The ST2/Interleukin-33 Axis in Hematologic Malignancies: The IL-33 Paradox

 ,
, 
Abstract
:1. Introduction
1.1. IL-33
1.2. The ST2 Receptors
1.3. IL-33 Cellular Targets
1.4. IL-33 Action in Nonhematologic Malignancies
2. IL-33 and Hematologic Malignancies
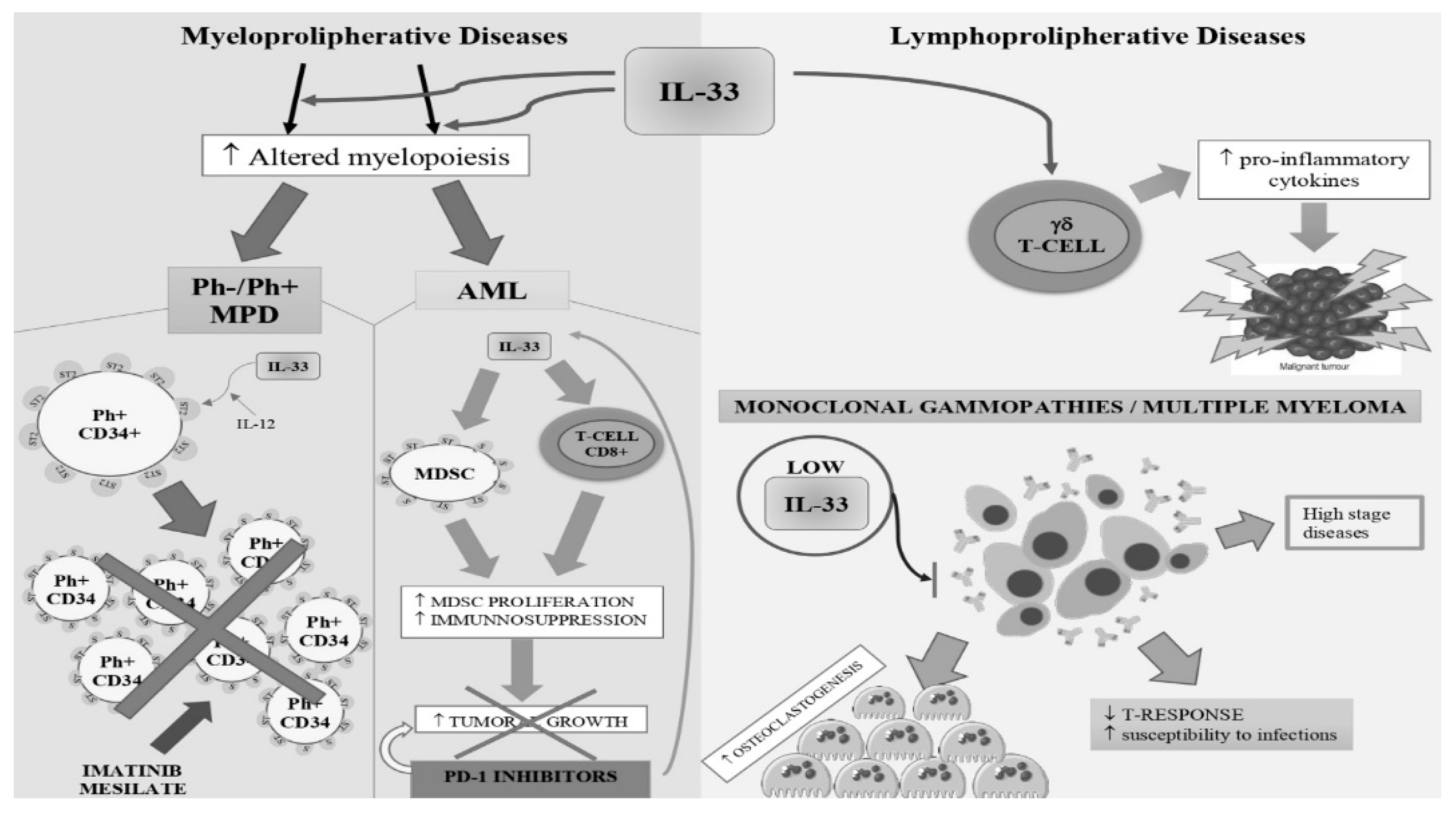
2.1. BCR-ABL1-Negative and Positive Myeloproliferative Neoplasms.
2.2. Acute Myeloid Leukemia
2.3. Lymphoproliferative Diseases
2.4. Monoclonal Gammopathies
3. Conclusions
3.1. Future Challenges and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Krumm, B.; Xiang, Y.; Deng, J. Structural biology of the IL-1 superfamily: Key cytokines in the regulation of immune and inflammatory responses. Protein Sci. 2014, 23, 526–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.-A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.-P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel ‘alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Gajardo Carrasco, T.; Morales, R.A.; Pérez, F.; Terraza, C.; Yáñez, L.; Campos-Mora, M.; Pino-Lagos, K. Alarmin’immunologists: IL-33 as a putative target for modulating T cell-dependent responses. Front. Immunol. 2015, 6, 232. [Google Scholar] [CrossRef]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.-P.; et al. Long-term IL-33–producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef]
- Gordon, E.D.; Simpson, L.J.; Rios, C.L.; Ringel, L.; Lachowicz-Scroggins, M.E.; Peters, M.C.; Wesolowska-Andersen, A.; Gonzalez, J.R.; MacLeod, H.J.; Christian, L.S.; et al. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc. Natl. Acad. Sci. USA 2016, 113, 8765–8770. [Google Scholar] [CrossRef] [Green Version]
- Lefrançais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Girard, J.-P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Nguyen, D.Q.; Falk, W.; Martin, M.U. Caspase 3 inactivates biologically active full length interleukin-33 as a classical cytokine but does not prohibit nuclear translocation. Biochem. Biophys. Res. Commun. 2010, 391, 1512–1516. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat. Commun. 2015, 6, 8327. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Bae, S.; Jhun, H.; Lee, S.; Choi, J.; Kang, T.; Kwak, A.; Hong, K.; Kim, E.; Jo, S.; et al. Identification of constitutively active interleukin 33 (IL-33) splice variant. J. Biol. Chem. 2011, 286, 20078–20086. [Google Scholar] [CrossRef]
- Tsuda, H.; Komine, M.; Karakawa, M.; Etoh, T.; Tominaga, S.; Ohtsuki, M. Novel splice variants of IL-33: Differential expression in normal and transformed cells. J. Investig. Dermatol. 2012, 132, 2661. [Google Scholar] [CrossRef]
- Smithgall, M.D.; Comeau, M.R.; Park Yoon, B.-R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1-and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Iijima, K.; Kobayashi, T.; Kephart, G.M.; McKenzie, A.N.; Kita, H. IL-33–responsive lineage−CD25+ CD44hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J. Immunol. 2012, 188, 1503–1513. [Google Scholar] [CrossRef]
