AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells

 ,
,
Abstract
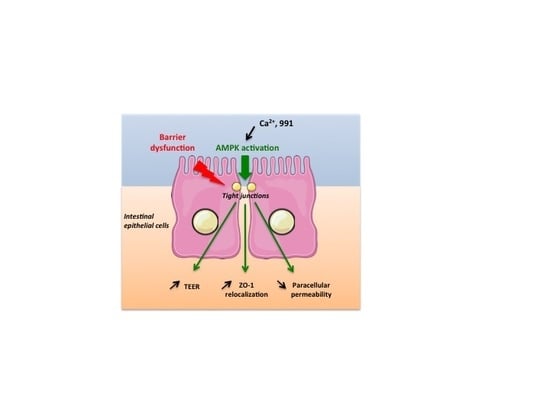
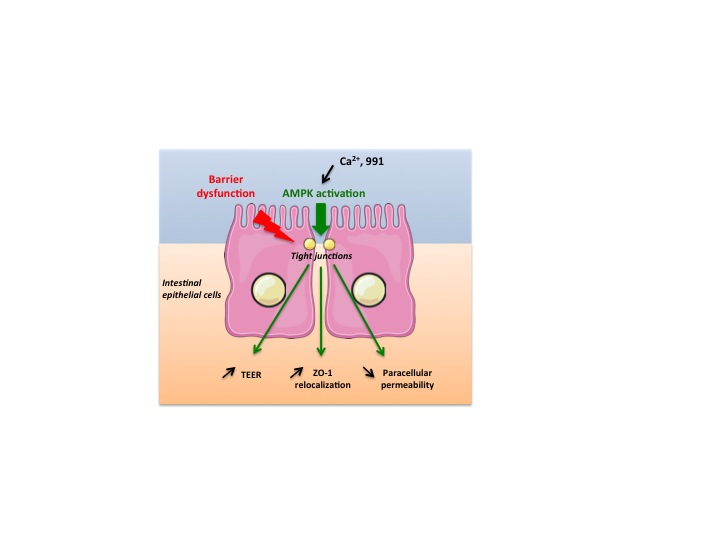
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
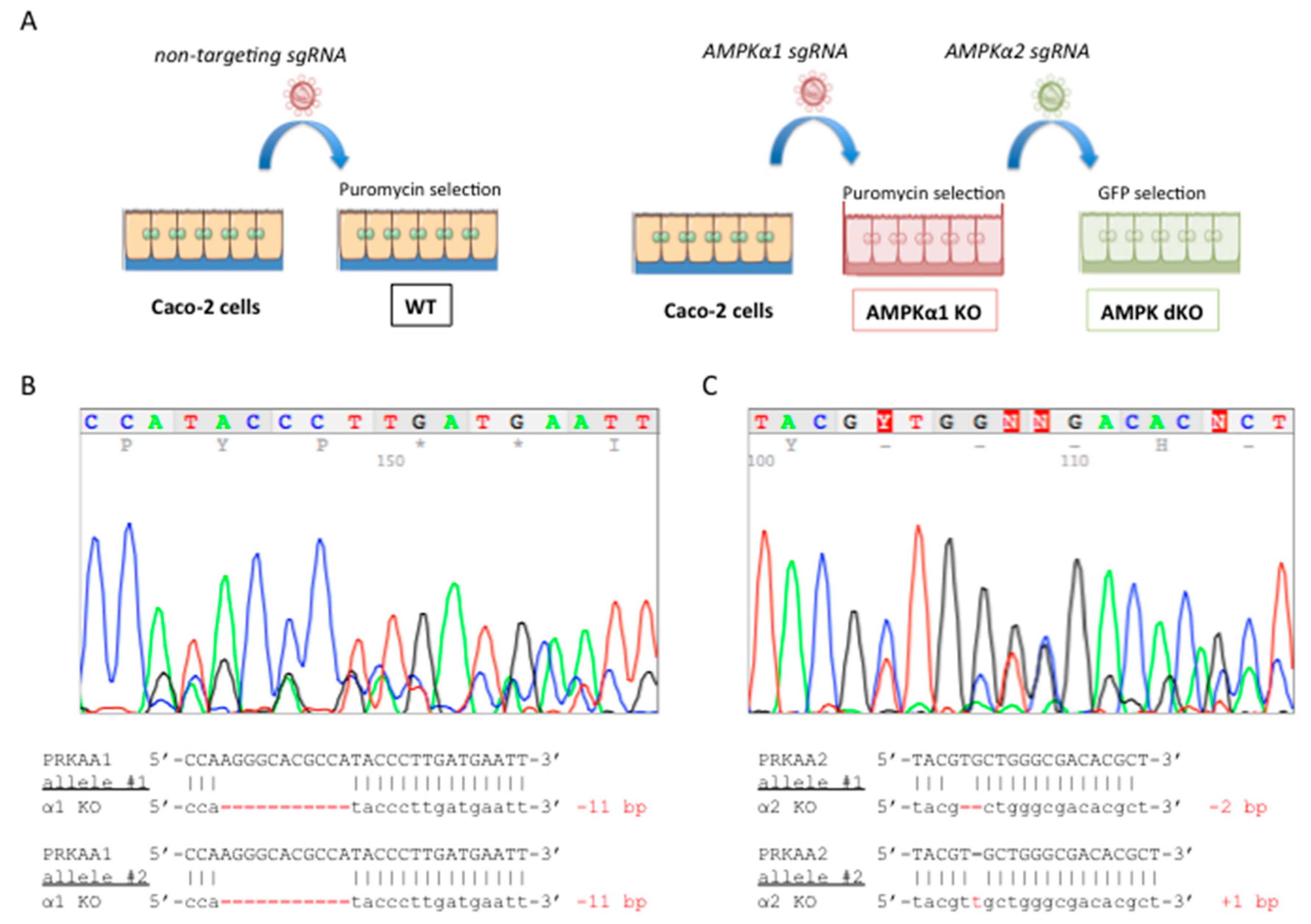
2.1. Deletion of both AMPKα1 and AMPKα2 is Necessary to Abrogate AMPK Signaling in Caco-2 Cells
2.2. Disruption of AMPK in Caco-2 cells does not Alter the Integrity of Tight Junction at Steady-State
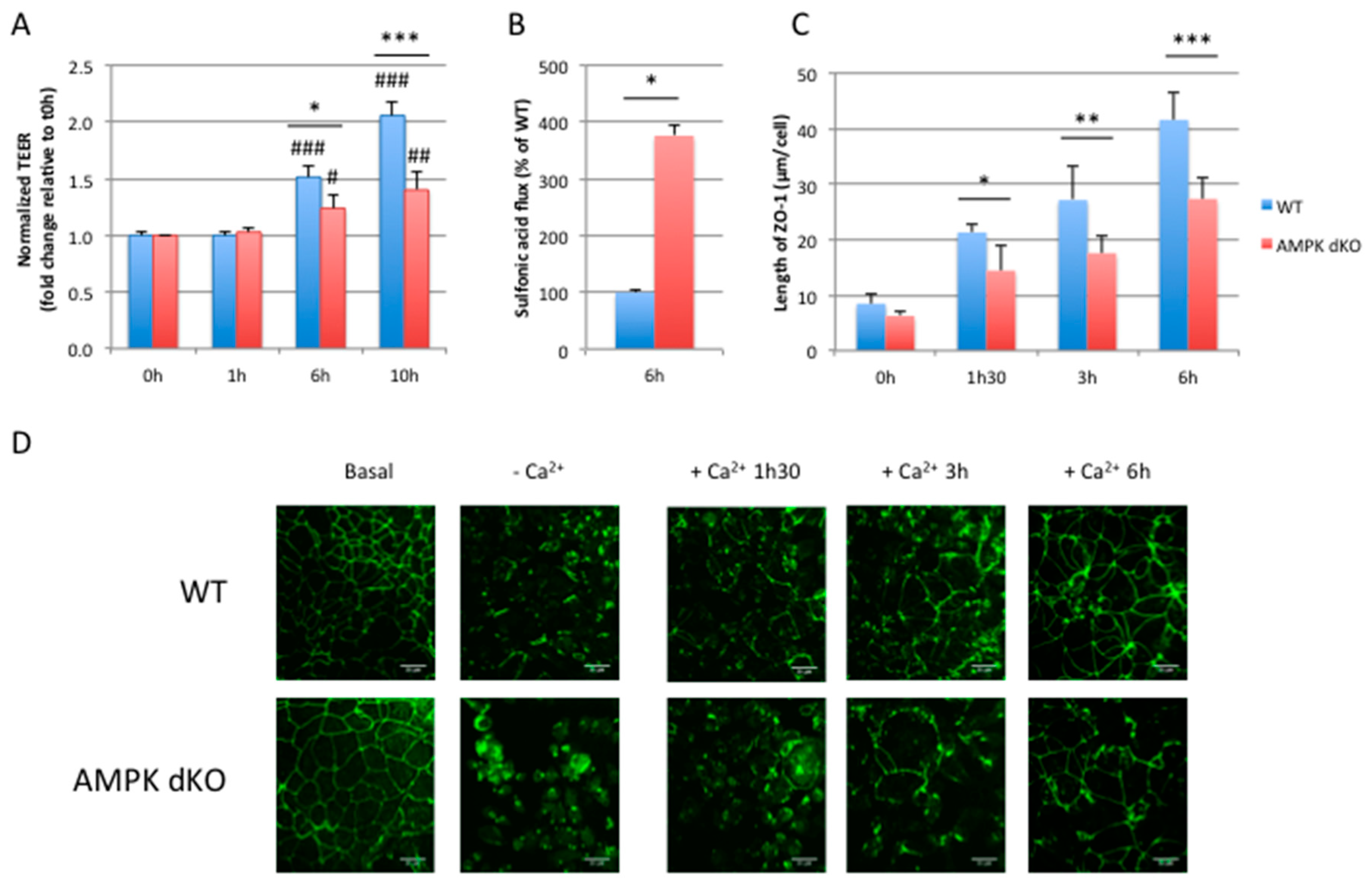
2.3. Deletion of AMPK Prevents Calcium-Induced Reassembly of Tight Junctions in Caco-2 Cells
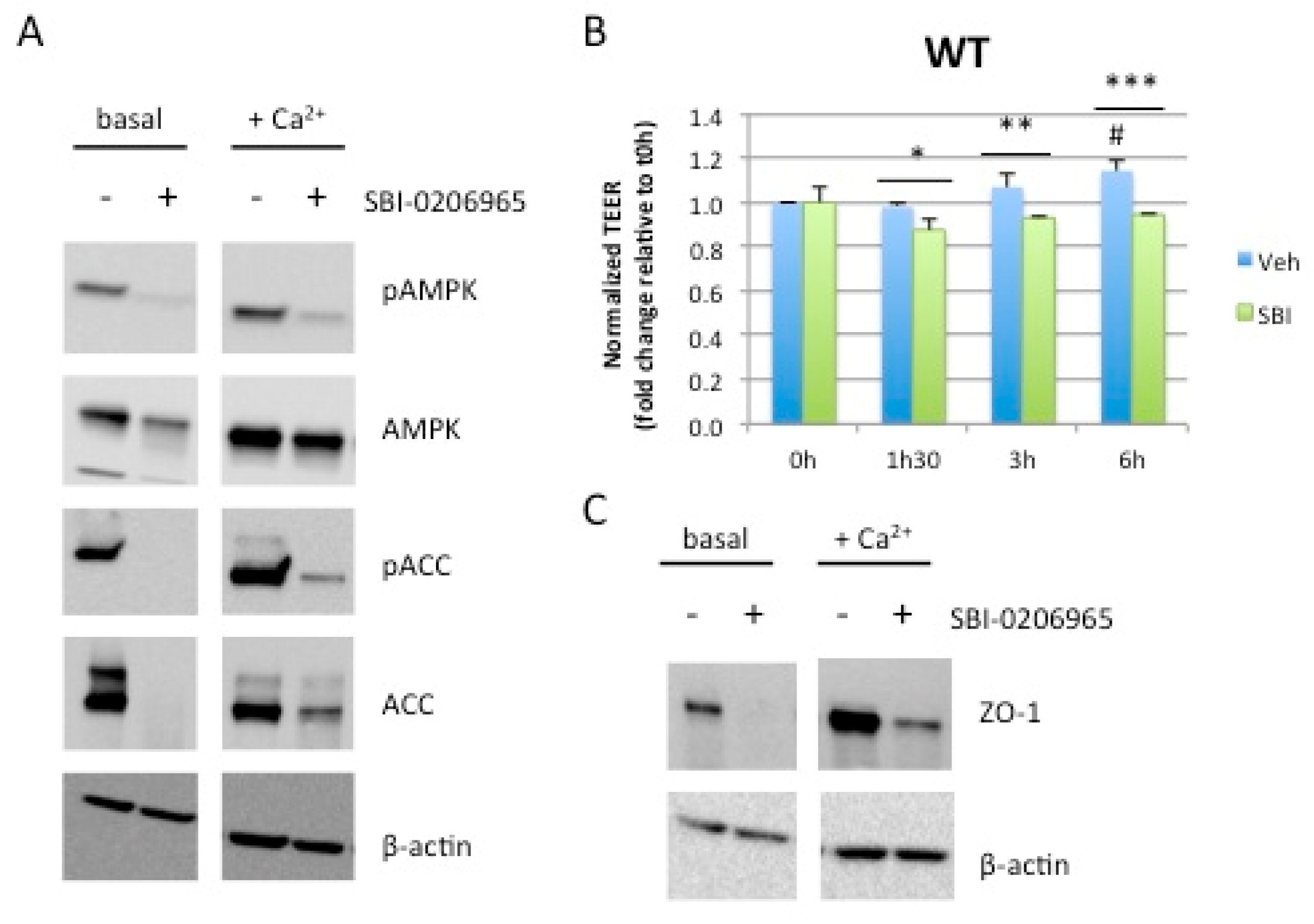
2.4. Acute Pharmacological Inhibition of AMPK Prevents Calcium-Induced Reassembly of Tight Junctions in Caco-2 Cells
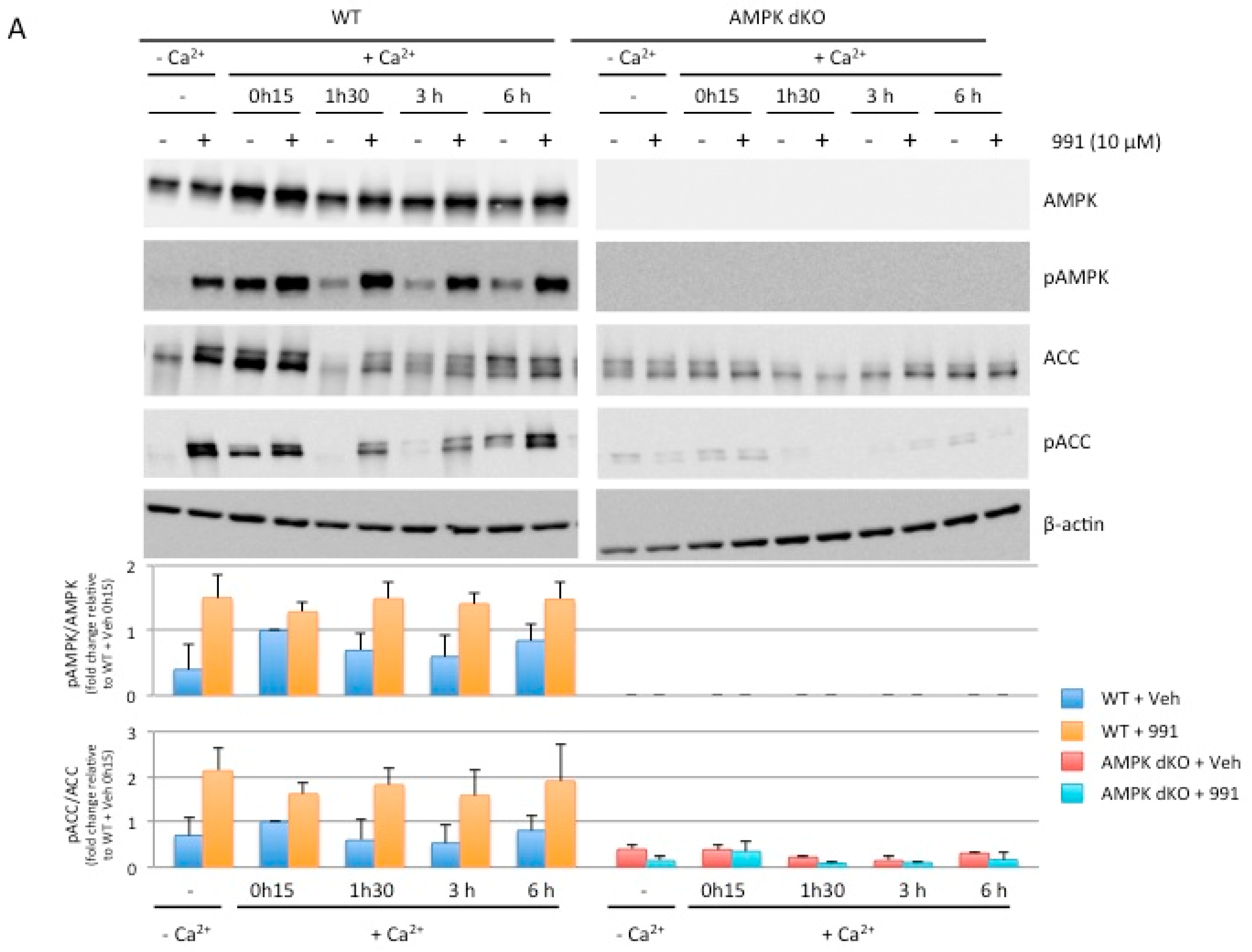
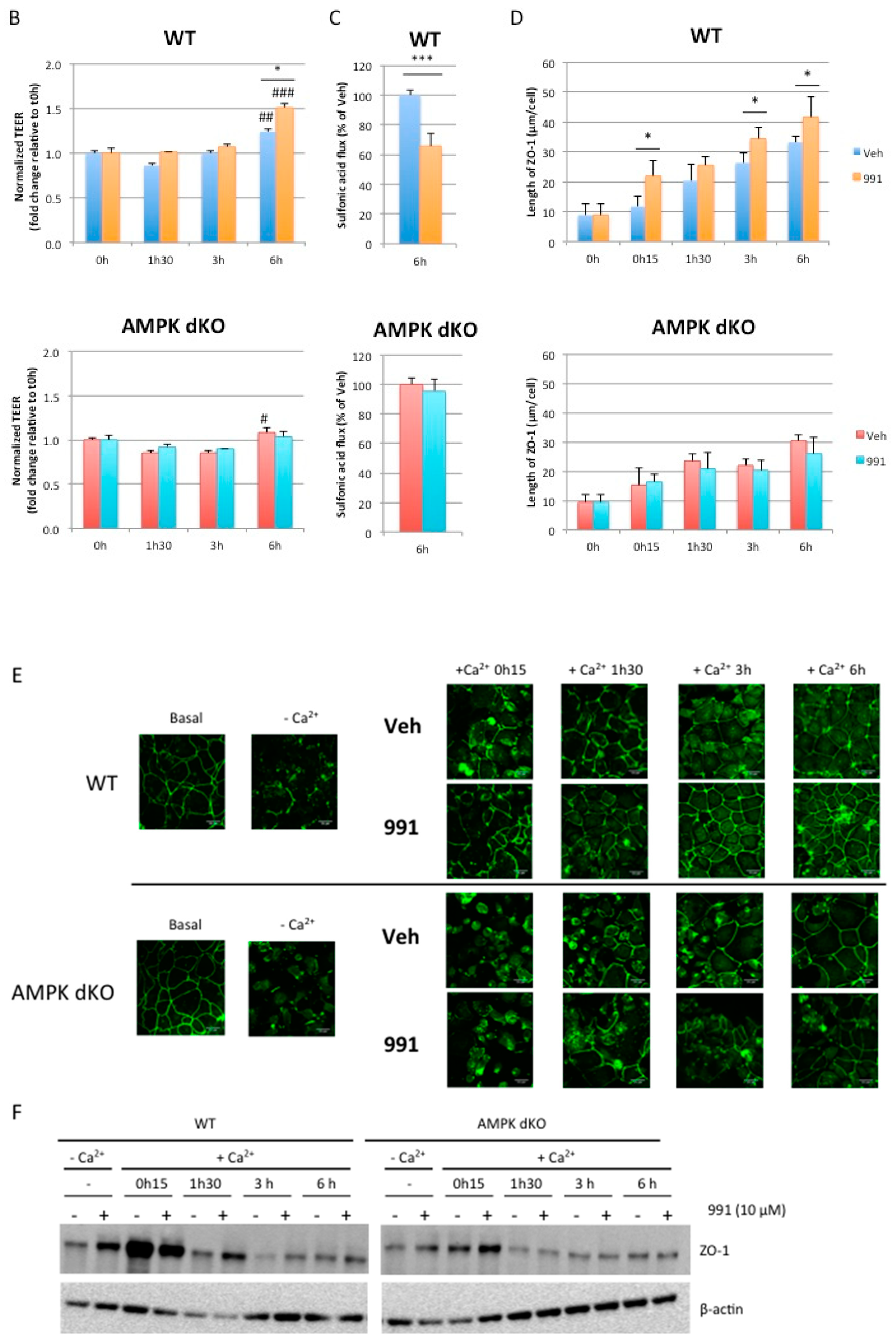
2.5. Pharmacological AMPK Activation Enhances the Reassembly of Tight Junctions
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Generation of AMPK Deficient Caco-2 Cells using CRISPR-Cas9 Gene Editing System
4.3. Cell Culture and Calcium Switch Assay
4.4. TEER and Paracellular Permeability Measurements
4.5. Immunofluorescence and Quantification of ZO-1
4.6. Electron Microscopy
4.7. Western Blotting
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl CoA carboxylase |
| ADaM | Allosteric drug and metabolite |
| AMPK | AMP-activated protein kinase |
