Elucidation of Novel Therapeutic Targets for Acute Myeloid Leukemias with RUNX1-RUNX1T1 Fusion

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Identification of RUNX1-RUNX1T1 Fusion-Positive AMLs
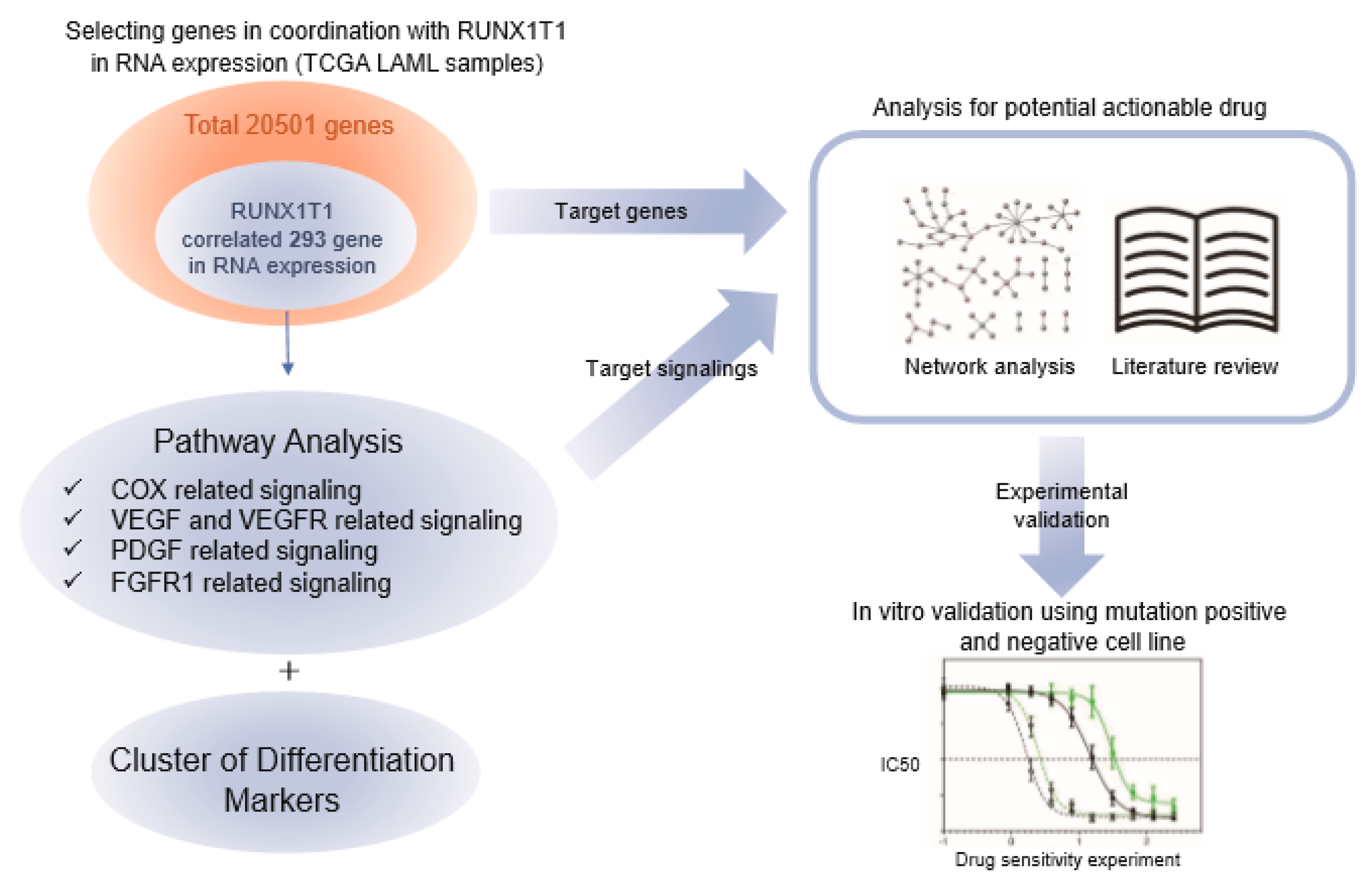
2.2. Genes and Pathways Specifically Altered in AMLs with RUNX1-RUNX1T1 Fusion
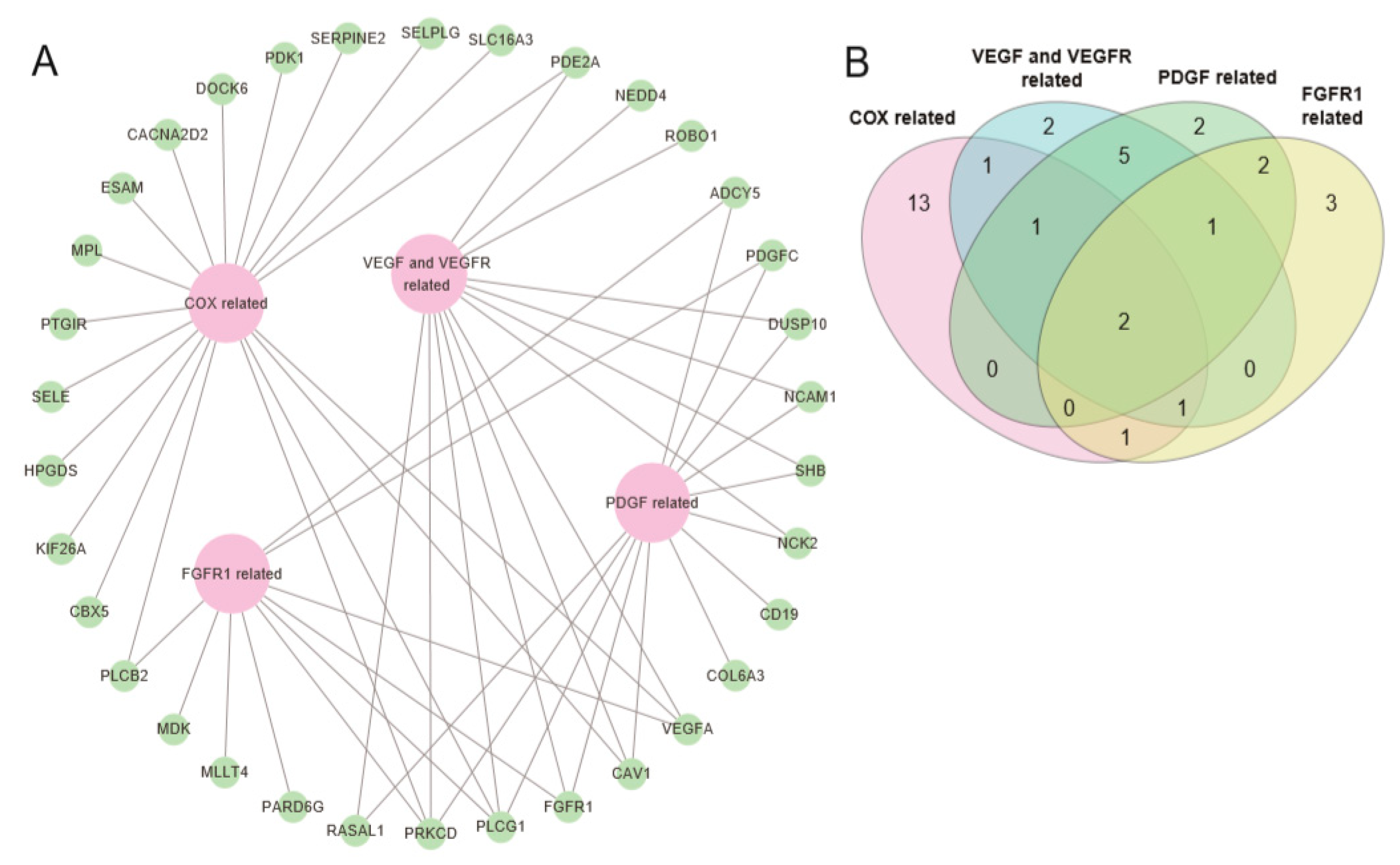
2.3. Pathway Prioritization by Targeting Possibility
2.4. Functional Validation of Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Inclusion Criteria
4.2. Over-Representation Pathway Analysis and Gene Set Enrichment Analysis
4.3. Network Analysis for Potential Actionable Drugs and Target Genes
4.4. Statistical Analysis and Visualization
4.5. Cell Culture
4.6. Receptor Tyrosine Kinase (RTK) Phosphorylation Array
4.7. Drug Sensitivity Test
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviation
| AML | acute myeloid leukemia |
| CML | chronic myeloid leukemia |
| TCGA | The Cancer Genome Atlas |
| FISH | fluorescence in situ hybridization |
| ORA | over-representation analysis |
| CPDB | ConsensusPathDB |
| GSEA | Gene Set Enrichment Analysis |
| FDR | false detection rate |
| FBS | fetal bovine serum |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| BCA | bicinchoninic acid |
| RTK | receptor tyrosine kinase |
| DMSO | Dimethyl sulfoxide |
| DRC | Dose-response curve |
| IC50 | half maximal inhibitory concentration |
References
- International classification of rodent tumours. Part I--The rat. 4. Haematopoietic system. IARC Sci. Publ. 1993, 1–27.
- Downing, J.R. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: Biology and clinical significance. Br. J. Haematol. 1999, 106, 296–308. [Google Scholar] [CrossRef]
- Licht, J.D. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene 2001, 20, 5660–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L.F.; Zhang, D.E. The 8;21 translocation in leukemogenesis. Oncogene 2004, 23, 4255–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spirin, P.V.; Lebedev, T.D.; Orlova, N.N.; Gornostaeva, A.S.; Prokofjeva, M.M.; Nikitenko, N.A.; Dmitriev, S.E.; Buzdin, A.A.; Borisov, N.M.; Aliper, A.M.; et al. Silencing AML1-ETO gene expression leads to simultaneous activation of both pro-apoptotic and proliferation signaling. Leukemia 2014, 28, 2222–2228. [Google Scholar] [CrossRef]