- Lefrançais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.-P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, K.; Takagi, T.; Tsukamoto, T.; Tetsuka, T.; Tominaga, S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. FEBS Lett. 1993, 318, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Hardman, C.; Ogg, G. Interleukin-33, friend and foe in type-2 immune responses. Curr. Opin. Immunol. 2016, 42, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Reikerstorfer, A.; Braselmann, S.; Graninger, P.; Busslinger, M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J. 1994, 13, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, S.; Kuroiwa, K.; Tago, K.; Iwahana, H.; Yanagisawa, K.; Komatsu, N. Presence and expression of a novel variant form of ST2 gene product in human leukemic cell line UT-7/GM. Biochem. Biophys. Res. Commun. 1999, 264, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Tago, K.; Noda, T.; Hayakawa, M.; Iwahana, H.; Yanagisawa, K.; Yashiro, T.; Tominaga, S. Tissue distribution and subcellular localization of a variant form of the human ST2 gene product, ST2V. Biochem. Biophys. Res. Commun. 2001, 285, 1377–1383. [Google Scholar] [CrossRef]
- Iwahana, H.; Hayakawa, M.; Kuroiwa, K.; Tago, K.; Yanagisawa, K.; Noji, S.; Tominaga, S. Molecular cloning of the chicken ST2 gene and a novel variant form of the ST2 gene product, ST2LV. Biochim. Biophys. Acta 2004, 1681, 1–14. [Google Scholar] [CrossRef]
- Oshikawa, K.; Yanagisawa, K.; Tominaga, S.; Sugiyama, Y. Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin. Exp. Allergy 2002, 32, 1520–1526. [Google Scholar] [CrossRef]
- Iwahana, H.; Yanagisawa, K.; Ito-Kosaka, A.; Kuroiwa, K.; Tago, K.; Komatsu, N.; Katashima, R.; Itakura, M.; Tominaga, S. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur. J. Biochem. 1999, 264, 397–406. [Google Scholar] [CrossRef]
- Leung, B.P.; Xu, D.; Culshaw, S.; McInnes, I.B.; Liew, F.Y. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J. Immunol. 2004, 173, 145–150. [Google Scholar] [CrossRef]
- Hayakawa, H.; Hayakawa, M.; Kume, A.; Tominaga, S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J. Biol. Chem. 2007, 282, 26369–26380. [Google Scholar] [CrossRef]
- Baba, Y.; Maeda, K.; Yashiro, T.; Inage, E.; Kasakura, K.; Suzuki, R.; Niyonsaba, F.; Hara, M.; Tanabe, A.; Ogawa, H.; et al. GATA2 Is a Critical Transactivator for the Human IL1RL1/ST2 Promoter in Mast Cells/Basophils OPPOSING ROLES FOR GATA2 and GATA1 IN HUMAN IL1RL1/ST2 GENE EXPRESSION. J. Biol. Chem. 2012, 287, 32689–32696. [Google Scholar] [CrossRef]
