Glucose-Sensing Transcription Factor MondoA/ChREBP as Targets for Type 2 Diabetes: Opportunities and Challenges


Abstract
:1. Introduction
2. Glucose Sensors: MondoA vs. ChREBP
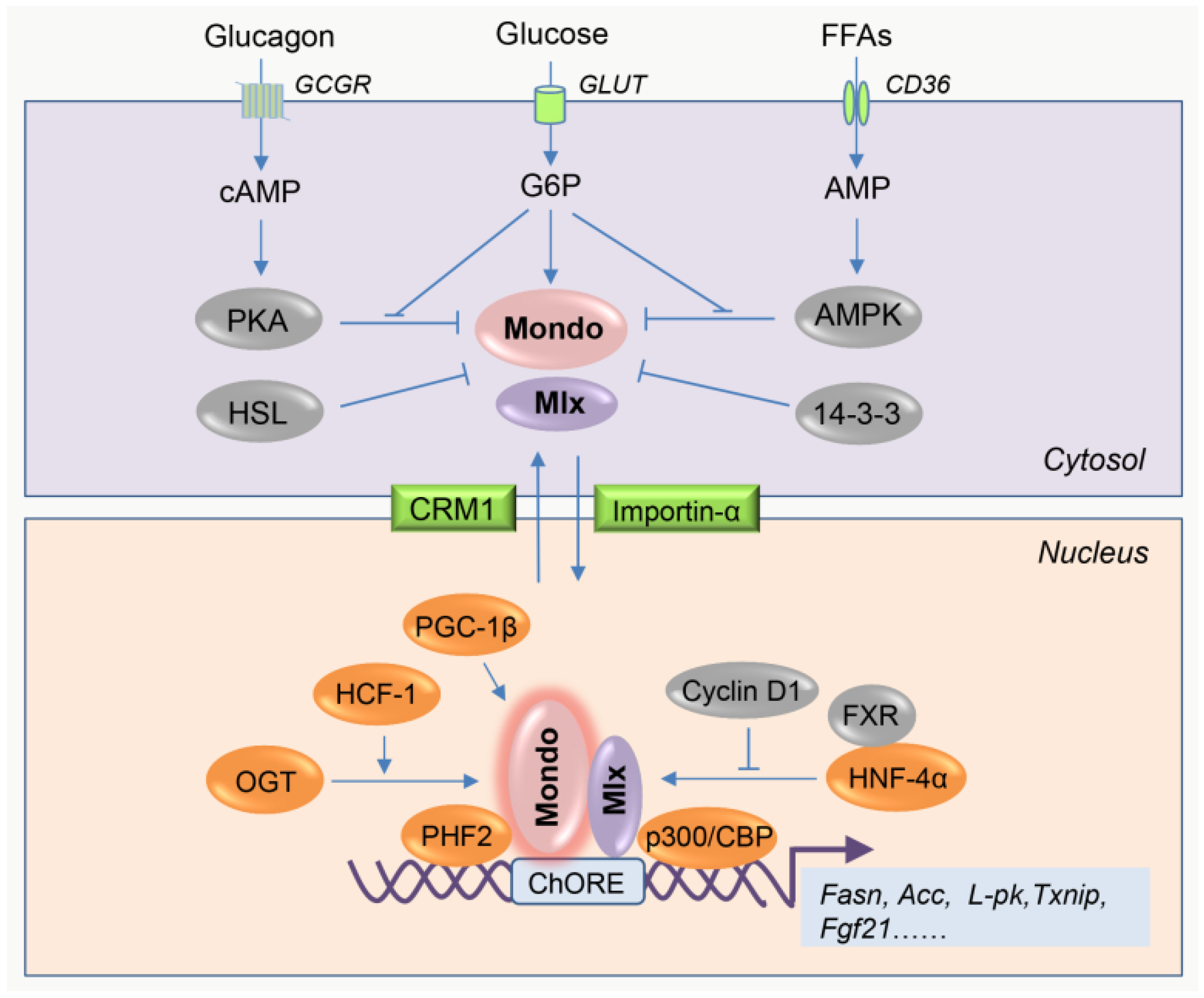
2.1. Overview of the Intrinsic Regulation of MondoA/ChREBP Activity
2.2. Differences between MondoA and ChREBP
3. Muscle MondoA: A Negative Regulator of Insulin Sensitivity
3.1. Role of MondoA in Muscle Glucose Metabolism
3.2. Role of Muscle MondoA in IR Development
4. Hepatic ChREBP: More Protector than Killer for Insulin Sensitivity
4.1. Promotion of IR Development by Hepatic ChREBP
4.2. Inhibition of IR Development by Hepatic ChREBP
5. Adipose ChREBP: A Master Regulator of Systemic Insulin Sensitivity
5.1. The Disturbed Expression of Adipose ChREBP during IR Development
5.2. Maintenance of Systemic Insulin Sensitivity by WAT ChREBP
5.3. BAT ChREBP is Dispensable for Systemic Insulin Sensitivity
6. Pancreatic ChREBP: A Double-Edged Sword for Insulin Production
6.1. Role of Pancreatic ChREBP in β-cell Adaptive Proliferation
6.2. Role of Pancreatic ChREBP in β-Cell Failure
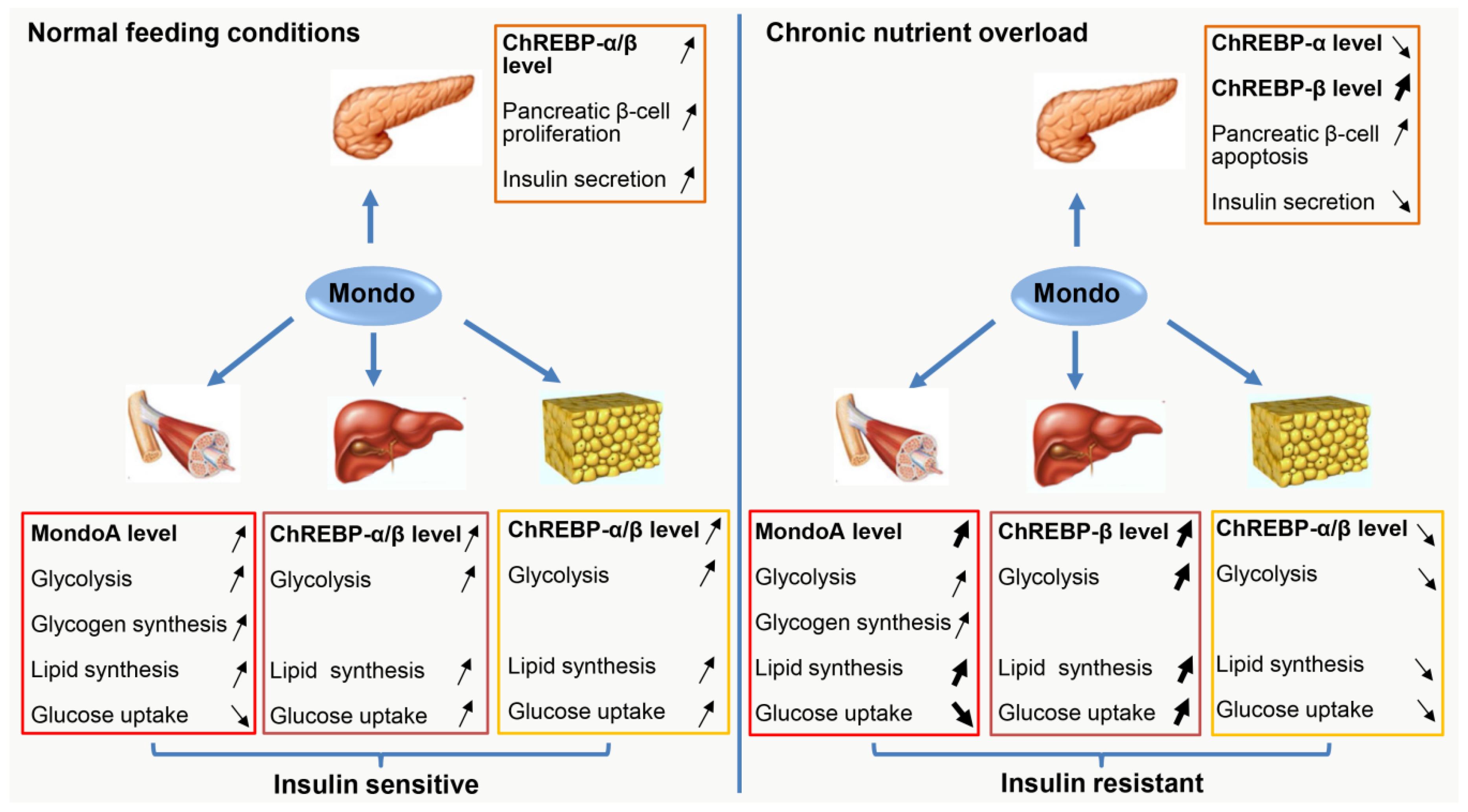
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACSL1 | acyl-CoA synthetase long chain family member 1 |
| AMPK | AMP-activated protein kinase |
| ARRDC4 | arrestin domain–containing 4 |
| AT | adipose tissue |
| BAT | brown adipose tissue |
| CBP | CREB-binding protein |
| ChoRE | carbohydrate response element |
| ChREBP | carbohydrate response element-binding protein |
| ChREBP-CA | constitutively active form of ChREBP |
| CRM1 | chromosomal region maintenance 1 |
| CDK4/6 | cyclin-dependent kinase (CDK) 4/6 |
| DNL | de novo lipogenesis |
| ELOVL5 | elongation of very long chain fatty acids protein 5 |
| FASN | fatty acid synthase |
| FGF21 | fibroblast growth factor 21 |
| Fru-2,6-P2 | fructose-2,6-bisphosphate |
| FXR | farnesoid X receptor |
| G6P | glucose-6-phosphate |
| GCGR | glucagon receptor |
| GFPT1/2 | glutamine-fructose-6-phosphate transaminase 1/2 |
| GLP-1 | glucagon-like peptide-1 |
| GLUT | glucose transporter |
| GSM | glucose sensing module |
| HCF-1 | host cell factor-1 |
| HFD | high-fat diet |
| HNF-4α | hepatocyte nuclear factor-4α |
| HOMA-IR | homeostasis model assessment of insulin resistance |
| HSL | hormone-sensitive lipase |
| IR | insulin resistance |
| MCP-1 | monocyte chemoattractant protein-1 |
| Mlx | Max-like protein x |
| MLXIP | Mlx-interacting protein |
| MLXIPL | Mlx-interacting protein-like |
| mTOR | mechanistic target of rapamycin |
| MUFAs | monounsaturated fatty acids |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| OGT | O-linked N-acetylglucosamine transferase |
| PAHSAs | palmitic acid esters of hydroxy stearic acids |
| PGC-1β | peroxisome proliferator-activated receptor-γ coactivator-1β |
| PHF2 | PHD finger protein 2 |
| PKA | protein kinase A |
| PPARα | peroxisome proliferator-activated receptor-α |
| PPARγ | peroxisome proliferator-activated receptor-γ |
| PPP1R3A | phosphoprotein phosphatase 1 regulatory subunit 3A |
| RetSat | retinol saturase |
| RORγ | retinoic acid receptor-related orphan receptor-γ |