- Krauth, M.T.; Eder, C.; Alpermann, T.; Bacher, U.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: Frequency and impact on clinical outcome. Leukemia 2014, 28, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Andrieu, V.; Radford-Weiss, I.; Troussard, X.; Chane, C.; Valensi, F.; Guesnu, M.; Haddad, E.; Viguier, F.; Dreyfus, F.; Varet, B.; et al. Molecular detection of t(8;21)/AML1-ETO in AML M1/M2: Correlation with cytogenetics, morphology and immunophenotype. Br. J. Haematol. 1996, 92, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.; Dharmadhikari, A.V.; Sait, S.N.; Deeb, G.; Wallace, P.K.; Thompson, J.E.; Wang, E.S.; Wetzler, M. CD19 expression in acute leukemia is not restricted to the cytogenetically aberrant populations. Leuk Lymphoma 2013, 54, 1517–1520. [Google Scholar] [CrossRef]
- Johnson, R.C.; Ma, L.; Cherry, A.M.; Arber, D.A.; George, T.I. B-cell transcription factor expression and immunoglobulin gene rearrangement frequency in acute myeloid leukemia with t(8;21)(q22;q22). Am. J. Clin. Pathol. 2013, 140, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ball, E.D.; Davis, R.B.; Griffin, J.D.; Mayer, R.J.; Davey, F.R.; Arthur, D.C.; Wurster-Hill, D.; Noll, W.; Elghetany, M.T.; Allen, S.L.; et al. Prognostic value of lymphocyte surface markers in acute myeloid leukemia. Blood 1991, 77, 2242–2250. [Google Scholar] [PubMed]
- Fortier, J.M.; Payton, J.E.; Cahan, P.; Ley, T.J.; Walter, M.J.; Graubert, T.A. POU4F1 is associated with t(8;21) acute myeloid leukemia and contributes directly to its unique transcriptional signature. Leukemia 2010, 24, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Dunne, J.; Gascoyne, D.M.; Lister, T.A.; Brady, H.J.; Heidenreich, O.; Young, B.D. AML1/ETO proteins control POU4F1/BRN3A expression and function in t(8;21) acute myeloid leukemia. Cancer Res. 2010, 70, 3985–3995. [Google Scholar] [CrossRef] [PubMed]