- Liu, X.; Hammel, M.; He, Y.; Tainer, J.A.; Jeng, U.-S.; Zhang, L.; Wang, S.; Wang, X. Structural insights into the interaction of IL-33 with its receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 14918–14923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, E.; van, L.P.; Samson, M.; Diem, S.; Barra, A.; Roga, S.; Gombert, J.M.; Schneider, E.; Dy, M.; Gourdy, P.; et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-γ production. Eur. J. Immunol. 2009, 39, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, W.V.; Fröhlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The alarmin interleukin-33 drives protective antiviral CD8+ T cell responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.-P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.-K.M.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010, 207, 651–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach, A.; Köhler, H.; Rappl, G.; Abken, H. Human CD4+ T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J. Immunol. 2006, 177, 5668–5675. [Google Scholar] [CrossRef]
- Komai-Koma, M.; Wang, E.; Kurowska-Stolarska, M.; Li, D.; McSharry, C.; Xu, D. Interleukin-33 promoting Th1 lymphocyte differentiation dependents on IL-12. Immunobiology 2016, 221, 412–417. [Google Scholar] [CrossRef]
- Peine, M.; Marek, R.M.; Löhning, M. IL-33 in T Cell Differentiation, Function, and Immune Homeostasis. Trends Immunol. 2016, 37, 321–333. [Google Scholar] [CrossRef]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Matta, B.M.; Lott, J.M.; Mathews, L.R.; Liu, Q.; Rosborough, B.R.; Blazar, B.R.; Turnquist, H.R. IL-33 Is an Unconventional Alarmin That Stimulates IL-2 Secretion by Dendritic Cells To Selectively Expand IL-33R/ST2+ Regulatory T Cells. J. Immunol. 2014, 193, 4010–4020. [Google Scholar] [CrossRef]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Frohlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Chen, L.; Souto, F.O.; Canasto-Chibuque, C.; Bongers, G.; Deshpande, M.; Harpaz, N.; Ko, H.M.; Kelley, K.; Furtado, G.C.; et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc (Min/+) mice. Sci. Rep. 2017, 7, 5520. [Google Scholar] [CrossRef] [PubMed]
- Meinicke, H.; Bremser, A.; Brack, M.; Akeus, P.; Pearson, C.; Bullers, S.; Hoffmeyer, K.; Stemmler, M.P.; Quiding-Jarbrink, M.; Izcue, A. Tumor-associated changes in intestinal epithelial cells cause local accumulation of KLRG1(+) GATA3(+) regulatory T cells in mice. Immunology 2017, 152, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ji, Y.; Wang, H.; Zhang, H.; Zhou, H. IL-33 Promotes the Development of Colorectal Cancer Through Inducing Tumor-Infiltrating ST2L(+) Regulatory T Cells in Mice. Technol. Cancer Res. Treat. 2018, 17, 1533033818780091. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, G.; Zhu, Y.; Liu, L.; Chen, E.; Turnquist, H.; Zhang, X.; Finn, O.J.; Chen, X.; Lu, B. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur. J. Immunol. 2011, 41, 3351–3360. [Google Scholar] [CrossRef]
- Gao, K.; Li, X.; Zhang, L.; Bai, L.; Dong, W.; Gao, K.; Shi, G.; Xia, X.; Wu, L.; Zhang, L. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett. 2013, 335, 463–471. [Google Scholar] [CrossRef]
