Twin CHCH Proteins, CHCHD2, and CHCHD10: Key Molecules of Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetic and Pathological Overlaps between ALS and FTD, but not between ALS–FTD and PD
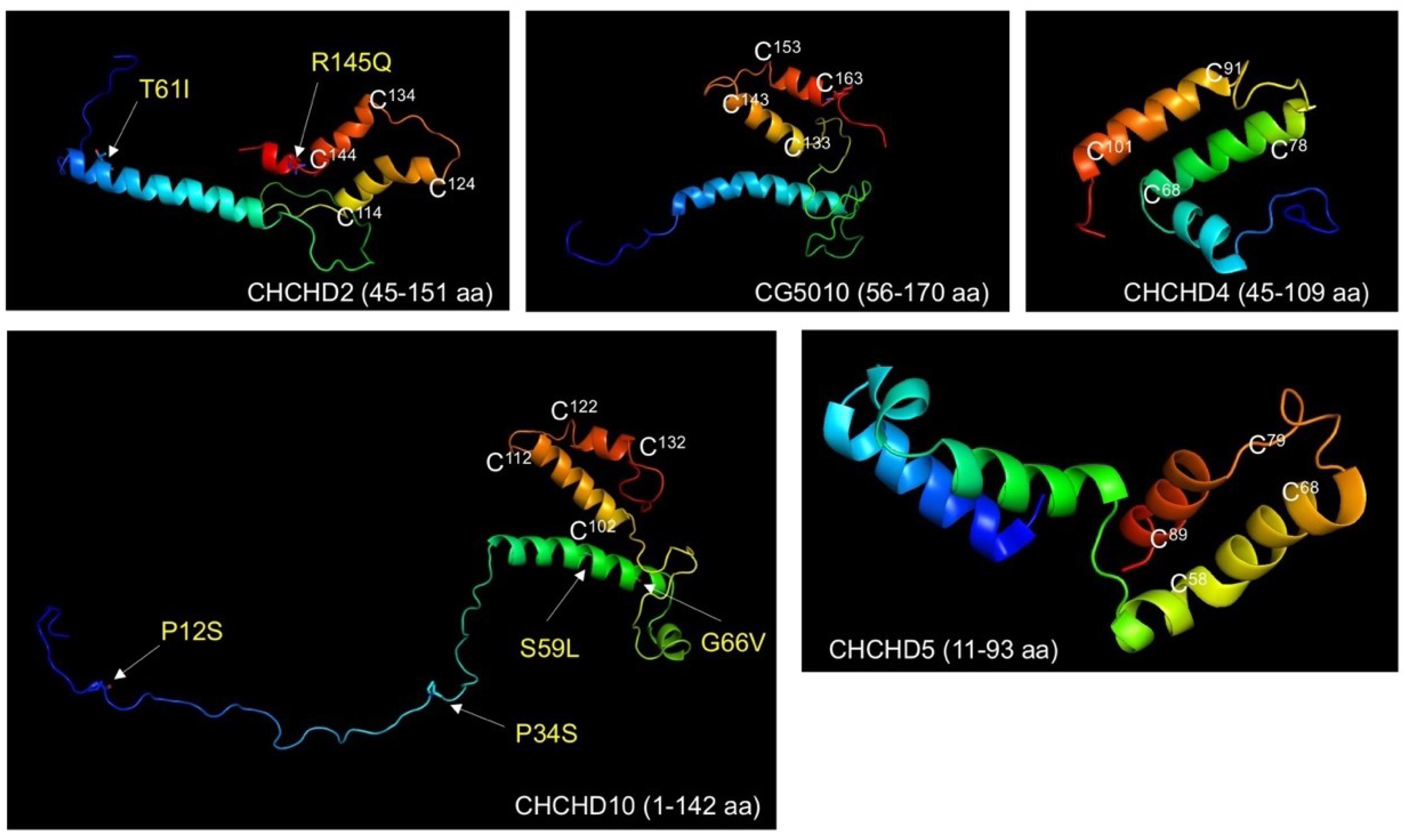
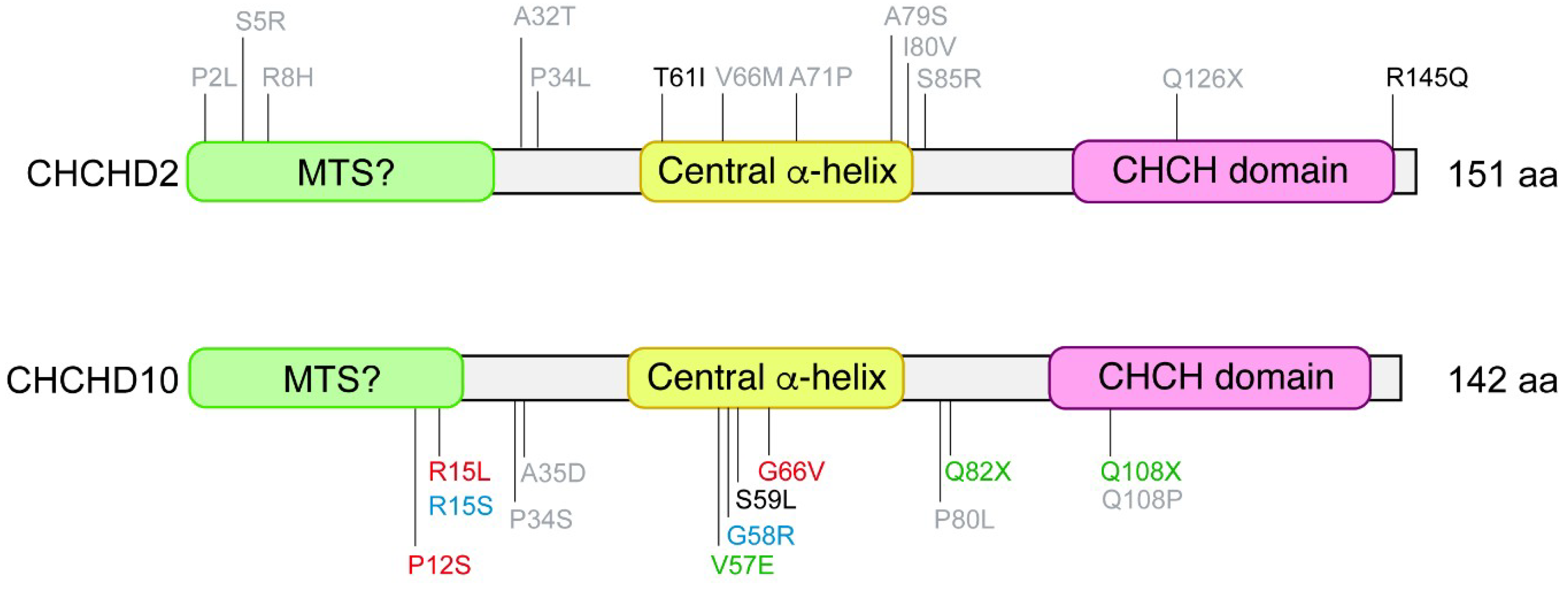
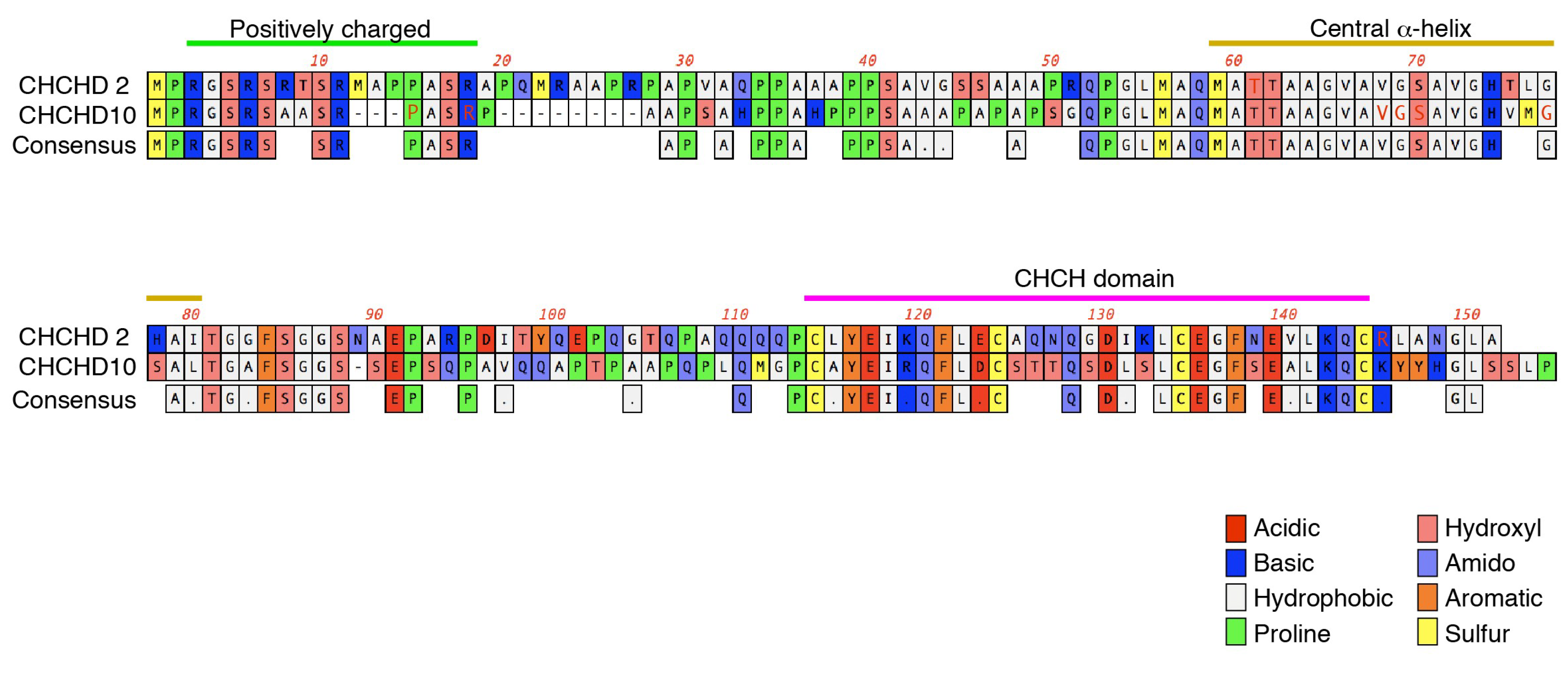
3. CHCHD2 Mutations Linked to PD
4. CHCHD10 Mutations Linked to ALS-FTD
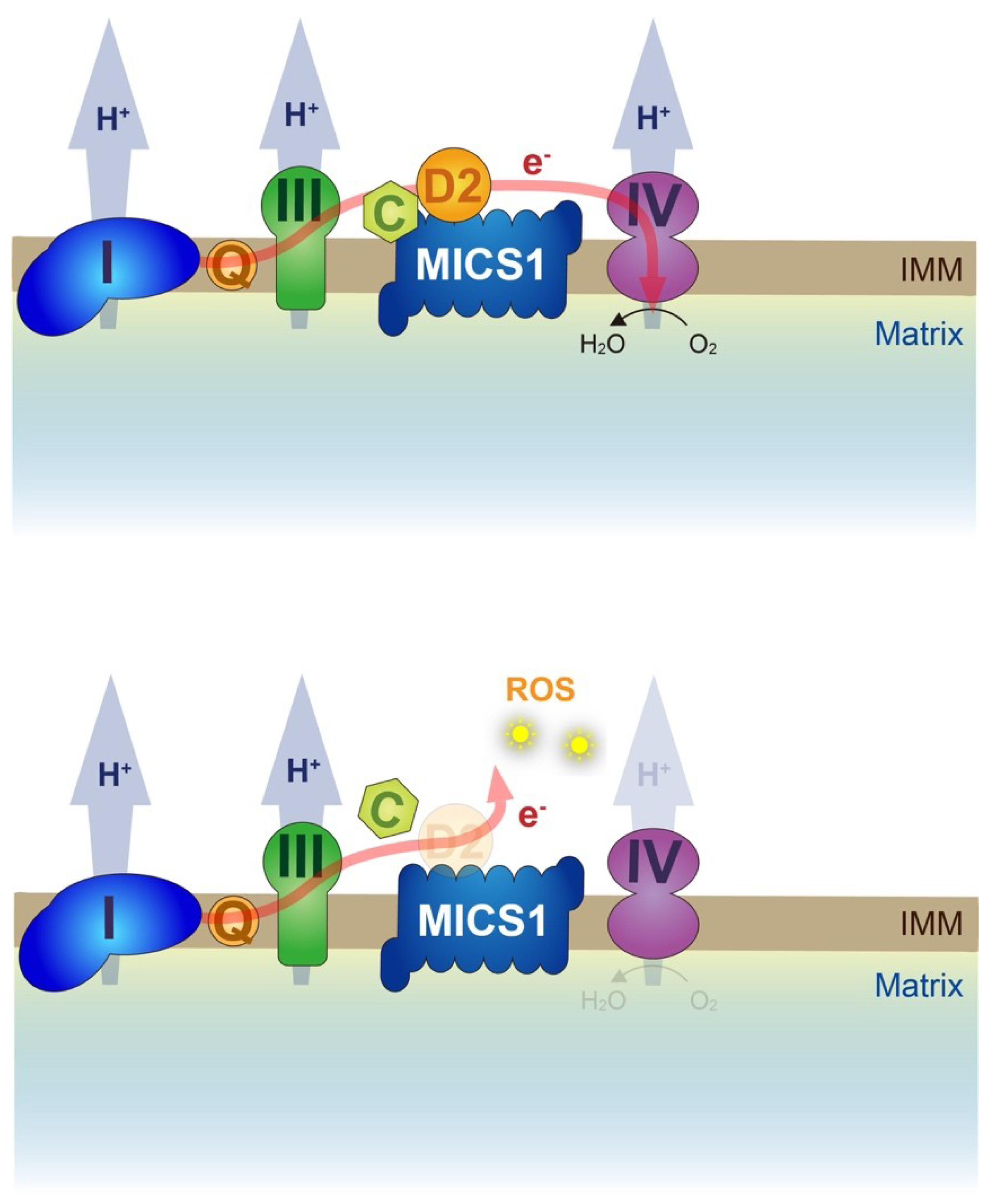
5. Twin Proteins, CHCHD2, and CHCHD10 Regulate OXPHOS
6. Submitochondrial Localization of Twin Proteins
7. Expressional Regulation and Tissue Expression of Twin Proteins
8. Binding Partners of CHCHD2 and CHCHD10
9. Pathogenesis caused by Twin CHCH Proteins
10. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| CHCH | Coiled-coil-helix-coiled-coil-helix |
| CMT2 | Charcot–Marie–Tooth disease type 2 |
| COX | Cytochrome c oxidase |
| DLB | Dementia with Lewy bodies |
| FTD | Frontotemporal lobe dementia |
| IMS | Mitochondrial intermembrane space |
| Mia40 | Mitochondrial intermembrane space import and assembly protein 40 kDa |
| MTS | Mitochondrial targeting sequence |
| MICOS | Mitochondrial contact site complex |
| OXPHOS | Oxidative phosphorylation |
| PD | Parkinson’s disease |
| ROS | Redox oxygen species |
| SMAJ | Jokela type spinal muscular atrophy |
| TDP-43 | TAR DNA binding protein of 43 kDa |
| ∆ψm | Mitochondrial membrane potential |
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef]
- Fischer, M.; Horn, S.; Belkacemi, A.; Kojer, K.; Petrungaro, C.; Habich, M.; Ali, M.; Kuttner, V.; Bien, M.; Kauff, F.; et al. Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol. Biol. Cell 2013, 24, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet. Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [Green Version]
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Ringholz, G.M.; Appel, S.H.; Bradshaw, M.; Cooke, N.A.; Mosnik, D.M.; Schulz, P.E. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005, 65, 586–590. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C. Clinical phenomenology and neuroimaging correlates in ALS-FTD. J. Mol. Neurosci. 2011, 45, 656–662. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138, 54–70. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef]
