STIM Proteins and Glutamate Receptors in Neurons: Role in Neuronal Physiology and Neurodegenerative Diseases


Abstract
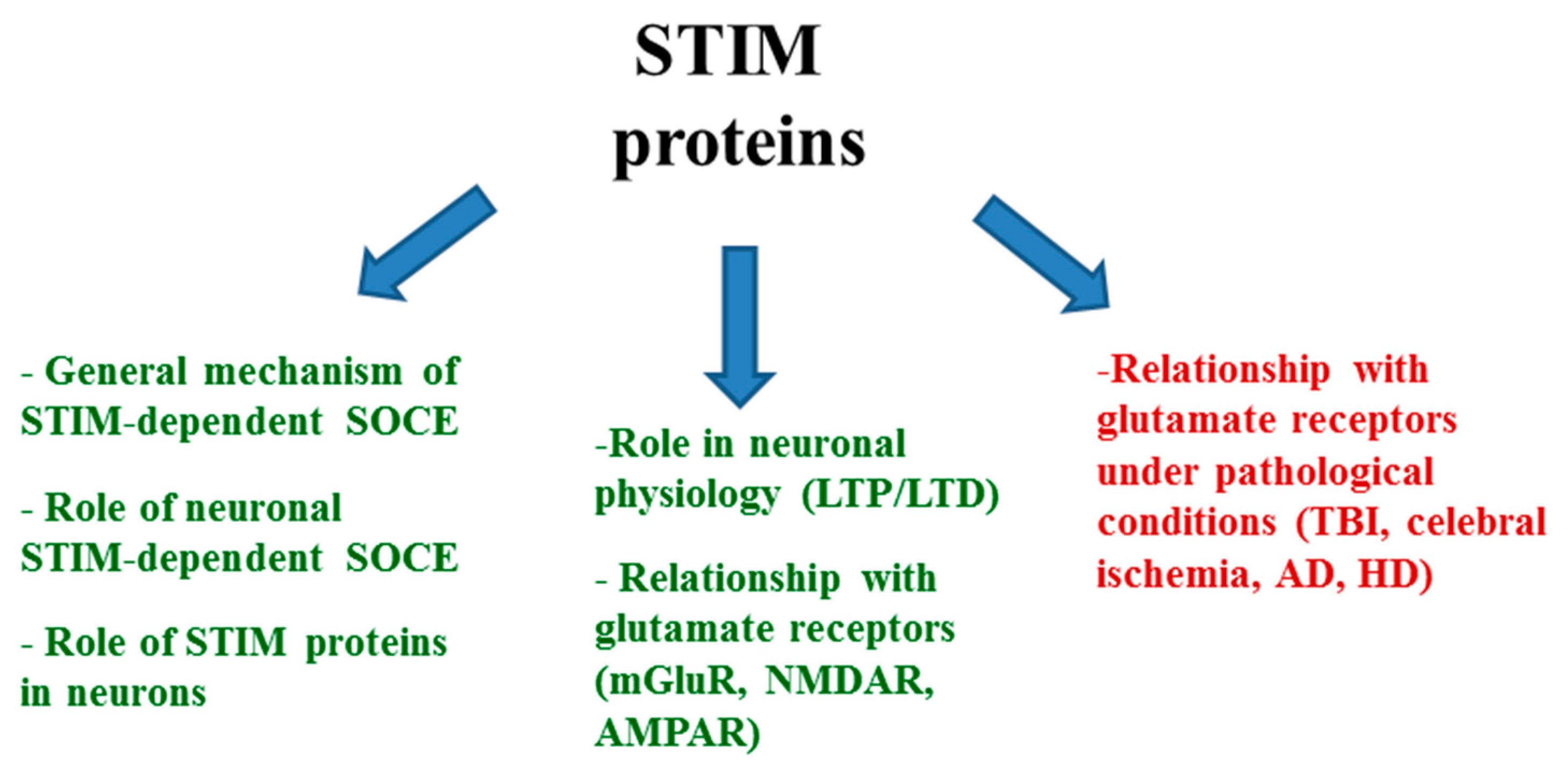
:1. Introduction
2. STIM Proteins under Physiological Conditions
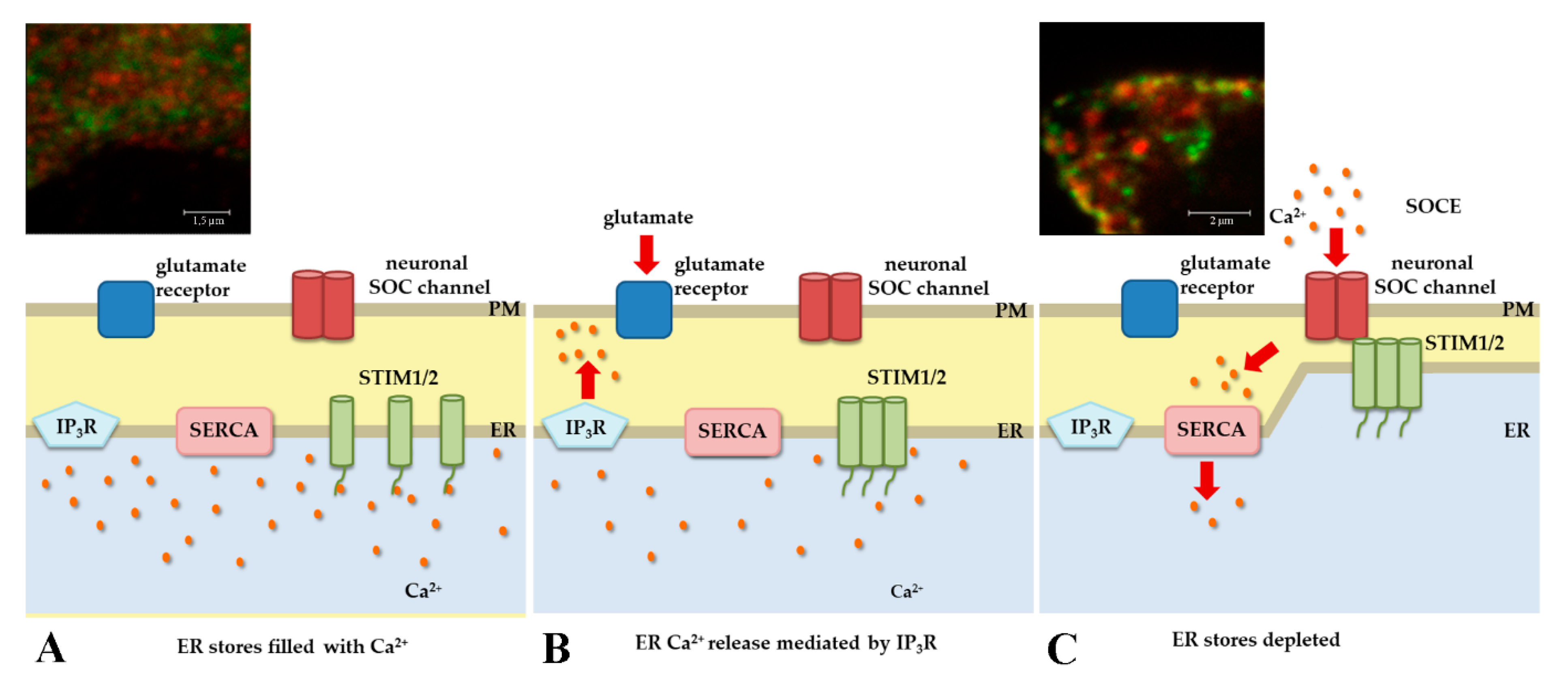
2.1. STIM Proteins and Intracellular Ca2+ Regulation
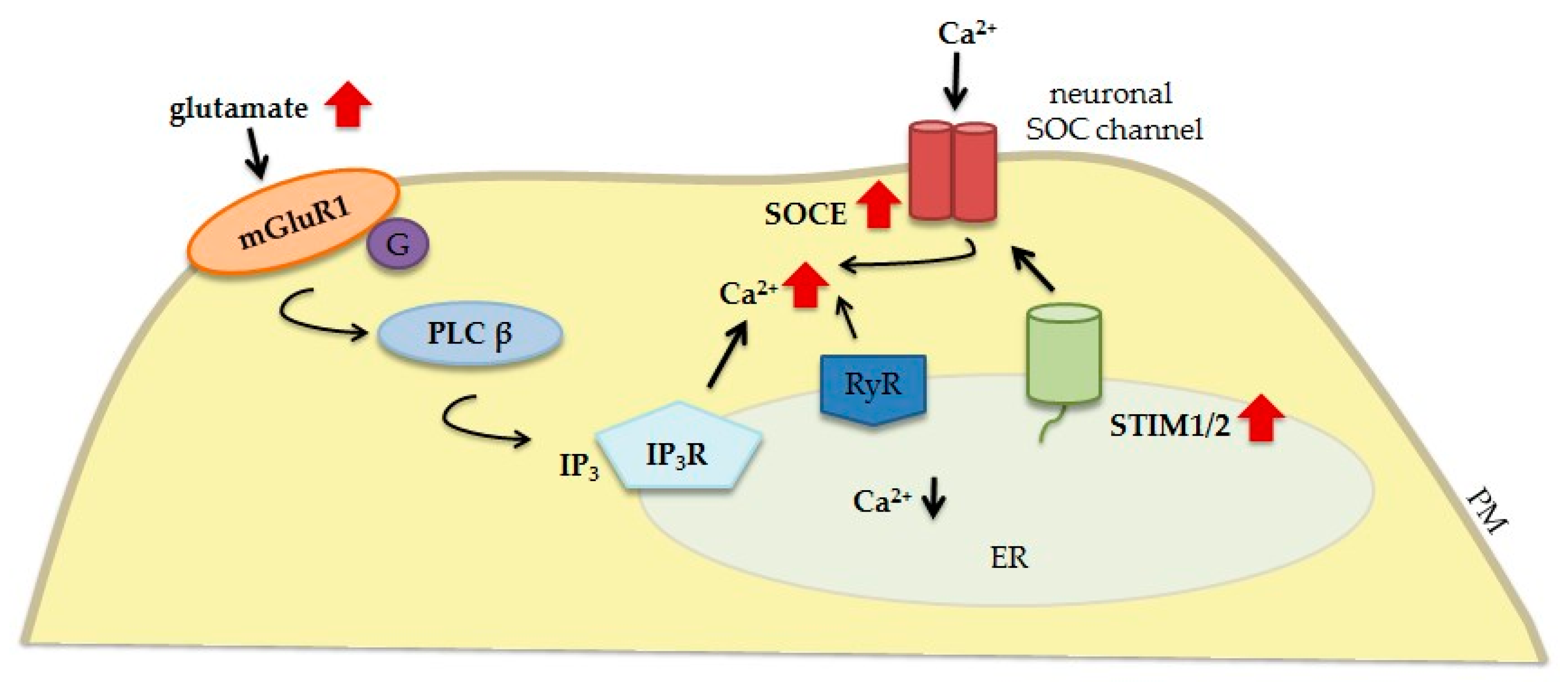
2.2. STIM Proteins and Their Relationship with Glutamate Receptors
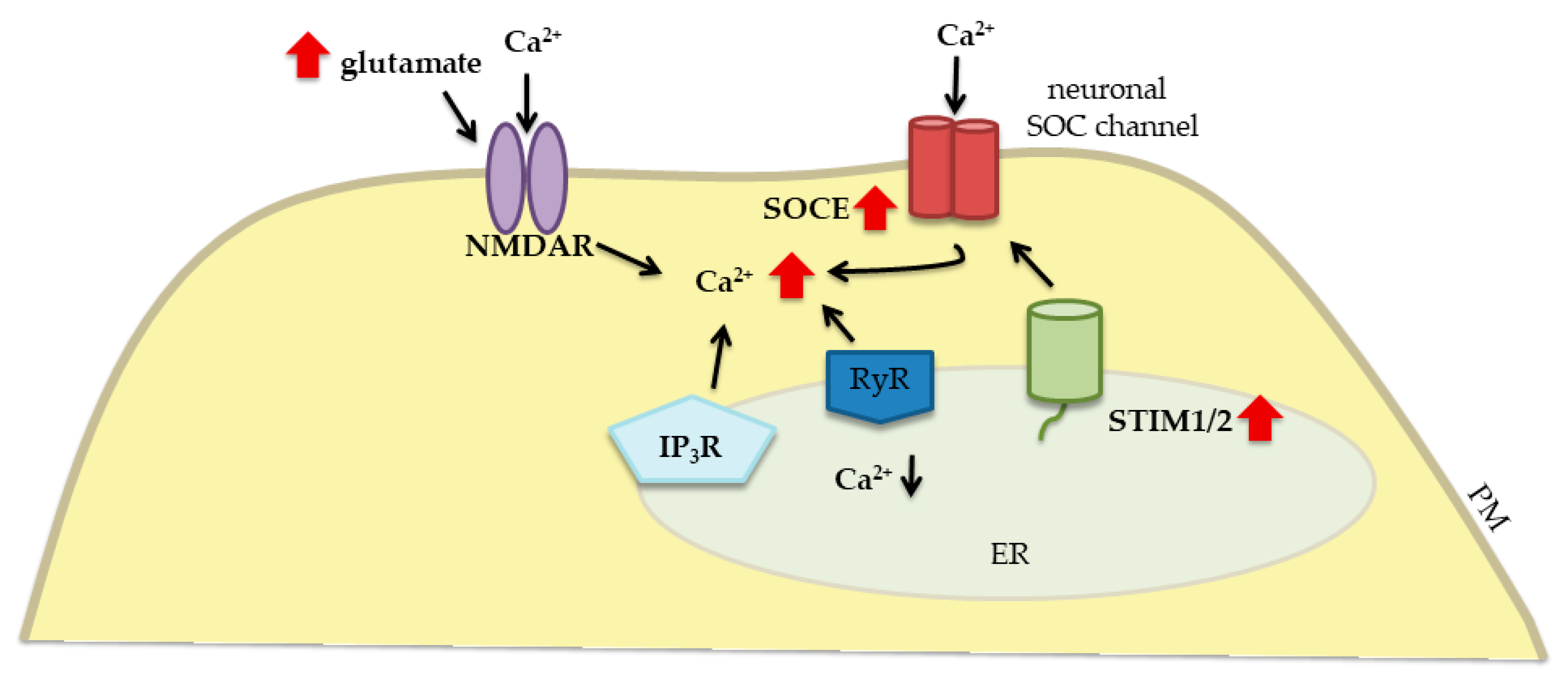
3. Relationship between STIM Proteins, Glutamate, and Glutamate Receptors under Pathological Conditions
3.1. Traumatic Brain Injury
3.2. Cerebral Ischemia
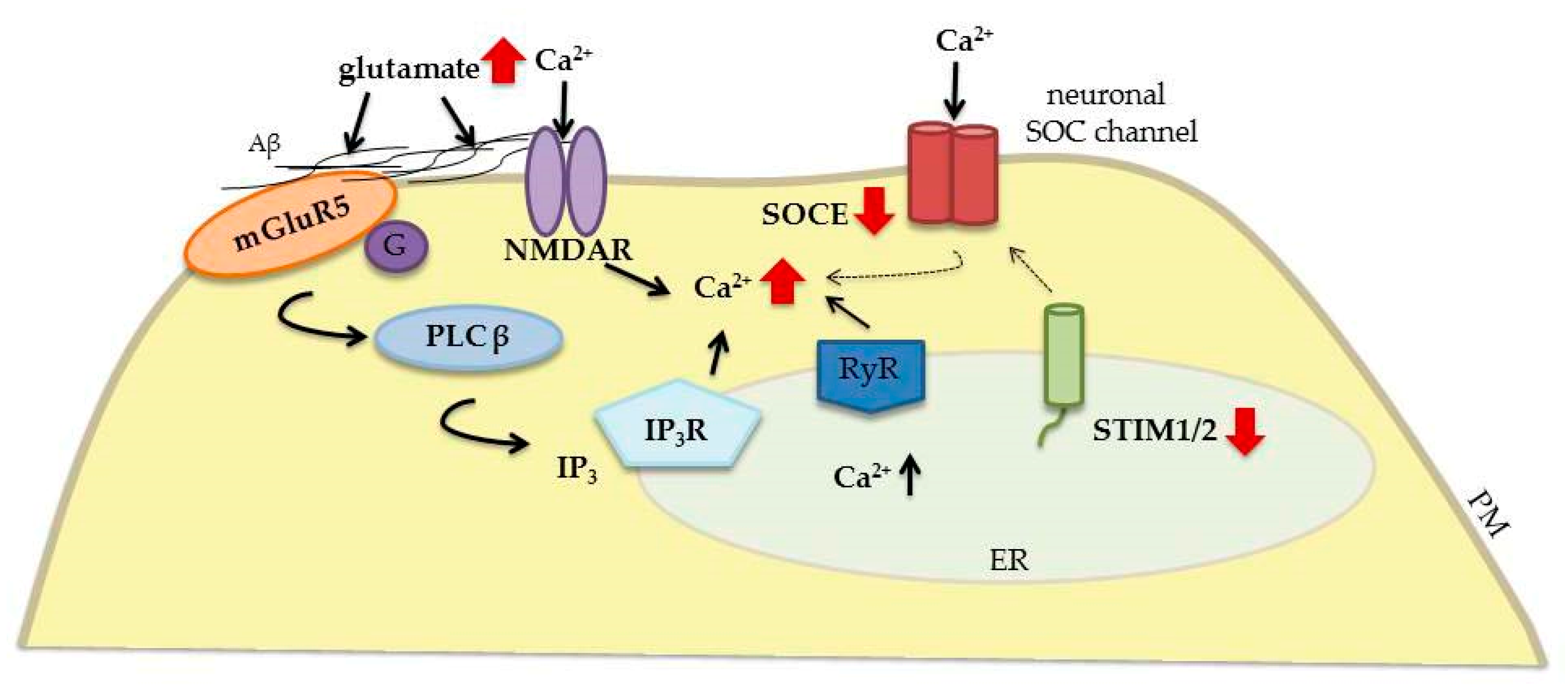
3.3. Alzheimer’s Disease
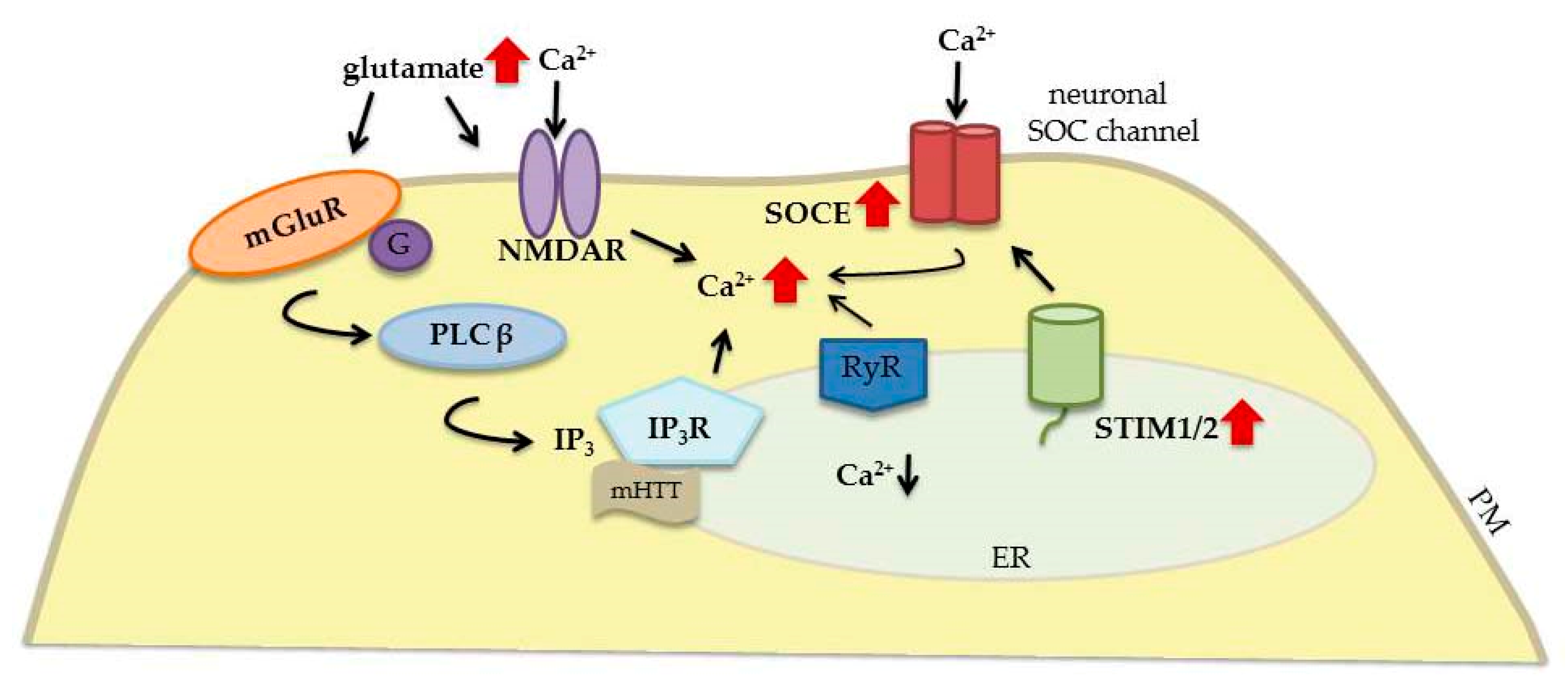
3.4. Huntington’s Disease
4. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AMPARs | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| APP | amyloid precursor protein |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CNS | central nervous system |
| ER | endoplasmic reticulum |
| HD | Huntington’s disease |
| HTT | huntingtin protein |
| IP3 | inositol trisphosphate |
| LTD | long-term depression |
| LTP | long-term potentiation |
| mGluRs | metabotropic glutamate receptors |
| MSNs | medium spiny neurons |
| NMDARs | N-methyl-D-aspartate receptors |
| PD | Parkinson’s disease |
| PNs | Purkinje neurons |
| PS1 | presenilin 1 |
| PS2 | presenilin 2 |
| RyRs | ryanodine receptors |
| SERCA | sarco-endoplasmic reticulum calcium-adenosine triphosphatase |
| SOCE | store-operated calcium entry |
| STIM | stromal interaction molecule |
| TBI | traumatic brain injury |
| TNI | traumatic neuronal injury |
| TRPC | transient receptor potential channel |
| VGCCs | voltage-gated calcium channels |
References