- Dunne, J.; Mannari, D.; Farzaneh, T.; Gessner, A.; van Delft, F.W.; Heidenreich, O.; Young, B.D.; Gascoyne, D.M. AML1/ETO and POU4F1 synergy drives B-lymphoid gene expression typical of t(8;21) acute myeloid leukemia. Leukemia 2012, 26, 1131–1135. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wheat, J.; Chen, X.; Jin, S.; Sadrzadeh, H.; Fathi, A.T.; Peterson, R.T.; Kung, A.L.; Sweetser, D.A.; et al. AML1-ETO mediates hematopoietic self-renewal and leukemogenesis through a COX/beta-catenin signaling pathway. Blood 2013, 121, 4906–4916. [Google Scholar] [CrossRef]
- Ter Elst, A.; Ma, B.; Scherpen, F.J.; de Jonge, H.J.; Douwes, J.; Wierenga, A.T.; Schuringa, J.J.; Kamps, W.A.; de Bont, E.S. Repression of vascular endothelial growth factor expression by the runt-related transcription factor 1 in acute myeloid leukemia. Cancer Res. 2011, 71, 2761–2771. [Google Scholar] [CrossRef]
- Lierman, E.; Cools, J. Recent breakthroughs in the understanding and management of chronic eosinophilic leukemia. Expert Rev. Anticancer. Ther. 2009, 9, 1295–1304. [Google Scholar] [CrossRef]
- Demoulin, J.B.; Essaghir, A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. 2014, 25, 273–283. [Google Scholar] [CrossRef]
- Asou, H.; Tashiro, S.; Hamamoto, K.; Otsuji, A.; Kita, K.; Kamada, N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood 1991, 77, 2031–2036. [Google Scholar] [PubMed]
- Matozaki, S.; Nakagawa, T.; Kawaguchi, R.; Aozaki, R.; Tsutsumi, M.; Murayama, T.; Koizumi, T.; Nishimura, R.; Isobe, T.; Chihara, K. Establishment of a myeloid leukaemic cell line (SKNO-1) from a patient with t(8;21) who acquired monosomy 17 during disease progression. Br. J. Haematol. 1995, 89, 805–811. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Demoulin, J.B.; Montano-Almendras, C.P. Platelet-derived growth factors and their receptors in normal and malignant hematopoiesis. Am. J. Blood Res. 2012, 2, 44–56. [Google Scholar]