- Lucarini, V.; Ziccheddu, G.; Macchia, I.; La Sorsa, V.; Peschiaroli, F.; Buccione, C.; Sistigu, A.; Sanchez, M.; Andreone, S.; D’Urso, M.T.; et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology 2017, 6, e1317420. [Google Scholar] [CrossRef]
- Jovanovic, I.; Radosavljevic, G.; Mitrovic, M.; Juranic, V.L.; McKenzie, A.N.J.; Arsenijevic, N.; Jonjic, S.; Lukic, M.L. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur. J. Immunol. 2011, 41, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682. [Google Scholar] [CrossRef]
- Wang, J.X.; Kaieda, S.; Ameri, S.; Fishgal, N.; Dwyer, D.; Dellinger, A.; Kepley, C.L.; Gurish, M.F.; Nigrovic, P.A. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proc. Natl. Acad. Sci. USA 2014, 111, 10281–10286. [Google Scholar] [CrossRef] [Green Version]
- Musolino, C.; Allegra, A.; Pioggia, G.; Gangemi, S. Immature myeloid-derived suppressor cells: A bridge between inflammation and cancer (Review). Oncol. Rep. 2017, 37, 671–683. [Google Scholar] [CrossRef]
- Xiao, P.; Wan, X.; Cui, B.; Liu, Y.; Qiu, C.; Rong, J.; Zheng, M.; Song, Y.; Chen, L.; He, J.; et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1063772. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Pecaric-Petkovic, T.; Didichenko, S.A.; Kaempfer, S.; Spiegl, N.; Dahinden, C.A. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood 2009, 113, 1526–1534. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Kim, S.; Karin, M. Role of TLR2-dependent inflammation in metastatic progression. Ann. N. Y. Acad. Sci. 2011, 1217, 191–206. [Google Scholar] [CrossRef]
- Wasmer, M.-H.; Krebs, P. The Role of IL-33-Dependent Inflammation in the Tumor Microenvironment. Front. Immunol. 2016, 7, 682. [Google Scholar] [CrossRef]
- Afferni, C.; Buccione, C.; Andreone, S.; Galdiero, M.R.; Varricchi, G.; Marone, G.; Mattei, F.; Schiavoni, G. The Pleiotropic Immunomodulatory Functions of IL-33 and Its Implications in Tumor Immunity. Front. Immunol. 2018, 9, 2601. [Google Scholar] [CrossRef]
- Lu, B.; Yang, M.; Wang, Q. Interleukin-33 in tumorigenesis, tumor immune evasion, and cancer immunotherapy. J. Mol. Med. 2016, 94, 535–543. [Google Scholar] [CrossRef]
- Casciaro, M.; Cardia, R.; Di Salvo, E.; Tuccari, G.; Ieni, A.; Gangemi, S. Interleukin-33 Involvement in Nonsmall Cell Lung Carcinomas: An Update. Biomolecules 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Feng, Y.; Yue, C.; Xu, B.; Chen, L.; Jiang, J.; Lu, B.; Zhu, Y. Lower expression level of IL-33 is associated with poor prognosis of pulmonary adenocarcinoma. PLoS ONE 2018, 13, e0193428. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, E.; Heo, J.S.; Bae, D.J.; Lee, J.U.; Lee, T.H.; Lee, H.J.; Chang, H.S.; Park, J.S.; Jang, A.S.; et al. Circulating IL-33 level is associated with the progression of lung cancer. Lung Cancer 2015, 90, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Hung, J.