| SAT | subcutaneous adipose tissue |
| SCD | stearoyl-CoA desaturase |
| SFAs | saturated fatty acids |
| TG | triglycerides |
| TNFα | tumor necrosis factor-α |
| TXNIP | thioredoxin-interacting protein |
| VAT | visceral adipose tissue |
| WAT | white adipose tissue |
| Xu5P | xylulose 5-phosphate |
| T2D | type 2 diabetes |
| ZBTB20 | zinc finger and BTB domain-containing protein 20 |
References
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Tancredi, M.; Rosengren, A.; Svensson, A.-M.; Kosiborod, M.; Pivodic, A.; Gudbjörnsdottir, S.; Wedel, H.; Clements, M.; Dahlqvist, S.; Lind, M. Excess mortality among persons with type 2 diabetes. N. Engl. J. Med. 2015, 373, 1720–1732. [Google Scholar] [CrossRef]
- Seuring, T.; Archangelidi, O.; Suhrcke, M. The economic costs of type 2 diabetes: A global systematic review. Pharmacoeconomics 2015, 33, 811–831. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism linking diabetes mellitus and obesity. Diabetes. Metab. Syndr. Obes. 2014, 7, 587. [Google Scholar] [CrossRef]
- Ley, S.H.; Hamdy, O.; Mohan, V.; Hu, F.B. Prevention and management of type 2 diabetes: Dietary components and nutritional strategies. Lancet 2014, 383, 1999–2007. [Google Scholar] [CrossRef]
- Connelly, J.; Kirk, A.; Masthoff, J.; MacRury, S. The use of technology to promote physical activity in type 2 diabetes management: A systematic review. Diabet. Med. 2013, 30, 1420–1432. [Google Scholar] [CrossRef]
- Kerru, N.; Singh-Pillay, A.; Awolade, P.; Singh, P. Current anti-diabetic agents and their molecular targets: A review. Eur. J. Med. Chem. 2018, 152, 436–488. [Google Scholar] [CrossRef]
- Ahn, B.; Wan, S.; Jaiswal, N.; Vega, R.B.; Ayer, D.E.; Titchenell, P.M.; Han, X.; Won, K.J.; Kelly, D.P. MondoA drives muscle lipid accumulation and insulin resistance. JCI Insight 2019, 5, 129119. [Google Scholar] [CrossRef]
- Vijayakumar, A.; Aryal, P.; Wen, J.; Syed, I.; Vazirani, R.P.; Moraes-Vieira, P.M.; Camporez, J.P.; Gallop, M.R.; Perry, R.J.; Peroni, O.D. Absence of carbohydrate response element binding protein in adipocytes causes systemic insulin resistance and impairs glucose transport. Cell Rep. 2017, 21, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Metukuri, M.R.; Zhang, P.; Basantani, M.K.; Chin, C.; Stamateris, R.E.; Alonso, L.C.; Takane, K.K.; Gramignoli, R.; Strom, S.C.; O’Doherty, R.M. ChREBP mediates glucose-stimulated pancreatic β-cell proliferation. Diabetes 2012, 61, 2004–2015. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef]
- Towle, H.C. Glucose as a regulator of eukaryotic gene transcription. Trends Endocrinol. Metab. 2005, 16, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Havula, E.; Hietakangas, V. Glucose sensing by ChREBP/MondoA–Mlx transcription factors. Semin. Cell Dev. Biol. 2012, 23, 640–647. [Google Scholar] [CrossRef]