- Kita, K.; Nakase, K.; Miwa, H.; Masuya, M.; Nishii, K.; Morita, N.; Takakura, N.; Otsuji, A.; Shirakawa, S.; Ueda, T.; et al. Phenotypical characteristics of acute myelocytic leukemia associated with the t(8;21)(q22;q22) chromosomal abnormality: Frequent expression of immature B-cell antigen CD19 together with stem cell antigen CD34. Blood 1992, 80, 470–477. [Google Scholar] [PubMed]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; Andre, F.; Soria, J.C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J. A New Cell-Cycle Target in Cancer - Inhibiting Cyclin D-Dependent Kinases 4 and 6. N. Engl. J. Med. 2016, 375, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ariza, A.; Lopez-Pedrera, C.; Aranda, E.; Barbarroja, N. VEGF targeted therapy in acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2011, 80, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Lai, C.; Karp, J.E.; Hourigan, C.S. Precision medicine for acute myeloid leukemia. Expert Rev. Hematol. 2016, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]
- Yamamoto, S.; Sakai, N.; Nakamura, H.; Fukagawa, H.; Fukuda, K.; Takagi, T. INOH: Ontology-based highly structured database of signal transduction pathways. Database (Oxford) 2011, 2011, bar052. [Google Scholar] [CrossRef]
- Kandasamy, K.; Mohan, S.S.; Raju, R.; Keerthikumar, S.; Kumar, G.S.; Venugopal, A.K.; Telikicherla, D.; Navarro, J.D.; Mathivanan, S.; Pecquet, C.; et al. NetPath: A public resource of curated signal transduction pathways. Genome Biol. 2010, 11, R3. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Melius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef] [PubMed]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478–D484. [Google Scholar] [CrossRef] [PubMed]
- Hewett, M.; Oliver, D.E.; Rubin, D.L.; Easton, K.L.; Stuart, J.M.; Altman, R.B.; Klein, T.E. PharmGKB: The Pharmacogenetics Knowledge Base. Nucleic Acids Res. 2002, 30, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Ma, H.W.; Zhao, X.M.; Goryanin, I. Compartmentalization of the Edinburgh Human Metabolic Network. BMC Bioinformatics 2010, 11, 393. [Google Scholar] [CrossRef]