-Y.; Lee, Y.-L.; Chen, F.-W.; Chang, K.-F.; Chang, W.-A.; Tsai, Y.-M.; Chong, I.-W.; Kuo, P.-L. Identification of novel gene expression signature in lung adenocarcinoma by using next-generation sequencing data and bioinformatics analysis. Oncotarget 2017, 8, 104831–104854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, S.M.; Rubner, C.; Kesselring, R.; Martin, M.; Griesshammer, E.; Ruemmele, P.; Stempfl, T.; Teufel, A.; Schlitt, H.J.; Fichtner-Feigl, S. Tumor-infiltrating, interleukin-33-producing effector-memory CD8(+) T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology 2015, 61, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Minaee, K.; Farsinejad, A.R.; Nemati, M.; Khosravimashizi, A.; Daneshvar, H.; Mohammadi, M.M.; Sheikhi, A.; Ghaderi, A. Evaluation of the circulating levels of IL-12 and IL-33 in patients with breast cancer: Influences of the tumor stages and cytokine gene polymorphisms. Iran J. Basic Med. Sci. 2015, 18, 1189–1198. [Google Scholar]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Mager, L.F.; Riether, C.; Schurch, C.M.; Banz, Y.; Wasmer, M.H.; Stuber, R.; Theocharides, A.P.; Li, X.; Xia, Y.; Saito, H.; et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J. Clin. Investig. 2015, 125, 2579–2591. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, S.; Allegra, A.; Profita, M.; Saitta, S.; Gerace, D.; Bonanno, A.; Alonci, A.; Petrungaro, A.; Russo, S.; Musolino, C. Decreased plasma levels of IL-33 could contribute to the altered function of Th2 lymphocytes in patients with polycythemia vera and essential thrombocythemia. Cancer Investig. 2013, 31, 212–213. [Google Scholar] [CrossRef]
- Arima, H.; Nishikori, M.; Otsuka, Y.; Kishimoto, W.; Izumi, K.; Yasuda, K.; Yoshimoto, T.; Takaori-Kondo, A. B cells with aberrant activation of Notch1 signaling promote Treg and Th2 cell-dominant T-cell responses via IL-33. Blood Adv. 2018, 2, 2282–2295. [Google Scholar] [CrossRef]
- Kabelitz, D.; Wesch, D.; Pitters, E.; Zoller, M. Characterization of tumor reactivity of human V gamma 9V delta 2 gamma delta T cells in vitro and in SCID mice in vivo. J. Immunol. 2004, 173, 6767–6776. [Google Scholar] [CrossRef]
- Duault, C.; Betous, D.; Bezombes, C.; Roga, S.; Cayrol, C.; Girard, J.P.; Fournie, J.J.; Poupot, M. IL-33-expanded human Vgamma9Vdelta2 T cells have anti-lymphoma effect in a mouse tumor model. Eur. J. Immunol. 2017, 47, 2137–2141. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, S.; Zha, X.; Yang, L.; Li, B.; Yu, Z.; Wang, L.; Li, Y. Expression feature of CD3, FcepsilonRIgamma, and Zap-70 in patients with chronic lymphocytic leukemia. Hematology 2012, 17, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, X.; Yang, Q.; Zhao, X.; Wen, W.; Li, G.; Lu, J.; Qin, W.; Qi, Y.; Xie, F.; et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J. Immunol. 