| CaMKKβ | Ca2+ calmodulin-activated protein kinase kinase beta |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/associated protein 9 endonuclease |
| FITC | Fluorescein-5-isothiocyanate |
| GFP | Green fluorescent protein |
| GIV | G-alpha interacting vesicle associated protein |
| LKB1 | Liver kinase B1 |
| MDCK | Madin-Darby canine kidney |
| MLC2 | Myosin II regulatory light chain |
| mTOR | Mammalian target of rapamycin |
| PKC | Proteins kinase C |
| SCFAs | Short chain fatty acids |
| TEER | Trans-epithelial electrical resistance |
| ZO-1 | Zonula occludens 1 |
References
- Anderson, J.M.; Van Itallie, C.M. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995, 269, G467-75. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, A. Molecular architecture of adherens junctions. Curr. Opin. Cell Biol. 2001, 13, 600–603. [Google Scholar] [CrossRef]
- Wodarz, A.; Nathke, I. Cell polarity in development and cancer. Nat. Cell Biol. 2007, 9, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. Intestinal permeability regulation by tight junction: Implication on inflammatory bowel diseases. Intest. Res. 2015, 13, 11–18. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Guo, H.; Zhang, C.S.; Lin, S.Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Guigas, B.; Viollet, B. Targeting AMPK: From Ancient Drugs to New Small-Molecule Activators. Exp. Suppl. 2016, 107, 327–350. [Google Scholar]