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [Green Version]
- Michael, A.R. Revisiting AMPA Receptors as an Antiepileptic Drug Target. Epilepsy Curr. 2011, 11, 56–63. [Google Scholar] [Green Version]
- Monaghan, D.T.; Jane, D.E. Biology of the NMDA Receptor, 1st ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; pp. 5–9. [Google Scholar]
- Niswender, C.M.; Jeffrey, C.P. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.; Karl, R.M.; Alexander, R.P.; Adelsberger, H.; Brill, M.S.; Rühlmann, C.; Ansel, A.; Sakimura, K.; Baba, Y.; Kurosaki, T.; et al. STIM1 controls neuronal Ca2 signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 2014, 82, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, A.M.; Madge, D.J.; Garthwaite, J. Synaptic activation of metabotropic glutamate receptors in the parallel Fibre-Purkinje cell pathway in rat cerebellar slices. Neuroscience 1994, 63, 911–915. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 Channels Are Required for Synaptic Transmission and Motor Coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Debanne, D.; Daoudal, G.; Sourdet, V.; Russier, M. Brain plasticity and ion channels. J. Physiol. Paris 2003, 97, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Youn, D.H. Differential roles of signal transduction mechanisms in long-term potentiation of excitatory synaptic transmission induced by activation of group I mGluRs in the spinal trigeminal subnucleus oralis. Brain Res. Bull 2014, 108, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ardiles, A.O.; Yang, S.; Tran, T.; Posada-Duque, R.; Valdivia, G.; Baek, M.; Chuang, Y.A.; Palacios, A.G.; Gallagher, M.; et al. Metabotropic Glutamate Receptors Induce a Form of LTP Controlled by Translation and Arc Signaling in the Hippocampus. J. Neurosci. 2016, 36, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Derkach, V.; Barria, A.; Soderling, T.R. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 3269–3274. [Google Scholar] [CrossRef]