2015, 194, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Dominguez, D.; Chen, S.; Fan, J.; Long, A.; Zhang, M.; Fang, D.; Zhang, Y.; Kuzel, T.M.; Zhang, B. Exogenous IL-33 overcomes T cell tolerance in murine acute myeloid leukemia. Oncotarget 2016, 7, 61069–61080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducassou, S.; Prouzet-Mauleon, V.; Deau, M.C.; Brunet de la Grange, P.; Cardinaud, B.; Soueidan, H.; Quelen, C.; Brousset, P.; Pasquet, J.M.; Moreau-Gaudry, F.; et al. MYB-GATA1 fusion promotes basophilic leukemia: Involvement of interleukin-33 and nerve growth factor receptors. J. Pathol. 2017, 242, 347–357. [Google Scholar] [CrossRef]
- Levescot, A.; Flamant, S.; Basbous, S.; Jacomet, F.; Féraud, O.; Anne Bourgeois, E.; Bonnet, M.-L.; Giraud, C.; Roy, L.; Barra, A.; et al. BCR-ABL–Induced Deregulation of the IL-33/ST2 Pathway in CD34(+) Progenitors from Chronic Myeloid Leukemia Patients. Cancer Res. 2014, 74, 2669–2676. [Google Scholar] [CrossRef]
- Tare, N.; Li, H.; Morschauser, A.; Cote-Sierra, J.; Ju, G.; Renzetti, L.; Lin, T.A. KU812 cells provide a novel in vitro model of the human IL-33/ST2L axis: Functional responses and identification of signaling pathways. Exp. Cell Res. 2010, 316, 2527–2537. [Google Scholar] [CrossRef]
- Saleh, H.; Eeles, D.; Hodge, J.M.; Nicholson, G.C.; Gu, R.; Pompolo, S.; Gillespie, M.T.; Quinn, J.M. Interleukin-33, a target of parathyroid hormone and oncostatin m, increases osteoblastic matrix mineral deposition and inhibits osteoclast formation in vitro. Endocrinology 2011, 152, 1911–1922. [Google Scholar] [CrossRef]
- Schulze, J.; Bickert, T.; Beil, F.T.; Zaiss, M.M.; Albers, J.; Wintges, K.; Streichert, T.; Klaetschke, K.; Keller, J.; Hissnauer, T.N.; et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J. Bone Miner. Res. 2011, 26, 704–717. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, Y.; Jiang, Y.; Qin, T.; Chen, J.; Chu, X.; Yi, Q.; Gao, S.; Wang, S. Dectin-1 signaling inhibits osteoclastogenesis via IL-33-induced inhibition of NFATc1. Oncotarget 2017, 8, 53366–53374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Richter, L.; Becker, M.; Amador, C.; Hyde, R.K. IL1RL1 is dynamically expressed on Cbfb-MYH11(+) leukemia stem cells and promotes cell survival. Sci. Rep. 2019, 9, 1729. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Mannucci, C.; Russo, S.; Alonci, A.; Maisano, V.; Calapai, G.; Gangemi, S. Possible Role of Interleukin-31/33 Axis in Imatinib Mesylate-Associated Skin Toxicity. Turk. J. Haematol. 2015, 32, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.; Corrales, L.; Kline, J. Targeting the innate immune system as immunotherapy for acute myeloid leukemia. Front. Oncol. 2015, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- Staveley-O’Carroll, K.; Sotomayor, E.; Montgomery, J.; Borrello, I.; Hwang, L.; Fein, S.; Pardoll, D.; Levitsky, H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA 1998, 95, 1178–1183. [Google Scholar] [CrossRef] [Green Version]
