Cyclobutane-Containing Scaffolds as Useful Intermediates in the Stereoselective Synthesis of Suitable Candidates for Biomedical Purposes: Surfactants, Gelators and Metal Cation Ligands

Abstract
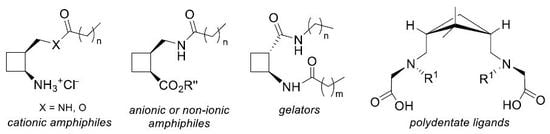
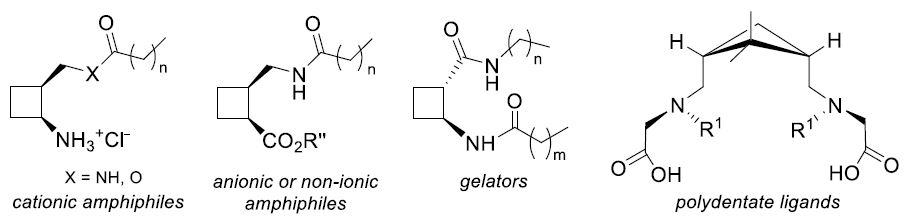
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Amphiphiles from 1,2-Disubstituted Cyclobutane Scaffolds: Surfactants and LMWG
2.2. Highly Rigid Polydentate Ligands From 1,3-disubstituted Cyclobutane Scaffolds
3. Materials and Methods
3.1. General Procedures
3.2. Experimental Section
3.2.1. Synthesis of tert-Butyl (1S,2R)-2-Hydroxymethylcyclobutane-1-Carboxylate (2)
3.2.2. Synthesis of tert-Butyl (1R,2S)-2-Tosyloxymethylcyclobutane-1-Carboxylate (3)
3.2.3. Synthesis of tert-Butyl (1S,2R)-2-Dodecanamidomethylcyclobutane-1-Carboxylate (5) through amine 4
3.2.4. Synthesis of (1S,2R)-2-Dodecanamidomethylcyclobutane-1-Carboxylic Acid (6)
3.2.5. Synthesis of tert-Butyl (1S,2R)-2-Hydroxymethylcyclobutane-1-Carbamate (8)
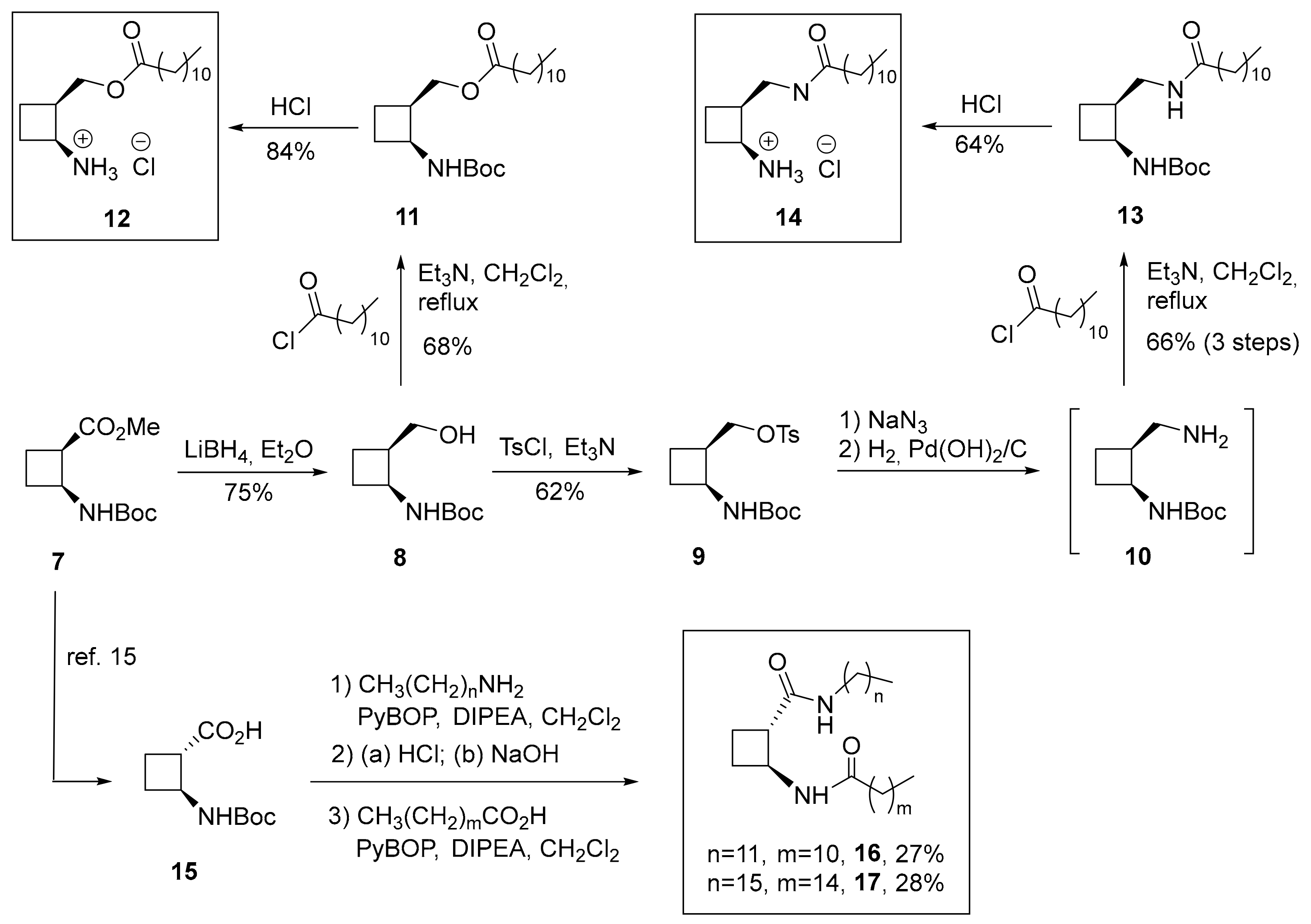
3.2.6. Synthesis of (1S,2R)-1-tert-Butyloxycarbonylaminocyclobutane-1-Methyl Dodecanoate (11)
3.2.7. Synthesis of (1S,2R)-2-Dodecanoyloxymethylcyclobutane-1-Ammonium Chloride (12)
3.2.8. Synthesis of tert-Butyl (1S,2R)-2-Tosyloxymethylcyclobutane-1-Carboxylate (9)
3.2.9. Synthesis of tert-Butyl (1S,2S)-2-Dodecanamidomethylcyclobutane-1-Carbamate (13) through diamine 10
3.2.10. Synthesis of (1S,2R)-2-Dodecanamidomethylcyclobutane-1-Ammonium Chloride (14)
3.2.11. Synthesis of Diamides 16 and 17. General Procedure
(1S,2S)-N-Dodecyl-2-Dodecanamidocyclobutane-1-Carboxamide (16)
(1S,2S)-N-Hexadecyl-2-Palmitamidocyclobutane-1-Carboxamide (17)
3.2.12. Synthesis of Tetra-tert-Butyl N,N,N’,N’-[((1S,3R)-2,2-Dimethylcyclobutane-1,3-diyl) Bis(Methylene)Bis(Azanetriyl)] Tetraacetate (20)
3.2.13. Synthesis of N,N,N’,N’-[((1S,3R)-2,2-dimethylcyclobutane-1,3-diyl) bis(methylene)-bis(azanetriyl)] tetraacetic acid (21)
3.2.14. Synthesis of Dimethyl 6,6′-[{2,2-Dimethylcyclobutane-1,3-diylbis[(2-tert-butoxy-2-oxoethyl)- azanemethylenediyl]}bis(methylene)] dipicolinate (24) through compound (23)
3.2.15. Synthesis of 6,6′-[{2,2-dimethylcyclobutane-1,3-diylbis[(carboxymethyl) azane methylene diyl]}- bis(methylene)] dipicolinic acid (25)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATR | Attenuated Total Reflection |
| CBAA | Cyclobutane Amino Acid |
| DDS | Drug Delivery Systems |
| DIPEA | Diisopropylethylamine |
| DMAP | Dimethylaminopyridine |
| DMF | Dimethylformamide |
| DNA | Deoxyribonucleic Acid |
| ESI | Electrospray Ionization |
| HRMS | High-Resolution Mass Spectrometry |