- Oh, M.C.; Derkach, V.A.; Guire, E.S.; Soderling, T.R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 2005, 281, 752–758. [Google Scholar] [CrossRef]
- Banke, T.G.; Bowie, D.; Lee, H.K.; Huganir, R.L.; Schousboe, A.; Traynelis, S.F. Control of GluR1 AMPA Receptor Function by cAMP-Dependent Protein Kinase. J. Neurosci. 2000, 20, 89–102. [Google Scholar] [CrossRef] [Green Version]
- Anggono, V.; Huganir, R.L. Regulation of AMPA Receptor Trafficking and Synaptic Plasticity. Curr. Opin. Neurobiol. 2012, 22, 461–469. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Lynch, M.A. Long-Term Potentiation and Memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Shi, S.H.; Esteban, J.A.; Piccini, A.; Poncer, J.C.; Malinow, R. Driving AMPA Receptors into Synapses by LTP and CaMKII: Requirement for GluR1 and PDZ Domain Interaction. Science 2000, 287, 2262–2267. [Google Scholar] [CrossRef]
- Zamanillo, D.; Sprengel, R.; Hvalby, O.; Jensen, V.; Burnashev, N.; Rozov, A.; Kaise, K.M.M. Importance of AMPA Receptors for Hippocampal Synaptic Plasticity But Not for Spatial Learning. Science 1999, 284, 1805–1811. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Homma, K.J.; Poo, M.M. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 2004, 44, 749–757. [Google Scholar] [CrossRef]
- Bear, M.F.; Connors, B.W.; Paradiso, M.A. Neuroscience: Exploring the Brain; Lippincott Williams and Wilkins: Hagerstwon, MD, USA, 2007; p. 718. [Google Scholar]
- Lüscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- González-Sánchez, P.; Del Arco, A.; Esteban, J.A.; Satrústegui, J. Store-Operated Calcium Entry Is Required for mGluR-Dependent Long Term Depression in Cortical Neurons. Front Cell Neurosci. 2017, 11, 363. [Google Scholar] [CrossRef]
- Reyes-Harde, M.; Empson, R.; Potter, B.V.L.; Galione, A.; Stanton, P.K. Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc. Natl. Acad. Sci. USA 1999, 96, 4061–4066. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Wang, Y.; Qin, Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef] [Green Version]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Potier, M.; Trebak, M. New development in the signaling mechanisms of the store-operated calcium entry pathway. Pflügers Archiv 2008, 457, 405–415. [Google Scholar] [CrossRef]
- Shim, A.H.; Tirado-Lee, L.; Prakriya, M. Structural and functional mechanisms of CRAC channel regulation. J. Mol. Biol. 2015, 427, 77–93. [Google Scholar] [CrossRef]
- Cahalan, M.D. STIMulating store-operated Ca2+ entry. Nat. Cell. Biol. 2009, 11, 669–677. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Majewski, L.; Kuznicki, J. SOCE in neurons: Signaling or just refilling? Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1940–1952. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Putney, J.W. Store-Operated Calcium Channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC4600950/ (accessed on 1 October 2015). [CrossRef] [Green Version]
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 900–906. [Google Scholar] [CrossRef]
- Emptage, N.J.; Reid, C.A.; Fine, A. Calcium Stores in Hippocampal Synaptic Boutons Mediate Short-Term Plasticity, Store-Operated Ca2+ Entry, and Spontaneous Transmitter Release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef]