- Richards, P.; Ourabah, S.; Montagne, J.; Burnol, A.-F.; Postic, C.; Guilmeau, S. MondoA/ChREBP: The usual suspects of transcriptional glucose sensing; implication in pathophysiology. Metabolism 2017, 70, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Havula, E.; Hietakangas, V. Sugar sensing by ChREBP/Mondo-Mlx—New insight into downstream regulatory networks and integration of nutrient-derived signals. Curr. Opin. Cell Biol. 2018, 51, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, A.; Aghajani, H.; Fallah, S.; Assadi, M.; Seifi, M. C771G (His241Gln) polymorphism of MLXIPL gene, TG levels and coronary artery disease: A case control study. Anatol. J. Cardiol. 2015, 15, 8. [Google Scholar] [CrossRef]
- Radovica, I.; Fridmanis, D.; Silamikelis, I.; Nikitina-Zake, L.; Klovins, J. Association between CETP, MLXIPL, and TOMM40 polymorphisms and serum lipid levels in a latvian population. Meta. Gene 2014, 2, 565–578. [Google Scholar] [CrossRef]
- Stagi, S.; Lapi, E.; Cecchi, C.; Chiarelli, F.; D’Avanzo, M.G.; Seminara, S.; de Martino, M. Williams-beuren syndrome is a genetic disorder associated with impaired glucose tolerance and diabetes in childhood and adolescence: New insights from a longitudinal study. Horm. Res. Paediatr. 2014, 82, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billin, A.N.; Eilers, A.L.; Coulter, K.L.; Logan, J.S.; Ayer, D.E. MondoA, a novel basic helix-loop-helix-leucine zipper transcriptional activator that constitutes a positive branch of a max-like network. Mol. Cell. Biol. 2000, 20, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Billin, A.N.; Eilers, A.L.; Queva, C.; Ayer, D.E. Mlx, a novel Max-like BHLHZip protein that interacts with the Max network of transcription factors. J. Biol. Chem. 1999, 274, 36344–36350. [Google Scholar] [CrossRef]
- Meroni, G.; Cairo, S.; Merla, G.; Messali, S.; Brent, R.; Ballabio, A.; Reymond, A. Mlx, a new Max-like BHLHZip family member: The center stage of a novel transcription factors regulatory pathway? Oncogene 2000, 19, 3266. [Google Scholar] [CrossRef]
- Stoeckman, A.K.; Ma, L.; Towle, H.C. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem. 2004, 279, 15662–15669. [Google Scholar] [CrossRef]
- Havula, E.; Teesalu, M.; Hyötyläinen, T.; Seppälä, H.; Hasygar, K.; Auvinen, P.; Orešič, M.; Sandmann, T.; Hietakangas, V. Mondo/ChREBP-Mlx-regulated transcriptional network is essential for dietary sugar tolerance in drosophila. PLoS Genet. 2013, 9, e1003438. [Google Scholar] [CrossRef]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP·Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA: Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferré, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2, 6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.L.; Al-Oanzi, Z.H.; Arden, C.; Tudhope, S.J.; Mann, J.; Kieswich, J.; Yaqoob, M.M.; Towle, H.C.; Agius, L. Glucose induces protein targeting to glycogen in hepatocytes by fructose 2, 6-bisphosphate-mediated recruitment of MondoA to the promoter. Mol. Cell. Biol. 2013, 33, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha-Molstad, H.; Saxena, G.; Chen, J.; Shalev, A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J. Biol. Chem. 2009, 284, 16898–16905. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Collier, J.J.; Scott, D.K. Camp opposes the glucose-mediated induction of the L-PK gene by preventing the recruitment of a complex containing ChREBP, HNF4alpha, and CBP. FASEB J. 2009, 23, 2855–2865. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.-R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P. Metabolite regulation of nuclear localization of carbohydrate-response element-binding protein (ChREBP) role of AMP as an allosteric inhibitor. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.-F.; Yang, X.; Lefebvre, T. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef]
