.C.; Hombach, A.A.; Abken, H. CD20-CD19 Bispecific CAR T Cells for the Treatment of B-Cell Malignancies. Hum. Gene Ther. 2017, 28, 1147–1157. [Google Scholar] [CrossRef]
- Davies, D.M.; Maher, J. Gated chimeric antigen receptor T-cells: The next logical step in reducing toxicity? Transl. Cancer Res. 2016, 5, S61–S65. [Google Scholar] [CrossRef]
- Wang, L.; Dou, M.; Ma, Q.; Yao, R.; Liu, J. Chimeric antigen receptor (CAR)-modified NK cells against cancer: Opportunities and challenges. Int. Immunopharmacol. 2019, 74, 105695. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, R.; Leuci, V.; Donini, C.; Cykowska, A.; Gammaitoni, L.; Medico, G.; Valabrega, G.; Aglietta, M.; Sangiolo, D. CAR-Based Strategies beyond T Lymphocytes: Integrative Opportunities for Cancer Adoptive Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2839. [Google Scholar] [CrossRef] [PubMed]
- Acharya, U.H.; Dhawale, T.; Yun, S.; Jacobson, C.A.; Chavez, J.C.; Ramos, J.D.; Appelbaum, J.; Maloney, D.G. Management of cytokine release syndrome and neurotoxicity in chimeric antigen receptor (CAR) T cell therapy. Expert Rev. Hematol. 2019, 12, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, B.C.; Le Luduec, J.B.; Forlenza, C.; Jakubowski, A.A.; Perales, M.A.; Young, J.W.; Hsu, K.C. Phase II Study of Haploidentical Natural Killer Cell Infusion for Treatment of Relapsed or Persistent Myeloid Malignancies Following Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 705–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Grote, S.; Seitz, C.M.; Diepold, S.; Buchner, M.; Baden, C.; Malenke, E.; Dieckmann, S.M.; Schwaemmle, H.; Mittelstaet, J.; Kaiser, A.; et al. Adapter Chimeric Antigen Receptor (aCAR)-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Universal Tumor Targeting. Blood 2018, 132, 3331. [Google Scholar] [CrossRef]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Investig. 2017, 127, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- de Weerdt, I.; Hofland, T.; Dobber, J.; Dubois, J.; Eldering, E.; Mobasher, M.; Croon-de Boer, F.; Hoogendoorn, M.; Velders, G.A.; van der Klift, M.; et al. First Evidence of Restoration of T and NK Cell Compartment after Venetoclax Treatment. Blood 2018, 132, 1860. [Google Scholar] [CrossRef]
- Itchaki, G.; Brown, J.R. Lenalidomide in the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Tonino, S.H.; Egle, A.; Ramsay, A.G. How does lenalidomide target the chronic lymphocytic leukemia microenvironment? Blood 2014, 124, 2184–2189. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Adams, M.; Carter, T.; Chen, R.; Muller, G.; Stirling, D.; Schafer, P.; Bartlett, J.B. lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin. Cancer Res. 2008, 14, 4650–4657. [Google Scholar] [CrossRef] [PubMed]
- Strati, P.; Takahashi, K.; Peterson, C.B.; Keating, M.J.; Thompson, P.A.; Daver, N.G.; Jain, N.; Burger, J.A.; Estrov, Z.; O’Brien, S.M.; et al. Efficacy and predictors of response of lenalidomide and rituximab in patients with treatment-naive and relapsed CLL. Blood Adv. 2019, 3, 1533–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kater, A.P.; van Oers, M.H.J.; van Norden, Y.; van der Straten, L.; Driessen, J.; Posthuma, W.F.M.; Schipperus, M.; Chamuleau, M.E.D.; Nijland, M.; Doorduijn, J.K.; et al. Feasibility and efficacy of addition of individualized-dose lenalidomide to chlorambucil and rituximab as first-line treatment in elderly and FCR-unfit patients with advanced chronic lymphocytic leukemia. Haematologica 2019, 104, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjonnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Tasken, K. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.S.; Roessner, P.M.; Scheffold, A.; Jebaraj, B.M.C.; Demerdash, Y.; Ozturk, S.; Lichter, P.; Stilgenbauer, S.; Seiffert, M. PI3Kdelta inhibition modulates regulatory and effector T-cell differentiation and function in chronic lymphocytic leukemia. Leukemia 2018. [Google Scholar] [CrossRef]