- Puschmann, A. New Genes Causing Hereditary Parkinson’s Disease or Parkinsonism. Curr. Neurol. Neurosci. Rep. 2017, 17, 66. [Google Scholar] [CrossRef]
- Yang, N.; Zhao, Y.; Liu, Z.; Zhang, R.; He, Y.; Zhou, Y.; Xu, Q.; Sun, Q.; Yan, X.; Guo, J.; et al. Systematically analyzing rare variants of autosomal-dominant genes for sporadic Parkinson’s disease in a Chinese cohort. Neurobiol. Aging 2018. Available online 20 November 2018. [Google Scholar] [CrossRef]
- Inoshita, T.; Cui, C.; Hattori, N.; Imai, Y. Regulation of membrane dynamics by Parkinson’s disease-associated genes. J. Genet. 2018, 97, 715–725. [Google Scholar] [CrossRef]
- Karch, C.M.; Wen, N.; Fan, C.C.; Yokoyama, J.S.; Kouri, N.; Ross, O.A.; Hoglinger, G.; Muller, U.; Ferrari, R.; Hardy, J.; et al. Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum. JAMA Neurol. 2018, 75, 860–875. [Google Scholar] [CrossRef]
- Jansen, I.E.; Bras, J.M.; Lesage, S.; Schulte, C.; Gibbs, J.R.; Nalls, M.A.; Brice, A.; Wood, N.W.; Morris, H.; Hardy, J.A.; et al. CHCHD2 and Parkinson’s disease. Lancet Neurol. 2015, 14, 678–679. [Google Scholar] [CrossRef]
- Muller, K.; Andersen, P.M.; Hubers, A.; Marroquin, N.; Volk, A.E.; Danzer, K.M.; Meitinger, T.; Ludolph, A.C.; Strom, T.M.; Weishaupt, J.H. Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 2014, 137, e309. [Google Scholar] [CrossRef]
- Chaussenot, A.; Le Ber, I.; Ait-El-Mkadem, S.; Camuzat, A.; de Septenville, A.; Bannwarth, S.; Genin, E.C.; Serre, V.; Auge, G.; French research network on, F.T.D.; et al. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 2014, 35, 2884 e2881–2884 e2884. [Google Scholar] [CrossRef]
- Dols-Icardo, O.; Nebot, I.; Gorostidi, A.; Ortega-Cubero, S.; Hernandez, I.; Rojas-Garcia, R.; Garcia-Redondo, A.; Povedano, M.; Llado, A.; Alvarez, V.; et al. Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 2015, 138, e400. [Google Scholar] [CrossRef]