| IR | Infrared |
| LMWG | Low Molecular-Weight Gelator |
| MS | Mass Spectrometry |
| NMR | Nuclear Magnetic Resonance |
| NPY | Neuropeptide Y |
| PyBOP | (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate |
| QTOF | Quadrupole Time-of-Flight |
| TEA | Triethylamine |
| TFA | Trifluoroacetic Acid |
| THF | Tetrahydrofuran |
| TLC | Thin layer chromatography |
References
- Sorrenti, A.; Illa, O.; Ortuño, R.M. Amphiphiles in aqueous solution: well beyond a soap bubble. Chem. Soc. Rev. 2013, 42, 8200–8219. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.S.; Lindman, B. DNA Interaction with Polymers and Surfactants; Dias, R.S., Lindman, B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Grueso, E.; Cerrillos, C.; Hidalgo, J.; Lopez-Cornejo, P. Compaction and Decompaction of DNA Induced by the Cationic Surfactant CTAB. Langmuir 2012, 28, 10968–10979. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.; Kermanshahi pour, A.; Beach, E.S.; Zimmerman, J.B. Derivation and Synthesis of Renewable Surfactants. Chem. Soc. Rev. 2012, 41, 1499–1518. [Google Scholar] [CrossRef] [PubMed]
- Escuder, B.; Miravet, J.F. Functional Molecular Gels; Escuder, B., Miravet, J.F., Eds.; RSC Soft Matter Series Royal Society of Chemistry: Cambridge, UK, 2014. [Google Scholar]
- Toth, S.É. Lacerda in Theranostics and Image Guided Drug Delivery; Thanou, M., Ed.; RSC: Cambridge, UK, 2018. [Google Scholar]
- Merbach, A.; Helm, L.; Tóth, É. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed.; Merbach, A., Helm, L., Tóth, É., Eds.; Wiley: Weinheim, Germany, 2013. [Google Scholar]
- Sorrenti, A.; Illa, O.; Pons, R.; Ortuño, R.M. Chiral Cyclobutane β-Amino Acid-Based Amphiphiles: Influence of cis/trans Stereochemistry on Solution Self-Aggregation and Recognition. Langmuir 2015, 31, 9608–9618. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, A.; Illa, O.; Ortuño, R.M.; Pons, R. Chiral Cyclobutane β-Amino-Acid Based Amphiphiles: Influence of cis/trans Stereochemistry on Condensed Phase and Monolayer Structure. Langmuir 2016, 32, 6977–6984. [Google Scholar] [CrossRef] [PubMed]
- Pi-Boleda, B.; Sorrenti, A.; Sans, M.; Illa, O.; Pons, R.; Branchadell, V.; Ortuño, R.M. Cyclobutane Scaffold in Bolaamphiphiles: Effect of Diastereoisomerism and Regiochemistry on Their Surface Activity Aggregate Structure. Langmuir 2018, 34, 11424–11432. [Google Scholar] [CrossRef] [PubMed]