- Fazekas, D.; Koltai, M.; Turei, D.; Modos, D.; Palfy, M.; Dul, Z.; Zsakai, L.; Szalay-Beko, M.; Lenti, K.; Farkas, I.J.; et al. SignaLink 2—a signaling pathway resource with multi-layered regulatory networks. BMC Syst. Biol. 2013, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.T.; Choi, Y.L.; Yun, J.W.; Kim, N.K.D.; Kim, S.Y.; Jeon, H.J.; Nam, J.Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun. 2017, 8, 1377. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- R Developement Core Team. R: A Language and Environment for Statistical Computing. 2016. Available online: http://www.R-project.org (accessed on 30 November 2017).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RUNX1-RUNX1T1 Fusion Positive (n = 7) | Normal Karyotype (n = 86) | Statistical Difference (U Test, Fisher’s Test) | ||
|---|---|---|---|---|
| Age at Diagnosis (Mean, †SD) | 44.6 ± 15.9 | 54.8 ± 16.9 | 0.001 | |
| Gender (male/total) | 3/7 | 42/86 | 1 | |
| Event of death | 3/7 | 59/86 | 0.217 | |
| Immunophenotype | CD19 | 4/4 | 3/28 | < 0.001 |
| CD33 | 3/4 | 72/77 | 0.269 | |
| CD34 | 7/7 | 42/63 | 0.094 | |
| CD45 | 2/2 | 10/10 | 1 | |
| CD56 | 2/2 | 7/33 | 0.061 | |
| CD117 | 7/7 | 73/73 | 1 | |
| HLA-DR | 6/6 | 44/52 | 0.581 | |
| NSE | 0/5 | 32/78 | 0.151 | |
| TdT | 1/2 | 1/4 | 1 | |
| Peripheral blood (median) | Blast | 67.0% | 71.5% | 0.002 |
| Hb (g/dL) | 9.3 | 9.7 | 0.004 | |
| Platelet (/µL) | 40K | 50.8K | 0.002 | |
| Bone marrow cellularity (median) | 70% | 88.3% | 0.001 | |
| ‡FAB classification | M0 | 0 | 5 | |
| M1 | 2 | 22 | ||
| M2 | 5 | 20 | ||
| M3 | 0 | 1 | ||
| M4 | 0 | 21 | ||
| M5 | 0 | 15 | ||
| M6 | 0 | 0 | ||
| M7 | 0 | 1 | ||
| §FLT3 mutation | 1/7 | 23/68 | 0.675 | |
| Pathway Name | Set Size | Candidates Contained | p-value | q-value | Pathway Source | Members_Input_Overlap | Potential Target Drug for Pathway |
|---|---|---|---|---|---|---|---|
| Signaling events mediated by VEGFR1 and VEGFR2 | 68 | 7 (10.3%) | 0.00003 | 0.0161 | PID | NCK2; NEDD4; PRKCD; PLCG1; VEGFA; SHB; CAV1 | VEGF/VEGFR inhibitors (bevacizumab, pazopanib) |
| Axon guidance | 459 | 17 (3.7%) | 0.00011 | 0.0214 | Reactome | ITGA9; ARHGEF12; NCK2; ROBO1; CRMP1; PLCG1; DUSP10; NRCAM; SHB; VEGFA; RASAL1; RHGAP39; COL6A3; LAMC1; PITPNA; NCAM1; FGFR1 | |