- Zhang, L.; Li, J.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2006, 103, 17272–17277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Cantley, L.C. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Kalsi, K.K.; Garnett, J.P.; Patkee, W.; Weekes, A.; Dockrell, M.E.; Baker, E.H.; Baines, D.L. Metformin attenuates the effect of Staphylococcus aureus on airway tight junctions by increasing PKCzeta-mediated phosphorylation of occludin. J. Cell Mol. Med. 2019, 23, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Castanares-Zapatero, D.; Bouleti, C.; Sommereyns, C.; Gerber, B.; Lecut, C.; Mathivet, T.; Horckmans, M.; Communi, D.; Foretz, M.; Vanoverschelde, J.L.; et al. Connection between cardiac vascular permeability, myocardial edema, and inflammation during sepsis: Role of the alpha1AMP-activated protein kinase isoform. Crit. Care Med. 2013, 41, e411-22. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Machida, T.; Kaneshima, S.; Matsuo, M.; Sakaguchi, S.; Takeshige, Y.; Yamauchi, A.; Kataoka, Y. Metformin induces up-regulation of blood-brain barrier functions by activating AMP-activated protein kinase in rat brain microvascular endothelial cells. Biochem. Biophy. Res. Commun. 2013, 433, 586–590. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J.; You, Q.; He, S.; Meng, Q.; Gao, J.; Wu, X.; Shen, Y.; Sun, Y.; Wu, X.; et al. Activating AMPK to Restore Tight Junction Assembly in Intestinal Epithelium and to Attenuate Experimental Colitis by Metformin. Front Pharmacol. 2018, 9, 761. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Zeng, L.; Lai, X.; Li, J.; Liu, L.; Lin, Q.; Chen, Y. Metformin protects against intestinal barrier dysfunction via AMPKalpha1-dependent inhibition of JNK signalling activation. J. Cell Mol. Med. 2018, 22, 546–557. [Google Scholar] [CrossRef]
- Xue, Y.; Zhang, H.; Sun, X.; Zhu, M.J. Metformin Improves Ileal Epithelial Barrier Function in Interleukin-10 Deficient Mice. PLoS ONE 2016, 11, e0168670. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Pieters, H.J.; Jonkers, D.M. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J Nutr. 2013, 143, 1872–1881. [Google Scholar] [CrossRef]
- Dite, T.A.; Langendorf, C.G.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.W.; Ling, N.X.Y.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenier, A.; Sujobert, P.; Olivier, S.; Guermouche, H.; Mondesir, J.; Kosmider, O.; Viollet, B.; Tamburini, J. Knockdown of Human AMPK Using the CRISPR/Cas9 Genome-Editing System. Methods Mol. Biol. 2018, 1732, 171–194. [Google Scholar] [PubMed] [Green Version]
- Harmel, E.; Grenier, E.; Bendjoudi Ouadda, A.; El Chebly, M.; Ziv, E.; Beaulieu, J.F.; Sane, A.; Spahis, S.; Laville, M.; Levy, E. AMPK in the small intestine in normal and pathophysiological conditions. Endocrinology 2014, 155, 873–888. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [Green Version]