- Kurzwelly, D.; Kruger, S.; Biskup, S.; Heneka, M.T. A distinct clinical phenotype in a German kindred with motor neuron disease carrying a CHCHD10 mutation. Brain 2015, 138, e376. [Google Scholar] [CrossRef]
- Perrone, F.; Nguyen, H.P.; Van Mossevelde, S.; Moisse, M.; Sieben, A.; Santens, P.; De Bleecker, J.; Vandenbulcke, M.; Engelborghs, S.; Baets, J.; et al. Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging 2017, 51, 177.e9–177.e16. [Google Scholar] [CrossRef]
- Auranen, M.; Ylikallio, E.; Shcherbii, M.; Paetau, A.; Kiuru-Enari, S.; Toppila, J.P.; Tyynismaa, H. CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol. Genet. 2015, 1, e1. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Poyhonen, M.; Kiuru-Enari, S.; Tienari, P.J.; Laaksovirta, H.; Toppila, J.; Ylikallio, E.; Tyynismaa, H.; Auranen, M. Intrafamilial clinical variability in individuals carrying the CHCHD10 mutation Gly66Val. Acta Neurol. Scand. 2016, 133, 361–366. [Google Scholar] [CrossRef]
- Penttila, S.; Jokela, M.; Saukkonen, A.M.; Toivanen, J.; Palmio, J.; Lahdesmaki, J.; Sandell, S.; Shcherbii, M.; Auranen, M.; Ylikallio, E.; et al. CHCHD10 mutations and motor neuron disease: The distribution in Finnish patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 272–277. [Google Scholar] [CrossRef]
- Ajroud-Driss, S.; Fecto, F.; Ajroud, K.; Lalani, I.; Calvo, S.E.; Mootha, V.K.; Deng, H.X.; Siddique, N.; Tahmoush, A.J.; Heiman-Patterson, T.D.; et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015, 16, 1–9. [Google Scholar] [CrossRef]
- Shi, C.H.; Mao, C.Y.; Zhang, S.Y.; Yang, J.; Song, B.; Wu, P.; Zuo, C.T.; Liu, Y.T.; Ji, Y.; Yang, Z.H.; et al. CHCHD2 gene mutations in familial and sporadic Parkinson’s disease. Neurobiol. Aging 2016, 38, 217.e9–217.e13. [Google Scholar] [CrossRef]
- Mao, C.Y.; Wu, P.; Zhang, S.Y.; Yang, J.; Liu, Y.T.; Zuo, C.T.; Zhuang, Z.P.; Shi, C.H.; Xu, Y.M. Brain glucose metabolism changes in Parkinson’s disease patients with CHCHD2 mutation based on F-18-FDG PET imaging. J. Neurol. Sci. 2016, 369, 303–305. [Google Scholar] [CrossRef]
- Ogaki, K.; Koga, S.; Heckman, M.G.; Fiesel, F.C.; Ando, M.; Labbe, C.; Lorenzo-Betancor, O.; Moussaud-Lamodiere, E.L.; Soto-Ortolaza, A.I.; Walton, R.L.; et al. Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders. Neurology 2015, 85, 2016–2025. [Google Scholar] [CrossRef]