| HuR stabilizes mRNA | 8 | 3 (37.5%) | 0.000121 | 0.0214 | Reactome | TNFSF13; ELAVL1; PRKCD | |
| Developmental Biology | 586 | 19 (3.3%) | 0.000245 | 0.0325 | Reactome | ITGA9; ARHGEF12; NCK2; ROBO1; CRMP1; PLCG1; NCAM1; NRCAM; SHB; VEGFA; TDGF1; RASAL1; ARHGAP39; COL6A3; LAMC1; PITPNA; DUSP10; CTNNA2; FGFR1 | |
| Celecoxib Pathway, Pharmacodynamics | 58 | 5 (8.6%) | 0.000984 | 0.077 | PharmGKB | CACNA2D2; PDK1; VEGFA; HPGDS; PTGIR | COX-2 inhibitors (celecoxib) |
| Role of second messengers in netrin-1 signaling | 4 | 2 (50.0%) | 0.00102 | 0.077 | Reactome | PLCG1; PITPNA | |
| Angiogenesis overview | 61 | 5 (8.2%) | 0.00124 | 0.077 | Wikipathways | ROBO1; PLCG1; VEGFA; SHB; FGFR1 | VEGF/VEGFR inhibitors (bevacizumab, pazopanib) |
| Focal adhesion - Homo sapiens (human) | 207 | 9 (4.3%) | 0.0017 | 0.077 | KEGG | ITGA9; PDGFC; ITGB4; CCND1; VEGFA; COL6A3; LAMC1; PARVG; CAV1 | |
| Beta1 integrin cell surface interactions | 66 | 5 (7.6%) | 0.00176 | 0.077 | PID | ITGA9; COL6A3; LAMC1; VEGFA; MDK | |
| S1P1 pathway | 19 | 3 (15.8%) | 0.00188 | 0.077 | PID | PLCB2; VEGFA; PLCG1 | |
| Cell surface interactions at the vascular wall | 101 | 6 (6.0%) | 0.00207 | 0.077 | Reactome | SELE; SLC16A3; SELPLG; PLCG1; ESAM; CAV1 | |
| Signaling by PDGF | 301 | 11 (3.7%) | 0.0021 | 0.077 | Reactome | ADCY5; PDGFC; NCK2; PRKCD; DUSP10; PLCG1; RASAL1; COL6A3; CD19; NCAM1; FGFR1 | PDGFR inhibitors (imatinib, ponatinib, sunitinib) |
| Hemostasis | 493 | 15 (3.0%) | 0.00216 | 0.077 | Reactome | SELE; SLC16A3; DOCK6; KIF26A; PLCG1; PTGIR; MPL; VEGFA; ESAM; SELPLG; CBX5; PDE2A; PRKCD; SERPINE2; CAV1 | |
| actions of nitric oxide in the heart | 42 | 4 (9.5%) | 0.00222 | 0.077 | BioCarta | PLCG1; PDE2A; VEGFA; CAV1 | |
| Thyroxine (Thyroid Hormone) Production | 6 | 2 (33.3%) | 0.00251 | 0.077 | Wikipathways | TPO; TRH | |
| Extracellular matrix organization | 264 | 10 (3.8%) | 0.00256 | 0.077 | Reactome | ITGA9; BMP4; ITGB4; HSPG2; COL14A1; FBLN2; COL6A3; LAMC1; NCAM1; FBLN5 | |