- Merla, G.; Howald, C.; Antonarakis, S.E.; Reymond, A. The subcellular localization of the ChoRE-binding protein, encoded by the Williams–Beuren syndrome critical region gene 14, is regulated by 14-3-3. Hum. Mol. Genet. 2004, 13, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, H.; Wynn, R.M.; Lee, W.-R.; Fukasawa, M.; Mizuguchi, H.; Gardner, K.H.; Repa, J.J.; Uyeda, K. Regulation of nuclear import/export of carbohydrate response element-binding protein (ChREBP) interaction of an alpha-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 2008, 283, 24899–24908. [Google Scholar] [CrossRef] [PubMed]
- Eilers, A.L.; Sundwall, E.; Lin, M.; Sullivan, A.A.; Ayer, D.E. A novel heterodimerization domain, CRM1, and 14-3-3 control subcellular localization of the MondoA-Mlx heterocomplex. Mol. Cell. Biol. 2002, 22, 8514–8526. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Nakagawa, T.; Wynn, R.M.; Chook, Y.M.; Miller, B.C.; Uyeda, K. Importin-alpha protein binding to a nuclear localization signal of carbohydrate response element-binding protein (ChREBP). J. Biol. Chem. 2011, 286, 28119–28127. [Google Scholar] [CrossRef]
- Meng, J.; Feng, M.; Dong, W.; Zhu, Y.; Li, Y.; Zhang, P.; Wu, L.; Li, M.; Lu, Y.; Chen, H. Identification of HNF-4α as a key transcription factor to promote ChREBP expression in response to glucose. Sci. Rep. 2016, 6, 23944. [Google Scholar] [CrossRef]
- Caron, S.; Samanez, C.H.; Dehondt, H.; Ploton, M.; Briand, O.; Lien, F.; Dorchies, E.; Dumont, J.; Postic, C.; Cariou, B. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol. Cell. Biol. 2013, 33, 2202–2211. [Google Scholar] [CrossRef]
- Chambers, K.T.; Chen, Z.; Lai, L.; Leone, T.C.; Towle, H.C.; Kralli, A.; Crawford, P.A.; Finck, B.N. PGC-1β and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol. Metab. 2013, 2, 194–204. [Google Scholar] [CrossRef]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [Green Version]
- Bricambert, J.; Alves-Guerra, M.-C.; Esteves, P.; Prip-Buus, C.; Bertrand-Michel, J.; Guillou, H.; Chang, C.J.; Vander Wal, M.N.; Canonne-Hergaux, F.; Mathurin, P. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 2018, 9, 2092. [Google Scholar] [CrossRef]
- Lane, E.A.; Choi, D.W.; Garcia-Haro, L.; Levine, Z.G.; Tedoldi, M.; Walker, S.; Danial, N.N. HCF-1 regulates de novo lipogenesis through a nutrient-sensitive complex with ChREBP. Mol. Cell 2019, 75, 357–371. [Google Scholar] [CrossRef]