- Puschmann, A.; Dickson, D.W.; Englund, E.; Wszolek, Z.K.; Ross, O.A. CHCHD2 and Parkinson's disease. Lancet. Neurol. 2015, 14, 679. [Google Scholar] [CrossRef] [Green Version]
- Koschmidder, E.; Weissbach, A.; Bruggemann, N.; Kasten, M.; Klein, C.; Lohmann, K. A nonsense mutation in CHCHD2 in a patient with Parkinson disease. Neurology 2016, 86, 577–579. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, Q.; An, R.; Zheng, J.; Tian, S.; Chen, Y.; Xu, Y. Mutational scanning of the CHCHD2 gene in Han Chinese patients with Parkinson’s disease and meta-analysis of the literature. Parkinsonism Relat. Disord. 2016, 29, 42–46. [Google Scholar] [CrossRef]
- Ikeda, A.; Matsushima, T.; Daida, K.; Nakajima, S.; Conedera, S.; Li, Y.; Yoshino, H.; Oyama, G.; Funayama, M.; Nishioka, K.; et al. A novel mutation of CHCHD2 p.R8H in a sporadic case of Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 34, 66–68. [Google Scholar] [CrossRef]
- Lee, R.G.; Sedghi, M.; Salari, M.; Shearwood, A.J.; Stentenbach, M.; Kariminejad, A.; Goullee, H.; Rackham, O.; Laing, N.G.; Tajsharghi, H.; et al. Early-onset Parkinson disease caused by a mutation in CHCHD2 and mitochondrial dysfunction. Neurol. Genet. 2018, 4, e276. [Google Scholar] [CrossRef]
- Liu, X.; Jiao, B.; Zhang, W.; Xiao, T.; Hou, L.; Pan, C.; Tang, B.; Shen, L. Identification of CHCHD2 mutations in patients with Alzheimer’s disease, amyotrophic lateral sclerosis and frontotemporal dementia in China. Mol. Med. Rep. 2018, 18, 461–466. [Google Scholar] [CrossRef]
- Che, X.Q.; Zhao, Q.H.; Huang, Y.; Li, X.; Ren, R.J.; Chen, S.D.; Guo, Q.H.; Wang, G. Mutation Screening of the CHCHD2 Gene for Alzheimer’s Disease and Frontotemporal Dementia in Chinese Mainland Population. J. Alzheimers Dis. 2018, 61, 1283–1288. [Google Scholar] [CrossRef]
- Nicoletti, G.; Gagliardi, M.; Procopio, R.; Iannello, G.; Morelli, M.; Annesi, G.; Quattrone, A. A new CHCHD2 mutation identified in a southern italy patient with multiple system atrophy. Parkinsonism Relat. Disord. 2018, 47, 91–93. [Google Scholar] [CrossRef]