| VEGFA-VEGFR2 Pathway | 266 | 10 (3.8%) | 0.00271 | 0.077 | Reactome | NCK2; PRKCD; NCAM1; RASAL1; PLCG1; VEGFA; SHB; CAV1; DUSP10; FGFR1 | VEGF/VEGFR inhibitors (bevacizumab, pazopanib) |
| Cell adhesion molecules (CAMs) - Homo sapiens (human) | 142 | 7 (4.9%) | 0.00275 | 0.077 | KEGG | ITGA9; NRCAM; SELE; CD34; SELPLG; ESAM; NCAM1 | |
| Signal transduction by L1 | 22 | 3 (13.6%) | 0.0029 | 0.077 | Reactome | ITGA9; NCAM1; FGFR1 | |
| PDGFR-alpha signaling pathway | 22 | 3 (13.6%) | 0.0029 | 0.077 | PID | PLCG1; SHB; CAV1 | PDGFR inhibitors (imatinib, ponatinib, sunitinib) |
| Proton Pump Inhibitor Pathway, Pharmacodynamics | 46 | 4 (8.7%) | 0.00311 | 0.0774 | PharmGKB | ADCY5; PLCB2; PLCG1; AKAP2 | |
| Laminin interactions | 23 | 3 (13.0%) | 0.0033 | 0.0774 | Reactome | HSPG2; ITGB4; LAMC1 | |
| Signaling by VEGF | 274 | 10 (3.7%) | 0.00335 | 0.0774 | Reactome | NCK2; PRKCD; NCAM1; RASAL1; PLCG1; VEGFA; SHB; CAV1; DUSP10; FGFR1 | VEGF/VEGFR inhibitors (bevacizumab, pazopanib) |
| Phospholipase C-mediated cascade: FGFR1 | 49 | 4 (8.2%) | 0.00392 | 0.0866 | Reactome | ADCY5; PLCG1; PRKCD; FGFR1 | FGFR1 inhibitors (dovitinib, ponatinib) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, J.W.; Bae, Y.K.; Cho, S.Y.; Koo, H.; Kim, H.-J.; Nam, D.-H.; Kim, S.-H.; Chun, S.; Joo, K.M.; Park, W.-Y. Elucidation of Novel Therapeutic Targets for Acute Myeloid Leukemias with RUNX1-RUNX1T1 Fusion. Int. J. Mol. Sci. 2019, 20, 1717. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071717
Yun JW, Bae YK, Cho SY, Koo H, Kim H-J, Nam D-H, Kim S-H, Chun S, Joo KM, Park W-Y. Elucidation of Novel Therapeutic Targets for Acute Myeloid Leukemias with RUNX1-RUNX1T1 Fusion. International Journal of Molecular Sciences. 2019; 20(7):1717. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071717
Chicago/Turabian StyleYun, Jae Won, Yoon Kyung Bae, So Yeong Cho, Harim Koo, Hee-Jin Kim, Do-Hyun Nam, Sun-Hee Kim, Sejong Chun, Kyeung Min Joo, and Woong-Yang Park. 2019. "Elucidation of Novel Therapeutic Targets for Acute Myeloid Leukemias with RUNX1-RUNX1T1 Fusion" International Journal of Molecular Sciences 20, no. 7: 1717. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071717