- Pi-Boleda, B.; Sans, M.; Campos, M.; Nolis, P.; Illa, O.; Estévez, J.C.; Branchadell, V.; Ortuño, R.M. Studies on cycloalkane-based organogelators: a new example of stochastic chiral symmetry-breaking induced by sonication. Chem. Eur. J. 2017, 23, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Pi-Boleda, B.; Campos, M.; Sans, M.; Basavilbaso, A.; Illa, O.; Branchadell, V.; Estévez, J.R.; Ortuño, R.M. Gaining insight into the influence of the hydroxyl groups on the gelling abilities of polyfunctional cyclohexane bisamides. Molecules 2019, 24, 352. [Google Scholar] [CrossRef] [PubMed]
- Porcar-Tost, O.; Pi-Boleda, B.; García-Anton, J.; Illa, O.; Ortuño, R.M. Cyclobutane-based peptides/terpyridine conjugates: Their use in metal catalysis and as functional organogelators. Tetrahedron 2018, 74, 7252–7260. [Google Scholar] [CrossRef]
- Izquierdo, S.; Rúa, F.; Sbai, A.; Parella, T.; Álvarez-Larena, A.; Branchadell, V.; Ortuño, R.M. (+)- and (−)-2-Aminocyclobutane-1-carboxylic acids and their incorporation into highly rigid β -peptides: Stereoselective synthesis and a structural study. J. Org. Chem. 2005, 70, 7963–7971. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Gauzy, C.; Yang, Y.; Roy, O.; Pereira, E.; Faure, S.; Aitken, D.J. [2+2] Photocycloadditions with chiral uracil derivatives: access to all four stereoisomers of 2-aminocyclobutanecarboxylic acid. Synthesis 2007, 2222–2232. [Google Scholar] [CrossRef]
- Torres, E.; Gorrea, E.; Da Silva, E.; Nolis, P.; Branchadell, V.; Ortuño, R.M. Prevalence of eight-membered hydrogen-bonded rings in some bis(cyclobutane) β-dipeptides with trans stereochemistry. Org. Lett. 2009, 11, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Gorrea, E.; Burusco, K.K.; Da Silva, E.; Nolis, P.; Rúa, F.; Boussert, S.; Díez-Pérez, I.; Dannenberg, S.; Izquierdo, S.; et al. Folding and self-assembling with β-oligomers based on (1R,2S)-2-aminocyclobutane-1-carboxylic acid. Org. Biomol. Chem. 2010, 8, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Gorrea, E.; Pohl, G.; Nolis, P.; Celis, S.; Burusco, K.K.; Branchadell, V.; Perczel, A.; Ortuño, R.M. Secondary structure of short β-peptides as the chiral expression of monomeric building units: A rational and predictive model. J. Org. Chem. 2012, 77, 9795–9806. [Google Scholar] [CrossRef] [PubMed]
- Gorrea, E.; Torres, E.; Nolis, P.; da Silva, E.; Amabilino, D.B.; Branchadell, V.; Ortuño, R.M. Self-assembly of chiral trans-cyclobutane containing β-dipeptides into ordered aggregates. Chem. Eur. J. 2011, 17, 4588–4597. [Google Scholar] [CrossRef] [PubMed]