- Johnson, J.O.; Glynn, S.M.; Gibbs, J.R.; Nalls, M.A.; Sabatelli, M.; Restagno, G.; Drory, V.E.; Chio, A.; Rogaeva, E.; Traynor, B.J. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 2014, 137, e311. [Google Scholar] [CrossRef]
- Ronchi, D.; Riboldi, G.; Del Bo, R.; Ticozzi, N.; Scarlato, M.; Galimberti, D.; Corti, S.; Silani, V.; Bresolin, N.; Comi, G.P. CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 2015, 138, e372. [Google Scholar] [CrossRef]
- Chio, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; Borghero, G.; et al. CHCH10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 2015, 36, 1767 e1763–1767 e1766. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, Z.; Zinman, L.; Bruni, A.C.; Maletta, R.G.; Curcio, S.A.; Rainero, I.; Rubino, E.; Pinessi, L.; Nacmias, B.; et al. Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 2015, 138, e380. [Google Scholar] [CrossRef]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghanel, M.; Exner, N.; Riemenschneider, H.; van der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef]
- Xiao, T.; Jiao, B.; Zhang, W.; Pan, C.; Wei, J.; Liu, X.; Zhou, Y.; Zhou, L.; Tang, B.; Shen, L. Identification of CHCHD10 Mutation in Chinese Patients with Alzheimer Disease. Mol. Neurobiol. 2017, 54, 5243–5247. [Google Scholar] [CrossRef]
- Longen, S.; Bien, M.; Bihlmaier, K.; Kloeppel, C.; Kauff, F.; Hammermeister, M.; Westermann, B.; Herrmann, J.M.; Riemer, J. Systematic analysis of the twin cx(9)c protein family. J. Mol. Biol. 2009, 393, 356–368. [Google Scholar] [CrossRef]
- Baughman, J.M.; Nilsson, R.; Gohil, V.M.; Arlow, D.H.; Gauhar, Z.; Mootha, V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009, 5, e1000590. [Google Scholar] [CrossRef]
- Martherus, R.S.; Sluiter, W.; Timmer, E.D.; VanHerle, S.J.; Smeets, H.J.; Ayoubi, T.A. Functional annotation of heart enriched mitochondrial genes GBAS and CHCHD10 through guilt by association. Biochem. Biophys. Res. Commun. 2010, 402, 203–208. [Google Scholar] [CrossRef]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef] [Green Version]