- Hanse, E.A.; Mashek, D.G.; Becker, J.R.; Solmonson, A.D.; Mullany, L.K.; Mashek, M.T.; Towle, H.C.; Chau, A.T.; Albrecht, J.H. Cyclin D1 inhibits hepatic lipogenesis via repression of carbohydrate response element binding protein and hepatocyte nuclear factor 4α. Cell Cycle 2012, 11, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mairal, A.; Ghilain, C.; Mouisel, E.; Benhamed, F.; Masri, B.; Recazens, E.; Denechaud, P.-D.; Tavernier, G. Interaction between hormone-sensitive lipase and ChREBP in fat cells controls insulin sensitivity. Nat. Metab. 2019, 1, 133. [Google Scholar] [CrossRef]
- Postic, C.; Ortega-Prieto, P. Carbohydrate sensing through the transcription factor ChREBP. Front. Genet. 2019, 10, 472. [Google Scholar]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx heterodimers are candidate sensors of cellular energy status: Mitochondrial localization and direct regulation of glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Gurmaches, J.; Tang, Y.; Jespersen, N.Z.; Wallace, M.; Calejman, C.M.; Gujja, S.; Li, H.; Edwards, Y.J.; Wolfrum, C.; Metallo, C.M. Brown fat AKT2 is a cold-induced kinase that stimulates ChREBP-mediated de novo lipogenesis to optimize fuel storage and thermogenesis. Cell Metab. 2018, 27, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tsatsos, N.G.; Towle, H.C. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J. Biol. Chem. 2005, 280, 12019–12027. [Google Scholar] [CrossRef] [PubMed]
- Tsatsos, N.G.; Davies, M.N.; O’Callaghan, B.L.; Towle, H.C. Identification and function of phosphorylation in the glucose-regulated transcription factor ChREBP. Biochem. J. 2008, 411, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Holland, W.; Sanchez-Ortiz, E.; Baskin, K.K.; Pearson, M.; Burgess, S.C.; Nelson, B.R.; Bassel-Duby, R.; Olson, E.N. A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism. Genes Dev. 2016, 30, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aune, D.; Norat, T.; Leitzmann, M.; Tonstad, S.; Vatten, L.J. Physical activity and the risk of type 2 diabetes: A systematic review and dose–response meta-analysis. Eur. J. Epidemiol. 2015, 30, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Soundarapandian, M.M.; Sessions, H.; Peddibhotla, S.; Roth, G.P.; Li, J.L.; Sugarman, E.; Koo, A.; Malany, S.; Wang, M.; et al. MondoA coordinately regulates skeletal myocyte lipid homeostasis and insulin signaling. J. Clin. Investig. 2016, 126, 3567–3579. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Chang, B.H.-J.; Kohjima, M.; Li, M.; Hwang, B.; Taegtmeyer, H.; Harris, R.A.; Chan, L. MondoA deficiency enhances sprint performance in mice. Biochem. J. 2014, 464, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E358–E364. [Google Scholar] [CrossRef] [PubMed]
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 6, 1381–1394. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.-D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [Green Version]
- Nuotio-Antar, A.M.; Poungvarin, N.; Li, M.; Schupp, M.; Mohammad, M.; Gerard, S.; Zou, F.; Chan, L. FABP4-Cre mediated expression of constitutively active ChREBP protects against obesity, fatty liver, and insulin resistance. Endocrinology 2015, 156, 4020–4032. [Google Scholar] [CrossRef]