- Celis, S.; Nolis, P.; Branchadell, V.; Illa, O.; Ortuño, R.M. Low-molecular-weight gelators consisting of hybrid cyclobutane-based peptides. Org. Biomol. Chem. 2013, 11, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Berlicki, Ł.; Kaske, M.; Gutiérrez-Abad, R.; Bernhardt, G.; Illa, O.; Ortuño, R.M.; Cabrele, C.; Buschauer, A.; Reiser, O. Replacement of Th32 and Gln34 in the C-terminal neuropeptide Y fragment 25-36 by cis-cyclobutane- and cis-cyclopentane β-amino acids shifts selectivity toward the Y4 receptor. J. Med. Chem. 2013, 56, 8422–8431. [Google Scholar] [CrossRef] [PubMed]
- Mayans, E.; Gargallo, A.; Illa, O.; Ortuño, R.M. Diastereodivergent synthesis of chiral cyclobutane scaffolds: 1,3-amino alcohols, 1,3-diamines and some derivatives. Eur. J. Org. Chem. 2013, 2013, 1425–1433. [Google Scholar] [CrossRef]
- Sans, M.; Illa, O.; Ortuño, R.M. Stereoselective synthesis of all stereoisomers of orthogonally protected cyclobutane-1,2-diamine and some chemoselective transformations. Org. Lett. 2012, 14, 2431–2433. [Google Scholar] [CrossRef]
- Andre, V.; Gras, M.; Awada, H.; Guillot, R.; Robin, S.; Aitken, D.J. A unified synthesis of all stereoisomers of (2-aminomethyl)cyclobutane-1-carboxylic acid. Tetrahedron 2013, 69, 3571–3576. [Google Scholar] [CrossRef]
- Awada, H.; Robin, S.; Guillot, R.; Yazbeck, O.; Naoufal, D.; Jaber, N.; Hachem, A.; Aitken, D.J. Practical syntheses of both enantiomers of the conformationally restricted GABA analogue cis-(2-aminocyclobutyl)acetic acid. Eur. J. Org. Chem. 2014, 2014, 7148–7155. [Google Scholar] [CrossRef]
- Ji, X.; Huang, H. Synthetic methods for 1,3-diamines. Org. Biomol. Chem. 2016, 14, 10557–10566. [Google Scholar] [CrossRef] [PubMed]
- Illa, O.; Porcar-Tost, O.; Robledillo, C.; Elvira, C.; Nolis, P.; Branchadell, V.; Reiser, O.; Ortuño, R.M. Stereoselectivity of Proline/Cyclobutane Amino Acid-Containing Peptide Organocatalysts for Asymmetric Aldol Additions: A Rationale. J. Org. Chem. 2018, 83, 350–363. [Google Scholar] [CrossRef]
- Gutiérrez-Abad, R.; Illa, O.; Ortuño, R.M. Synthesis of chiral cyclobutane containing C3-symmetric peptide dendrimers. Org. Lett. 2010, 12, 3148–3151. [Google Scholar] [CrossRef]
- Gorrea, E.; Carbajo, D.; Gutiérrez-Abad, R.; Illa, O.; Royo, M.; Ortuño, R.M. Searching for new cell-penetrating agents: hybrid cyclobutane-proline γ, γ -peptides. Org. Biomol. Chem. 2012, 10, 4050–4057. [Google Scholar] [CrossRef]
- Kiss, L.; Fülöp, F. Synthesis of carbocyclic and heterocyclic β-aminocarboxylic acids. Chem. Rev. 2014, 114, 1116–1169. [Google Scholar] [CrossRef] [PubMed]
- Park, J.D.; Allphin, N.L., Jr.; Choi, S.K.; Settine, R.L.; Hedrick, G.W. Hydroxyalky and Olefinic Substituted gem-Dimethylcyclobutanes. Ind. Eng. Chem. Prod. Res. Dev. 1965, 4, 149–153. [Google Scholar] [CrossRef]