- Baba, A.; Yasui, T.; Fujisawa, S.; Yamada, R.X.; Yamada, M.K.; Nishiyama, N.; Matsuki, N.; Ikegaya, Y. Activity-Evoked Capacitative Ca2+ Entry: Implications in Synaptic Plasticity. J. Neurosci. 2003, 23, 7737–7741. [Google Scholar] [CrossRef]
- Samtleben, S.; Wachter, B.; Blum, R. Store-operated calcium entry compensates fast ER calcium loss in resting hippocampal neurons. Cell Calcium 2015, 58, 147–159. [Google Scholar] [CrossRef]
- Klejman, M.E.; Gruszczynska-Biegala, J.; Skibinska-Kijek, A.; Wisniewska, M.B.; Misztal, K.; Blazejczyk, M.; Bojarski, L.; Kuznicki, J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem. Int. 2009, 54, 49–55. [Google Scholar] [CrossRef]
- Gruszczynska-Biegala, J.; Pomorski, P.; Wisniewska, M.B.; Kuznicki, J. Differential Roles for STIM1 and STIM2 in Store-Operated Calcium Entry in Rat Neurons. PLoS ONE 2011, 6, e19285. [Google Scholar] [CrossRef]
- Venkiteswaran, G.; Hasan, G. Intracellular Ca2+ signaling and store-operated Ca2+ entry are required in Drosophila neurons for flight. Proc. Natl. Acad. Sci. USA 2009, 106, 10326–10331. [Google Scholar] [CrossRef] [PubMed]
- Skibinska-Kijek, A.; Wisniewska, M.B.; Gruszczynska-Biegala, J.; Methner, A.; Kuznicki, J. Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol. Exp. (Wars). 2009, 69, 413–428. [Google Scholar]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, W.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Kuznicki, J. Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J. Neurochem. 