- Poungvarin, N.; Lee, J.; Yechoor, V.; Li, M.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.; Saha, P.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Hunt, L.C.; Xu, B.; Finkelstein, D.; Fan, Y.; Carroll, P.A.; Cheng, P.F.; Eisenman, R.N.; Demontis, F. The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev. 2015, 29, 2475–2489. [Google Scholar] [CrossRef] [Green Version]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2011, 4, 177–197. [Google Scholar]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The subtle balance between lipolysis and lipogenesis: A critical point in metabolic homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84. [Google Scholar] [CrossRef]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef]
- Abdul-Wahed, A.; Guilmeau, S.; Postic, C. Sweet sixteenth for ChREBP: Established roles and future goals. Cell Metab. 2017, 26, 324–341. [Google Scholar] [CrossRef]
- Iizuka, K. The role of carbohydrate response element–binding protein in the development of liver diseases. In Dietary Interventions in Liver Disease; Watson, R.R., Preedy, V.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 263–274. [Google Scholar]
- Stamatikos, A.D.; Da Silva, R.P.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Jacobs, R.L.; Paton, C.M. Tissue specific effects of dietary carbohydrates and obesity on ChREBPα and ChREBPβ expression. Lipids 2016, 51, 95–104. [Google Scholar] [CrossRef]
- del Pozo, C.H.; Vesperinas-García, G.; Rubio, M.-Á.; Corripio-Sánchez, R.; Torres-García, A.J.; Obregon, M.-J.; Calvo, R.M. ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. BBA Mol. Cell Biol. 2011, 1811, 1194–1200. [Google Scholar] [CrossRef] [Green Version]
- Eissing, L.; Scherer, T.; Tödter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Hepatic overexpression of dominant negative Mlx improves metabolic profile in diabetes-prone C57BL/6J mice. Biochem. Biophys. Res. Commun. 2009, 379, 499–504. [Google Scholar] [CrossRef]
- Heidenreich, S.; Witte, N.; Weber, P.; Goehring, I.; Tolkachov, A.; von Loeffelholz, C.; Döcke, S.; Bauer, M.; Stockmann, M.; Pfeiffer, A.F. Retinol saturase coordinates liver metabolism by regulating ChREBP activity. Nat. Commun. 2017, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhou, L.; Zhang, H.; Chen, R.; Zhang, Y.; Li, L.; Lu, J.Y.; Jiang, H.; Liu, D.; Qi, S.; et al. Regulation of hepatic lipogenesis by the zinc finger protein Zbtb20. Nat. Commun. 2017, 8, 14824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Régnier, M.; Fouché, E.; Lukowicz, C.; Cauzac, M. A specific ChREBP and PPARα cross-talk is required for the glucose-mediated FGF21 response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as modulator of metabolism in health and disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef]
- Townsend, K.L.; Tseng, Y.H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xiaoli, A.; Yang, F. Regulation and metabolic significance of de novo lipogenesis in adipose tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Pedersen, D.J.; Henchey, E.; Henriques, F.S.; Danai, L.V.; Shen, Y.; Yenilmez, B.; Jung, D.; Kim, J.K.; Lodhi, I.J. Adipocyte lipid synthesis coupled to neuronal control of thermogenic programming. Mol. Metab. 2017, 6, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Kursawe, R.; Caprio, S.; Giannini, C.; Narayan, D.; Lin, A.; D’Adamo, E.; Shaw, M.; Pierpont, B.; Cushman, S.W.; Shulman, G.I. Decreased transcription of ChREBP-α/β isoforms in abdominal subcutaneous adipose tissue of obese adolescents with prediabetes or early type 2 diabetes: Associations with insulin resistance and hyperglycemia. Diabetes 2013, 62, 837–844. [Google Scholar] [CrossRef]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef]