- Cereijido, M.; Robbins, E.S.; Dolan, W.J.; Rotunno, C.A.; Sabatini, D.D. Polarized monolayers formed by epithelial cells on a permeable and translucent support. J. Cell Biol. 1978, 77, 853–880. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Baas, A.F.; Kuipers, J.; van der Wel, N.N.; Batlle, E.; Koerten, H.K.; Peters, P.J.; Clevers, H.C. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 2004, 116, 457–466. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Q.; Rogers, C.J.; Du, M.; Zhu, M.J. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017, 24, 819–831. [Google Scholar] [CrossRef]
- Guigas, B.; Sakamoto, K.; Taleux, N.; Reyna, S.M.; Musi, N.; Viollet, B.; Hue, L. Beyond AICA riboside: In search of new specific AMP-activated protein kinase activators. IUBMB Life 2009, 61, 18–26. [Google Scholar] [CrossRef]
- Olivier, S.; Foretz, M.; Viollet, B. Promise and challenges for direct small molecule AMPK activators. Biochem. Pharmacol. 2018, 153, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Nunez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Suzuki, T.; Seth, A.; Samak, G.; Rao, R. Protein kinase Czeta phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem. J. 2011, 437, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Elias, B.C.; Seth, A.; Shen, L.; Turner, J.R.; Giorgianni, F.; Desiderio, D.; Guntaka, R.; Rao, R. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc. Natl. Acad. Sci. USA 2009, 106, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Wu, X.; Wang, K.; Wang, W.; Wang, Y.; Li, Z.; Liu, J.; Li, L.; Peng, L. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCbeta2. Int. J. Mol. Sci. 2016, 17, 1696. [Google Scholar] [CrossRef]
- Lee, J.H.; Koh, H.; Kim, M.; Kim, Y.; Lee, S.Y.; Karess, R.E.; Lee, S.H.; Shong, M.; Kim, J.M.; Kim, J.; et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 2007, 447, 1017–1020. [Google Scholar] [CrossRef]
- Zhang, L.; Jouret, F.; Rinehart, J.; Sfakianos, J.; Mellman, I.; Lifton, R.P.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase (AMPK) activation and glycogen synthase kinase-3beta (GSK-3beta) inhibition induce Ca2+-independent deposition of tight junction components at the plasma membrane. J. Biol. Chem. 2011, 286, 16879–16890. [Google Scholar] [CrossRef]
- Rowart, P.; Wu, J.; Caplan, M.J.; Jouret, F. Implications of AMPK in the Formation of Epithelial Tight Junctions. Int. J. Mol. Sci. 2018, 19, 2040. [Google Scholar] [CrossRef]
- Yano, T.; Matsui, T.; Tamura, A.; Uji, M.; Tsukita, S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol. 2013, 203, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Aznar, N.; Patel, A.; Rohena, C.C.; Dunkel, Y.; Joosen, L.P.; Taupin, V.; Kufareva, I.; Farquhar, M.G.; Ghosh, P. AMP-activated protein kinase fortifies epithelial tight junctions during energetic stress via its effector GIV/Girdin. Elife 2016, 5. [Google Scholar] [CrossRef]
- Ghosh, P.; Swanson, L.; Sayed, I.M.; Mittal, Y.; Lim, B.B.; Ibeawuchi, S.-R.; Foretz, M.; Viollet, B.; Sahoo, D.; Das, S. A Signaling Pathway to Detect and Repair the Leaky Gut barrier: Implications in Aging and Cancer. Unpublished work. 2019. [Google Scholar]
- Elamin, E.; Masclee, A.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Pieters, H.J.; Dekker, J.; Jonkers, D. Fatty acid ethyl esters induce intestinal epithelial barrier dysfunction via a reactive oxygen species-dependent mechanism in a three-dimensional cell culture model. PLoS ONE 2013, 8, e58561. [Google Scholar] [CrossRef] [PubMed]
- Muanprasat, C.; Wongkrasant, P.; Satitsri, S.; Moonwiriyakit, A.; Pongkorpsakol, P.; Mattaveewong, T.; Pichyangkura, R.; Chatsudthipong, V. Activation of AMPK by chitosan oligosaccharide in intestinal epithelial cells: Mechanism of action and potential applications in intestinal disorders. Biochem Pharmacol 2015, 96, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jin, X.; Chen, Y.; Song, Z.; Jiang, X.; Hu, F.; Conlon, M.A.; Topping, D.L. Polyphenol-Rich Propolis Extracts Strengthen Intestinal Barrier Function by Activating AMPK and ERK Signaling. Nutrients 2016, 8, 272. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Du, M.; Navarre, D.A.; Zhu, M.J. Purple Potato Extract Promotes Intestinal Epithelial Differentiation and Barrier Function by Activating AMP-Activated Protein Kinase. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kunitake, Y.; Hirasaki, N.; Tanaka, M.; Matsui, T. Theaflavins enhance intestinal barrier of Caco-2 Cell monolayers through the expression of AMP-activated protein kinase-mediated Occludin, Claudin-1, and ZO-1. Biosci. Biotechnol. Biochem. 2015, 79, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivier, S.; Leclerc, J.; Grenier, A.; Foretz, M.; Tamburini, J.; Viollet, B. AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 5171. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205171
Olivier S, Leclerc J, Grenier A, Foretz M, Tamburini J, Viollet B. AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells. International Journal of Molecular Sciences. 2019; 20(20):5171. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205171
Chicago/Turabian StyleOlivier, Séverine, Jocelyne Leclerc, Adrien Grenier, Marc Foretz, Jérôme Tamburini, and Benoit Viollet. 2019. "AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells" International Journal of Molecular Sciences 20, no. 20: 5171. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205171