2013, 126, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Parvez, S.; Beck, A.; Peinelt, C.; Soboloff, J.; Lis, A.; Monteilh-Zoller, M.; Gill, D.L.; Fleig, A.; Penner, R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008, 22, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Subedi, K.P.; Ong, H.L.; Son, G.Y.; Liu, X.; Ambudkar, I.S. STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 2018, 23, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.A.; Leech, C.A.; Kopp, R.F.; Roe, M.W. Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry. Int. J. Mol. Sci. 2018, 19, 1522. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.; Feske, S.; White, C.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, A.; Shum, A.K.; McBride, H.J.; Kessler, J.A.; Feske, S.; Miller, R.J.; Prakriya, M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 2014, 34, 9107–9123. [Google Scholar] [CrossRef]
- Gopurappilly, R.; Deb, B.K.; Chakraborty, P.; Hasan, G. Stable STIM1 Knockdown in Self-Renewing Human Neural Precursors Promotes Premature Neural Differentiation. Front. Mol. Neurosci. 2018, 11, 178. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC Channel Activator STIM1 Binds and Inhibits L-Type Voltage-Gated Calcium Channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca2+ entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef]
- Dittmer, P.J.; Wild, A.R.; Dell’Acqua, M.L.; Sather, W.A. STIM1 Ca2+ Sensor Control of L-type Ca2+-Channel-Dependent Dendritic Spine Structural Plasticity and Nuclear Signaling. Cell Rep. 2017, 19, 321–334. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Selvaraj, S.; Sukumaran, P.; Lei, S.; Birnbaumer, L.; Singh, B.B. Inhibition of L-Type Ca2+ Channels by TRPC1-STIM1 Complex Is Essential for the Protection of Dopaminergic Neurons. J. Neurosci. 2017, 37, 3364–3377. [Google Scholar] [CrossRef]