- Sotomayor, E.M.; Borrello, I.; Rattis, F.M.; Cuenca, A.G.; Abrams, J.; Staveley-O’Carroll, K.; Levitsky, H.I. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood 2001, 98, 1070–1077. [Google Scholar] [CrossRef]
- Chen, X.; Kline, D.E.; Kline, J. Peripheral T-cell tolerance in hosts with acute myeloid leukemia. Oncoimmunology 2013, 2, e25445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, X.; Liu, X.; Kline, D.E.; Teague, R.M.; Gajewski, T.F.; Kline, J. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J. Clin. Investig. 2013, 123, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Cassel, S.L.; Joly, S.; Sutterwala, F.S. The NLRP3 inflammasome: A sensor of immune danger signals. Semin. Immunol. 2009, 21, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jeno, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef]
- Hebbeler, A.M.; Cairo, C.; Cummings, J.S.; Pauza, C.D. Individual Vgamma2-Jgamma1.2+ T cells respond to both isopentenyl pyrophosphate and Daudi cell stimulation: Generating tumor effectors with low molecular weight phosphoantigens. Cancer Immunol. Immunother. 2007, 56, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Musolino, C.; Allegra, A.; Profita, M.; Alonci, A.; Saitta, S.; Bonanno, A.; Gerace, D.; Calabro, L.; Gangemi, S. Reduction in IL-33 plasma levels might be involved in T cell dysregulation in chronic lymphocytic leukemia. Acta Hematol. 2014, 131, 165–166. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Dmoszynska, A.; Rolinski, J.; Wasik, E. T type 1/type 2 subsets balance in B-cell chronic lymphocytic leukemia—the three-color flow cytometry analysis. Leuk. Res. 2002, 26, 657–660. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Profita, M.; Alonci, A.; Saitta, S.; Russo, S.; Bonanno, A.; Innao, V.; Gangemi, S. Reduced IL-33 plasma levels in multiple myeloma patients are associated with more advanced stage of disease. Br. J. Haematol. 2013, 160, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hong, S.; Wezeman, M.; Qian, J.; Yang, J.; Yi, Q. Dendritic cell vaccine but not idiotype-KLH protein vaccine primes therapeutic tumor-specific immunity against multiple myeloma. Front. Biosci. 2007, 12, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Peña, M.M.O. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Chen, L.J.; Zheng, X.; Shen, Y.P.; Zhu, Y.B.; Li, Q.; Chen, J.; Xia, R.; Zhou, S.M.; Wu, C.P.; Zhang, X.G.; et al. Higher numbers of T-bet(+) intratumoral lymphoid cells correlate with better survival in gastric cancer. Cancer Immunol. Immunother. 2013, 62, 553–561. [Google Scholar] [CrossRef]