- Forgács, A.; Giovenzana, G.B.; Botta, M.; Brücher, E.; Tóth, I.; Baranyai, Z. Influence of gem-dimethyl substitution on the stability, kinetics and relaxometric properties of PDTA complexes. Eur. J. Inorg. Chem. 2012, 2012, 2074–2086. [Google Scholar] [CrossRef]
- Gale, E.M.; Kenton, N.; Caravan, P. [Gd(CyPic3A)((H2O)2]−: A stable bis(aquated) and high-relaxivity Gd(III) complex. Chem. Commun. 2013, 49, 8060–8062. [Google Scholar] [CrossRef]
- Tircsó, G.; Regueiro-Figueroa, M.; Nagy, V.; Garda, Z.; Garai, T.; Kálmán, F.K.; Esteban-Gómez, D.; Tóth, É.; Platas-Iglesias, C. Approaching the kinetic inertness of macrocyclic gadolinium(III)-based MRI contrast agents with highly rigid open-chain derivatives. Chem. Eur. J. 2016, 22, 896–901. [Google Scholar] [CrossRef]
- Thiele, N.A.; Brown, V.; Kelly, J.M.; Amor-Coarasa, A.; Jermilova, U.; MacMillan, S.N.; Nikolopoulou, A.; Ponnala, S.; Ramogida, C.F.; Robertson, A.K.H.; et al. An eighteen-membered macrocyclic ligand for actinium-225 targeted alpha therapy. Angew. Chem. Int. Ed. 2017, 56, 14712–14717. [Google Scholar] [CrossRef] [PubMed]
- Thiele, N.A.; MacMillan, S.N.; Wilson, J.J. Rapid dissolution of BaSO4 by macropa, an 18-membered macrocycle with high affinity for Ba2+. J. Am. Chem. Soc. 2018, 140, 17071–17078. [Google Scholar] [CrossRef] [PubMed]
- Kálmán, F.K.; Végh, A.; Regueiro-Figueroa, M.; Tóth, É.; Platas-Iglesias, C.; Tircsó, G. H4octapa: highly stable complexation of lanthanide(III) ions and copper(II). Inorg. Chem. 2015, 54, 2345–2356. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illa, O.; Serra, A.; Ardiaca, A.; Herrero, X.; Closa, G.; Ortuño, R.M. Cyclobutane-Containing Scaffolds as Useful Intermediates in the Stereoselective Synthesis of Suitable Candidates for Biomedical Purposes: Surfactants, Gelators and Metal Cation Ligands. Int. J. Mol. Sci. 2019, 20, 4333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184333
Illa O, Serra A, Ardiaca A, Herrero X, Closa G, Ortuño RM. Cyclobutane-Containing Scaffolds as Useful Intermediates in the Stereoselective Synthesis of Suitable Candidates for Biomedical Purposes: Surfactants, Gelators and Metal Cation Ligands. International Journal of Molecular Sciences. 2019; 20(18):4333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184333
Chicago/Turabian StyleIlla, Ona, Albert Serra, Agustí Ardiaca, Xavier Herrero, Guillem Closa, and Rosa M. Ortuño. 2019. "Cyclobutane-Containing Scaffolds as Useful Intermediates in the Stereoselective Synthesis of Suitable Candidates for Biomedical Purposes: Surfactants, Gelators and Metal Cation Ligands" International Journal of Molecular Sciences 20, no. 18: 4333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184333