- Kuang, X.; Liuab, Y.; Changab, Y.; Zhouab, J.; Zhang, H.; Li, Y.; Quab, J.; Wua, S. Inhibition of store-operated calcium entry by sub-lethal levels of proteasome inhibition is associated with STIM1/STIM2 degradation. Cell Calcium 2016, 59, 172–180. [Google Scholar] [CrossRef]
- Keil, J.M.; Shen, Z.; Briggs, S.P.; Patrick, G.N. Regulation of STIM1 and SOCE by the Ubiquitin-Proteasome System (UPS). PLoS ONE 2010, 5, e13465. [Google Scholar] [CrossRef]
- Ng, A.N.; Krogh, M.; Toresson, H. Dendritic EGFP-STIM1 activation after type I metabotropic glutamate and muscarinic acetylcholine receptor stimulation in hippocampal neuron. J. Neurosci. Res. 2011, 89, 1235–1244. [Google Scholar] [CrossRef]
- Gruszczynska-Biegala, J.; Sladowska, M.; Kuznicki, J. AMPA Receptors Are Involved in Store-Operated Calcium Entry and Interact with STIM Proteins in Rat Primary Cortical Neurons. Front. Cell. Neurosci. 2016, 10, 251. [Google Scholar] [CrossRef]
- Garcia-Alvarez, G.; Lu, B.; Yap, K.A.F.; Wong, L.C.; Thevathasanm, J.V.; Lim, L.; Ji, F.; Tan, K.W.; Mancuso, J.J.; Tang, W.; et al. STIM2 regulates PKA-dependent phosphorylation and trafficking of AMPARs. Mol. Biol. Cell 2015, 26, 1141–1159. [Google Scholar] [CrossRef] [Green Version]
- Yap, K.A.F.; Shetty, M.S.; Garcia-Alvarez, G.; Lu, B.; Alagappan, D.; Oh-Hora, M.; Sajikumar, S.; Fivaz, M. STIM2 regulates AMPA receptor trafficking and plasticity at hippocampal synapses. Neurobiol. Learn. Mem. 2017, 138, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Takamiya, K.; He, K.; Song, L.; Huganir, R.L. Specific Roles of AMPA Receptor Subunit GluR1 (GluA1) Phosphorylation Sites in Regulating Synaptic Plasticity in the CA1 Region of Hippocampus. J. Neurophysiol. 2010, 103, 479–489. [Google Scholar] [CrossRef]