- Shen, J.X.; Liu, J.; Zhang, G.J. Interleukin-33 in Malignancies: Friends or Foes? Front. Immunol. 2018, 9, 3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, C.; O’Grady, K.; Lavelle, E.C.; Fallon, P.G. Interleukin 33: An innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur. J. Immunol. 2016, 46, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegra, A.; Penna, G.; Innao, V.; Greve, B.; Maisano, V.; Russo, S.; Musolino, C. Vaccination of multiple myeloma: Current strategies and future prospects. Crit. Rev. Oncol. Hematol. 2015, 96, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Russo, S.; Gerace, D.; Calabro, L.; Maisano, V.; Innao, V.; Musolino, C. Vaccination strategies in lymphoproliferative disorders: Failures and successes. Leuk. Res. 2015, 39, 1006–1019. [Google Scholar] [CrossRef]
- Villarreal, D.O.; Wise, M.C.; Walters, J.N.; Reuschel, E.L.; Choi, M.J.; Obeng-Adjei, N.; Yan, J.; Morrow, M.P.; Weiner, D.B. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res. 2014, 74, 1789–1800. [Google Scholar] [CrossRef]
- Kojima, R.; Scheller, L.; Fussenegger, M. Nonimmune cells equipped with T-cell-receptor-like signaling for cancer cell ablation. Nat. Chem. Biol. 2018, 14, 42–49. [Google Scholar] [CrossRef]
- Ramadan, A.M.; Daguindau, E.; Rech, J.C.; Chinnaswamy, K.; Zhang, J.; Hura, G.L.; Griesenauer, B.; Bolten, Z.; Robida, A.; Larsen, M.; et al. From proteomics to discovery of first-in-class ST2 inhibitors active in vivo. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
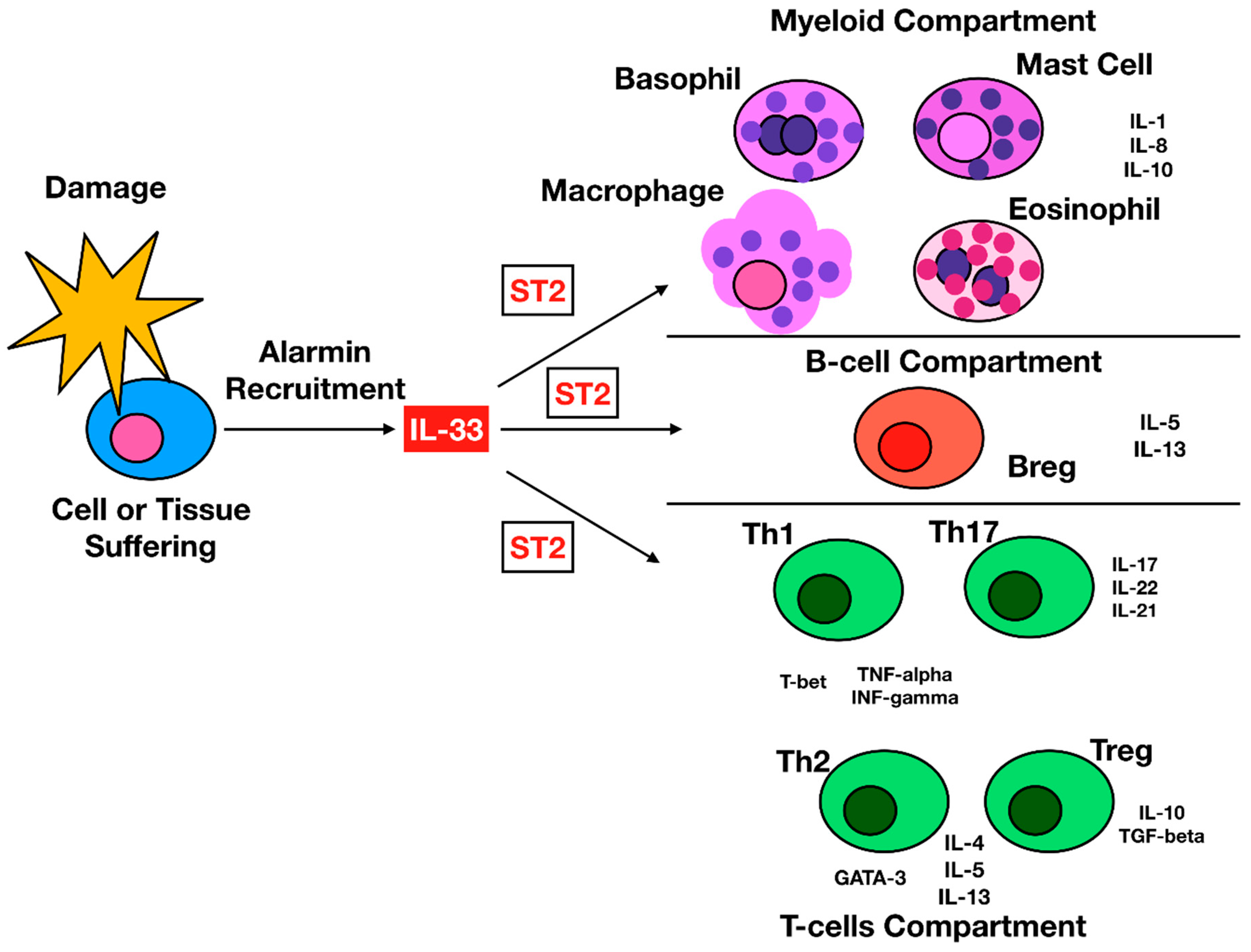




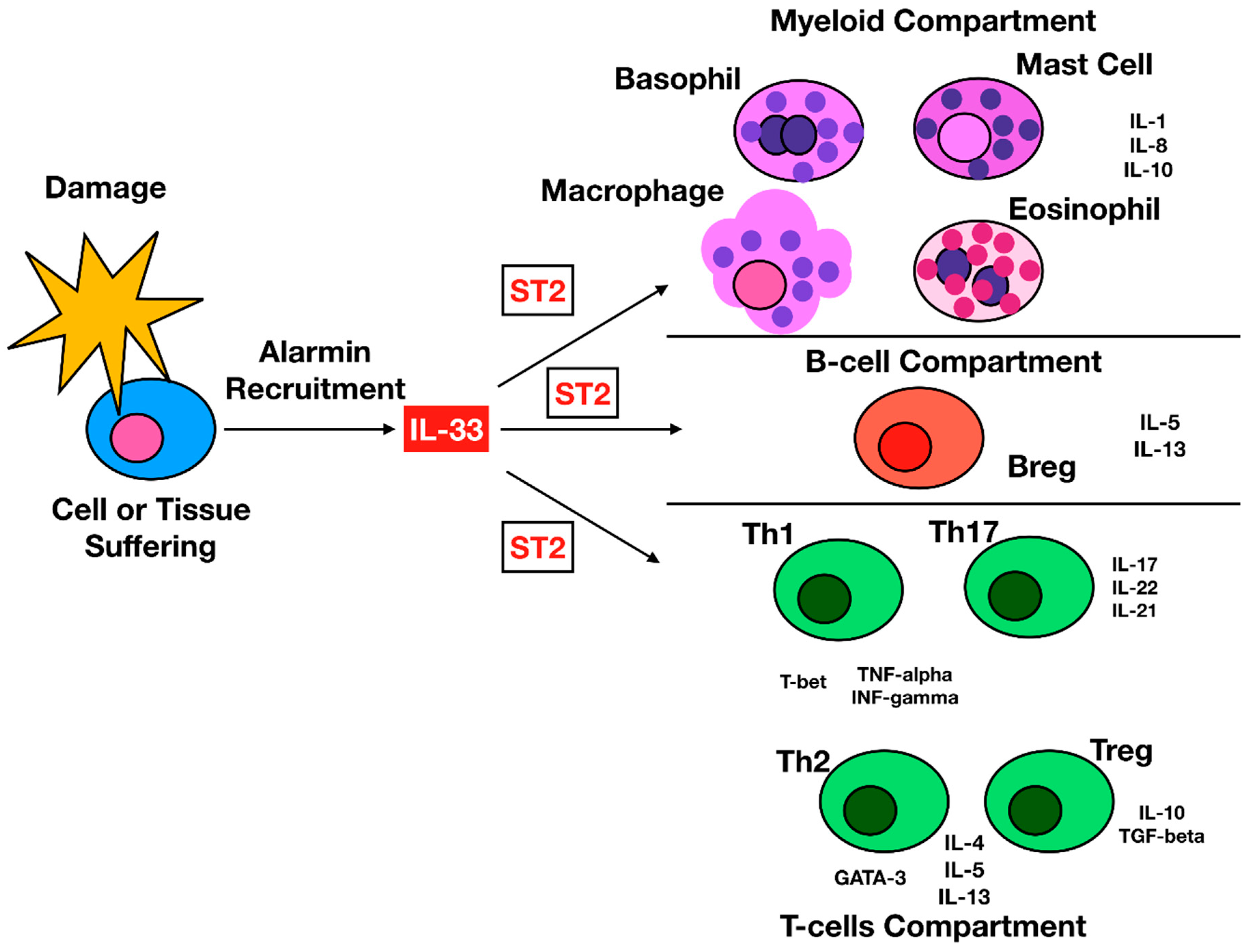{kind=link}
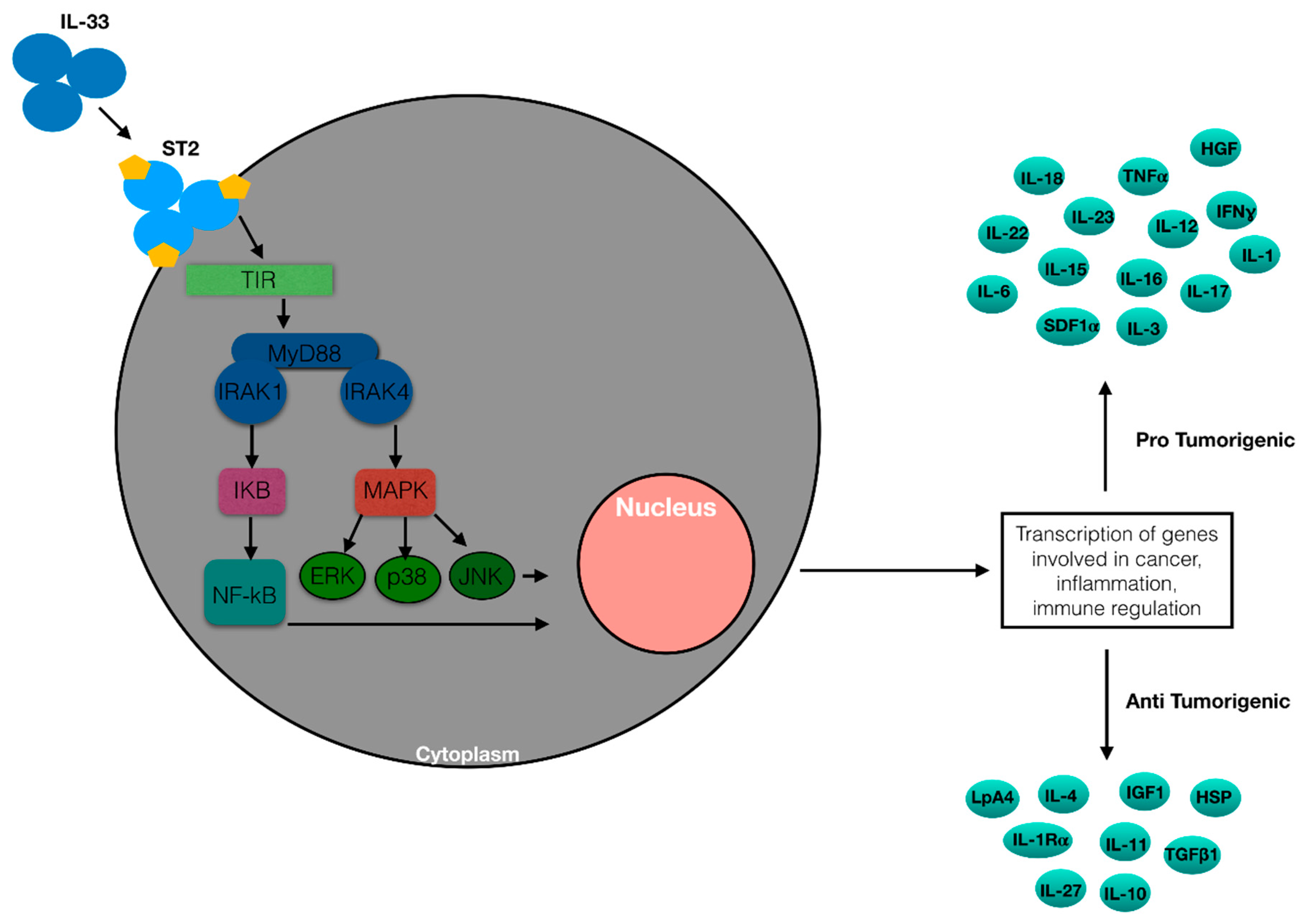{kind=link}
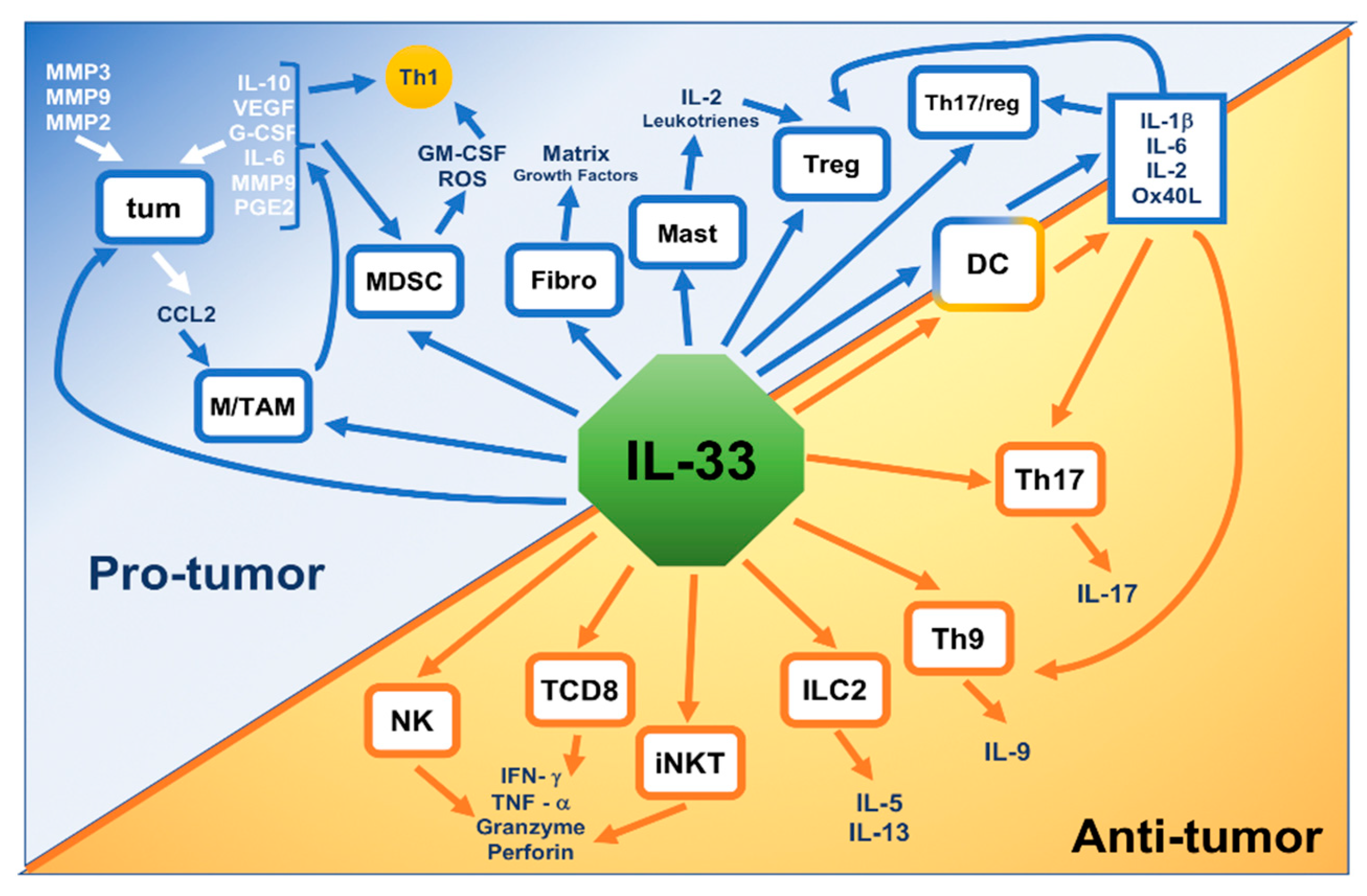{kind=link}
{kind=link}
| Disease | Model Used | Pro/Anti |
|---|---|---|
| BCR-ABL1-negative myeloproliferative neoplasms neoplasms [68,69] | Cell lines Animal models | pro |
| Lymphomas [70,71,72,73] | Cell lines Mouse model Patients tissues | pro/anti |
| Acute myeloid leukemia [52,74,75,76] | Cell lines Animal models | anti |
| Chronic myeloid leukemia [68,77,78] | Cell lines Animal models | pro |
| MM bone disease [79,80,81] | Cell lines Patients tissues | anti |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Innao, V.; Tartarisco, G.; Pioggia, G.; Casciaro, M.; Musolino, C.; Gangemi, S. The ST2/Interleukin-33 Axis in Hematologic Malignancies: The IL-33 Paradox. Int. J. Mol. Sci. 2019, 20, 5226. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205226
Allegra A, Innao V, Tartarisco G, Pioggia G, Casciaro M, Musolino C, Gangemi S. The ST2/Interleukin-33 Axis in Hematologic Malignancies: The IL-33 Paradox. International Journal of Molecular Sciences. 2019; 20(20):5226. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205226
Chicago/Turabian StyleAllegra, Alessandro, Vanessa Innao, Gennaro Tartarisco, Giovanni Pioggia, Marco Casciaro, Caterina Musolino, and Sebastiano Gangemi. 2019. "The ST2/Interleukin-33 Axis in Hematologic Malignancies: The IL-33 Paradox" International Journal of Molecular Sciences 20, no. 20: 5226. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205226