- Witte, N.; Muenzner, M.; Rietscher, J.; Knauer, M.; Heidenreich, S.; Nuotio-Antar, A.M.; Graef, F.A.; Fedders, R.; Tolkachov, A.; Goehring, I. The glucose sensor ChREBP links de novo lipogenesis to PPARγ activity and adipocyte differentiation. Endocrinology 2015, 156, 4008–4019. [Google Scholar] [CrossRef]
- Katz, L.S.; Xu, S.; Ge, K.; Scott, D.K.; Gershengorn, M.C. T3 and glucose coordinately stimulate ChREBP-mediated Ucp1 expression in brown adipocytes from male mice. Endocrinology 2017, 159, 557–569. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Kaisanlahti, A.; Glumoff, T. Browning of white fat: Agents and implications for beige adipose tissue to type 2 diabetes. J. Physiol. Biochem. 2019, 75, 1–10. [Google Scholar] [CrossRef]
- Xia, B.; Cai, G.H.; Yang, H.; Wang, S.P.; Mitchell, G.A.; Wu, J.W. Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice. PLoS Genet. 2017, 13, e1007110. [Google Scholar] [CrossRef]
- Roder, P.V.; Wu, B.B.; Liu, Y.X.; Han, W.P. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed]
- Honka, H.; Hannukainen, J.C.; Tarkia, M.; Karlsson, H.; Saunavaara, V.; Salminen, P.; Soinio, M.; Mikkola, K.; Kudomi, N.; Oikonen, V. Pancreatic metabolism, blood flow, and β-cell function in obese humans. J. Clin. Endocrinol. Metab. 2014, 99, E981–E990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kumar, A.; Katz, L.S.; Li, L.; Paulynice, M.; Herman, M.A.; Scott, D.K. Induction of the ChREBPβ isoform is essential for glucose-stimulated β-cell proliferation. Diabetes 2015, 64, 4158–4170. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.F.; Madsen, J.G.S.; Frafjord, K.Ø.; la Cour Poulsen, L.; Salö, S.; Boergesen, M.; Loft, A.; Larsen, B.D.; Madsen, M.S.; Holst, J.J. Integrative genomics outlines a biphasic glucose response and a ChREBP-RORγ axis regulating proliferation in β cells. Cell Rep. 2016, 16, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Katz, L.S.; Schulz, A.M.; Kim, M.; Honig, L.B.; Li, L.; Davenport, B.; Homann, D.; Garcia-Ocaña, A.; Herman, M.A. Activation of Nrf2 is required for normal and ChREBPα-augmented glucose-stimulated β-cell proliferation. Diabetes 2018, 67, 1561–1575. [Google Scholar] [CrossRef]
- Chau, G.C.; Im, D.U.; Kang, T.M.; Bae, J.M.; Kim, W.; Pyo, S.; Moon, E.-Y.; Um, S.H. mTOR controls ChREBP transcriptional activity and pancreatic β cell survival under diabetic stress. J. Cell Biol. 2017, 216, 2091–2105. [Google Scholar] [CrossRef]
- Jing, G.; Chen, J.; Xu, G.; Shalev, A. Islet ChREBP-β is increased in diabetes and controls ChREBP-α and glucose-induced gene expression via a negative feedback loop. Mol. Metab. 2016, 5, 1208–1215. [Google Scholar] [CrossRef]