- Sharma, S.; Rai, K.R. Chronic lymphocytic leukemia (CLL) treatment: So many choices, such great options. Cancer 2019, 125, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.; Ubelhart, R.; Schubert, M.L.; Fan, F.; He, B.; Hoffmann, J.M.; Wang, L.; Wang, S.; Gong, W.; Neuber, B.; et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Hofland, T.; De Weerdt, I.; ter Burg, J.; de Boer, R.; Tannheimer, S.; Tonino, S.H.; Kater, A.P.; Eldering, E. Dissection of the effects of JAK and BTK inhibitors on the functionality of healthy and malignant lymphocytes. J. Immunol. 2019, 3. [Google Scholar] [CrossRef]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Sivina, M.; Robins, H.; Yusko, E.; Vignali, M.; O’Brien, S.; Keating, M.J.; Ferrajoli, A.; Estrov, Z.; Jain, N.; et al. Ibrutinib Therapy Increases T Cell Repertoire Diversity in Patients with Chronic Lymphocytic Leukemia. J. Immunol. 2017, 198, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Hirayama, A.V.; Hay, K.A.; Li, D.; Lymp, J.; Sheih, A.; Purushe, J.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; et al. Comparison of Efficacy and Toxicity of CD19-Specific Chimeric Antigen Receptor T-Cells Alone or in Combination with Ibrutinib for Relapsed and/or Refractory CLL. Blood 2018, 132, 299. [Google Scholar] [CrossRef]
- Gill, S.I.; Vides, V.; Frey, N.V.; Metzger, S.; O’Brien, M.; Hexner, E.; Mato, A.R.; Lacey, S.F.; Melenhorst, J.J.; Pequignot, E.; et al. Prospective Clinical Trial of Anti-CD19 CAR T Cells in Combination with Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia Shows a High Response Rate. Blood 2018, 132, 298. [Google Scholar] [CrossRef]


{kind=link}
| CD4 T Cells | CD8 T Cells | NK Cells | |
|---|---|---|---|
| Absolute numbers | Increased | Increased | Increased |
| Differentiation | Naïve↓ Effector ↑ TH1 ↑ TH2 Tfh ↑ Treg ↑ | Naïve↓ Effector ↑ | Increased maturation |
| Cytokine production | High | High | Low |
| Proliferation | Low | Low | Low |
| Cytotoxicity | / | Low | Natural cytotoxicity: low ADCC: normal |
| Exhaustion markers | High | High | Inconsistent |
| Treatment | Target | Phase | # patients | ORR | Reference |
|---|---|---|---|---|---|
| Nivolumab | PD-1 | 2 | 3 | 0 | [9] |
| Pembrolizumab | PD-1 | 2 | 16 | 0 | [57] |
| Nivolumab + ibrutinib | PD-1 | 2 | 36 | 61 * | [58] |
| In vitro studies | |||||
| Anti-TIGIT-Fc | TIGIT | - | - | - | [23] |
| Monalizumab | CD94/NKG2A | - | - | - | [49] |
| Construct Type | Target | Effector | Reference |
|---|---|---|---|
| Bi-specific single-chain antibody | CD19xCD3 | T cells | [62,63] |
| Bi-specific single-chain Fc-Fv | CD19xCD3 | T cells | [64] |
| DART | CD19xCD3 | T cells | [65,66] |
| DART | CD20xCD3 | T cells | [67] |
| Bi-specific single-chain antibody | ROR1xCD3 | T cells | [68] |
| Bi-specific single-chain Fc-Fv | ROR1xCD3 | T cells | [69] |
| Bi-specific single-chain antibody | CD19xCD19xNKG2D | NK cells | [70] |
| Tri-specific single-chain antibody | CD19xCD33xNKG2D | NK cells | [70] |
| Bi-specific single-chain antibody | CD19xCD16 | NK cells | [71] |
| Tri-specific single-chain antibody | CD19xCD22xCD16 | NK cells | [71] |
| Target | Co-stimulation Domain | Phase | No. of patients | % ORR/CR | Reference |
|---|---|---|---|---|---|
| CD19 | 4-1BB | 1/2 | 24 | 71/21 | [75] |
| CD19 | 4-1BB | 1/2 | 14 | 57/29 | [76] |
| CD19 | CD28z | 1 | 16 | 38/12 | [78] |
| BCR κ chains | CD28z | 1 | 2 | 0/0 | [82] |
| In vitro studies | |||||
| CD20 | CD28z | / | / | / | [83] |
| CD37 | 4-1BB | / | / | / | [84] |
| BCR Fc µ chains | CD28z | / | / | / | [85] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofland, T.; Eldering, E.; Kater, A.P.; Tonino, S.H. Engaging Cytotoxic T and NK Cells for Immunotherapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2019, 20, 4315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174315
Hofland T, Eldering E, Kater AP, Tonino SH. Engaging Cytotoxic T and NK Cells for Immunotherapy in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences. 2019; 20(17):4315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174315
Chicago/Turabian StyleHofland, Tom, Eric Eldering, Arnon P. Kater, and Sanne H. Tonino. 2019. "Engaging Cytotoxic T and NK Cells for Immunotherapy in Chronic Lymphocytic Leukemia" International Journal of Molecular Sciences 20, no. 17: 4315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174315