- Burstein, S.R.; Valsecchi, F.; Kawamata, H.; Bourens, M.; Zeng, R.; Zuberi, A.; Milner, T.A.; Cloonan, S.M.; Lutz, C.; Barrientos, A.; et al. In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Human Mol. Genet. 2018, 27, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef] [PubMed]
- Darshi, M.; Trinh, K.N.; Murphy, A.N.; Taylor, S.S. Targeting and import mechanism of coiled-coil helix coiled-coil helix domain-containing protein 3 (ChChd3) into the mitochondrial intermembrane space. J. Biol. Chem. 2012, 287, 39480–39491. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.C.; Bannwarth, S.; Lespinasse, F.; Ortega-Vila, B.; Fragaki, K.; Itoh, K.; Villa, E.; Lacas-Gervais, S.; Jokela, M.; Auranen, M.; et al. Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol. Dis. 2018, 119, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Straub, I.R.; Janer, A.; Weraarpachai, W.; Zinman, L.; Robertson, J.; Rogaeva, E.; Shoubridge, E.A. Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Human Mol. Genet. 2018, 27, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, B.P.; Nguyen, D.; Liu, Y.T.; Marani, M.; Hench, J.; Benit, P.; Kozjak-Pavlovic, V.; Rustin, P.; Frank, S.; et al. CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Human Mol. Genet. 2018, 27, 3881–3900. [Google Scholar] [CrossRef]
- Zhou, W.; Ma, D.; Sun, A.X.; Tran, H.D.; Ma, D.L.; Singh, B.K.; Zhou, J.; Zhang, J.; Wang, D.; Zhao, Y.; et al. PD-linked CHCHD2 mutations impair CHCHD10 and MICOS complex leading to mitochondria dysfunction. Human Mol. Genet. in press. [CrossRef]
- Zetterstrom, R.H.; Solomin, L.; Jansson, L.; Hoffer, B.J.; Olson, L.; Perlmann, T. Dopamine neuron agenesis in Nurr1-deficient mice. Science 1997, 276, 248–250. [Google Scholar] [CrossRef]
- Kadkhodaei, B.; Ito, T.; Joodmardi, E.; Mattsson, B.; Rouillard, C.; Carta, M.; Muramatsu, S.; Sumi-Ichinose, C.; Nomura, T.; Metzger, D.; et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci. 2009, 29, 15923–15932. [Google Scholar] [CrossRef]