- Boergesen, M.; la Cour Poulsen, L.; Schmidt, S.F.; Frigerio, F.; Maechler, P.; Mandrup, S. ChREBP mediates glucose repression of peroxisome proliferator-activated receptor α expression in pancreatic β-cells. J. Biol. Chem. 2011, 286, 13214–13225. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces β-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef]
- Chen, J.; Saxena, G.; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and β-cell apoptosis. Diabetes 2008, 57, 938–944. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Saurina, J.; Sentellas, S. Liquid chromatography coupled to mass spectrometry for metabolite profiling in the field of drug discovery. Expert Opin. Drug Dis. 2019, 14, 469–483. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Characteristic | MondoA | ChREBP |
|---|---|---|
| Other names | MLXIP | MLXIPL, MondoB, WBSCR14 |
| Coding gene location (Homo sapiens) | chromosome 12q24.31 | chromosome 7q11.23 |
| Isoforms | MondoA | ChREBP-α and ChREBP-β |
| Protein weight (Homo sapiens) | 919 AA | 852 AA and 675AA |
| Primary enriched tissues | skeletal muscle | liver, adipose tissue |
| Basal subcellular localization | outer mitochondrial membrane | cytosol |
| Major downstream pathways | glycolysis | lipogenesis |
| Mouse Models | Context | Body Weight | Fat Mass | Hepatic Steatosis | Insulin Sensitivity | Reference |
|---|---|---|---|---|---|---|
| MondoA global knockout | Standard diet | = | ND | ND | = | [64] |
| High-fat diet | NA | NA | ||||
| MondoA muscle-specific knockout | Standard diet | = | ND | ND | = | [11] |
| High-fat diet | = | ND | ND |  | [11] | |
| ChREBP global knockout | Standard diet | = |  | = | | [55] |
| Standard diet in ob/ob mice background | | | | | [65] | |
| ChREBP liver-specific knockout | Standard diet | = | | = | | [66] |
| High-fat diet | = | = | = | | [66] | |
| High-carbohydrate diet | | | | | [66] | |
| ChREBP liver-specific overexpression | Standard diet | = | | | = | [67] |
| High-fat diet | = | | | | [67] | |
| ChREBP AT-specific knockout | Standard diet | = | = | | | [12] |
| High-fat diet | = | = | = | | [12] | |
| ChREBP AT-specific overexpression | Standard diet | | | = | = | [68] |
| High-fat diet | | | | | [68] | |
| ChREBP pancreatic β cell-specific overexpression | Standard diet | | ND | ND | | [69] |
| High-fat diet | NA | NA | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Yang, H.; Zhou, L.; Yang, F. Glucose-Sensing Transcription Factor MondoA/ChREBP as Targets for Type 2 Diabetes: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 5132. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205132
Song Z, Yang H, Zhou L, Yang F. Glucose-Sensing Transcription Factor MondoA/ChREBP as Targets for Type 2 Diabetes: Opportunities and Challenges. International Journal of Molecular Sciences. 2019; 20(20):5132. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205132
Chicago/Turabian StyleSong, Ziyi, Hao Yang, Lei Zhou, and Fajun Yang. 2019. "Glucose-Sensing Transcription Factor MondoA/ChREBP as Targets for Type 2 Diabetes: Opportunities and Challenges" International Journal of Molecular Sciences 20, no. 20: 5132. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205132