- Kadkhodaei, B.; Alvarsson, A.; Schintu, N.; Ramskold, D.; Volakakis, N.; Joodmardi, E.; Yoshitake, T.; Kehr, J.; Decressac, M.; Bjorklund, A.; et al. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 2360–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Gomez-Duran, A.; Saretzki, G.; Jin, S.; Tilgner, K.; Melguizo-Sanchis, D.; Anyfantis, G.; Al-Aama, J.; Vallier, L.; Chinnery, P.; et al. The mitochondrial protein CHCHD2 primes the differentiation potential of human induced pluripotent stem cells to neuroectodermal lineages. J. Cell Biol. 2016, 215, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Nayak, R.R.; Kearns, M.; Spielman, R.S.; Cheung, V.G. Coexpression network based on natural variation in human gene expression reveals gene interactions and functions. Genome Res. 2009, 19, 1953–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Huttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Huttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Purandare, N.; Somayajulu, M.; Huttemann, M.; Grossman, L.I.; Aras, S. The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): Partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem. 2018, 293, 6517–6529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aras, S.; Arrabi, H.; Purandare, N.; Huttemann, M.; Kamholz, J.; Zuchner, S.; Grossman, L.I. Abl2 kinase phosphorylates Bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 440–448. [Google Scholar] [CrossRef]
- Liu, Y.; Clegg, H.V.; Leslie, P.L.; Di, J.; Tollini, L.A.; He, Y.; Kim, T.H.; Jin, A.; Graves, L.M.; Zheng, J.; et al. CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ. 2015, 22, 1035–1046. [Google Scholar] [CrossRef]
- Oka, T.; Sayano, T.; Tamai, S.; Yokota, S.; Kato, H.; Fujii, G.; Mihara, K. Identification of a novel protein MICS1 that is involved in maintenance of mitochondrial morphology and apoptotic release of cytochrome c. Mol. Biol. Cell 2008, 19, 2597–2608. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragoszewski, P.; Gornicka, A.; Sztolsztener, M.E.; Chacinska, A. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol. Cell. Biol. 2013, 33, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Vellanki, R.N.; Coyaud, E.; Ignatchenko, V.; Li, L.; Krieger, J.R.; Taylor, P.; Tong, J.; Pham, N.A.; Liu, G.; et al. CHCHD2 Is Coamplified with EGFR in NSCLC and Regulates Mitochondrial Function and Cell Migration. Mol. Cancer Res. 2015, 13, 1119–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, M.; Uchiumi, T.; Takazaki, S.; Okuno, B.; Nomura, M.; Yoshida, S.; Kanki, T.; Kang, D. p32/gC1qR is indispensable for fetal development and mitochondrial translation: Importance of its RNA-binding ability. Nucleic Acids Res. 2012, 40, 9717–9737. [Google Scholar] [CrossRef]
- Lutas, A.; Wahlmark, C.J.; Acharjee, S.; Kawasaki, F. Genetic Analysis in Drosophila Reveals a Role for the Mitochondrial Protein P32 in Synaptic Transmission. G3-Genes Genom. Genet. 2012, 2, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. Mitochondrial Regulation by PINK1-Parkin Signaling. ISRN Cell Biol. 2012, 2012, 926160. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Human Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Wang, H.; Luo, H.; Zhang, S.; Xu, H.; Zhang, S.; Rosenblum, J.; Wang, Z.; Zhang, Q.; Tang, M.; et al. CHCHD10 is involved in the development of Parkinson’s disease caused by CHCHD2 loss-of-function mutation p.T61I. Neurobiol. Aging 2018, 75, 38–41. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imai, Y.; Meng, H.; Shiba-Fukushima, K.; Hattori, N. Twin CHCH Proteins, CHCHD2, and CHCHD10: Key Molecules of Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Int. J. Mol. Sci. 2019, 20, 908. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040908
Imai Y, Meng H, Shiba-Fukushima K, Hattori N. Twin CHCH Proteins, CHCHD2, and CHCHD10: Key Molecules of Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. International Journal of Molecular Sciences. 2019; 20(4):908. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040908
Chicago/Turabian StyleImai, Yuzuru, Hongrui Meng, Kahori Shiba-Fukushima, and Nobutaka Hattori. 2019. "Twin CHCH Proteins, CHCHD2, and CHCHD10: Key Molecules of Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia" International Journal of Molecular Sciences 20, no. 4: 908. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040908