- Lee, H.K.; Takamiya, K.; Han, J.S.; Man, H.; Kim, C.H.; Rumbaugh, G.; Yu, S.; Ding, L.; He, C.; Petralia, R.S.; et al. Phosphorylation of the AMPA Receptor GluR1 Subunit Is Required for Synaptic Plasticity and Retention of Spatial Memory. Cell 2003, 112, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alvarez, G.; Shetty, M.S.; Lu, B.; Yap, K.A.; Oh-Hora, M.; Sajikumar, S.; Bichler, Z.; Fivaz, M. Impaired spatial memory and enhanced long-term potentiation in mice with forebrain-specific ablation of the Stim genes. Front. Behav. Neurosci. 2015, 9, 180. [Google Scholar] [CrossRef]
- Ryu, C.; Jang, D.C.; Jung, D.; Kim, Y.G.; Shim, H.G.; Ryu, H.H.; Lee, Y.S.; Linden, D.J.; Worley, P.F.; Kim, S.J. STIM1 Regulates Somatic Ca2+ Signals and Intrinsic Firing Properties of Cerebellar Purkinje Neurons. J. Neurosci. 2017, 37, 8876–8894. [Google Scholar] [CrossRef]
- Korkotian, E.; Oni-Biton, E.; Segal, M. The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 2017, 595, 125–140. [Google Scholar] [CrossRef]
- Korkotian, E.; Frotscher, M.; Segal, M. Synaptopodin regulates spine plasticity: Mediation by calcium stores. J. Neurosci. 2014, 34, 11641–11651. [Google Scholar] [CrossRef]
- Majewski, Ł.; Maciąg, F.; Boguszewski, P.M.; Wasilewska, I.; Wiera, G.; Wójtowicz, T.; Mozrzymas, J.; Kuznicki, J. Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1071–1087. [Google Scholar] [CrossRef]
- Oliet, S.H.R.; Malenka, R.C.; Nicoll, R.A. Two Distinct Forms of Long-Term Depression Coexist in CA1 Hippocampal Pyramidal Cells. Neuron 1997, 18, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Bolshakov, V.Y.; Siegelbaum, S.A. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 1994, 264, 1148–1152. [Google Scholar] [CrossRef]
- Khatri, N.; Pareek, V.; Thankur, M.; Kumar, S. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Algattas, H.; Huang, J.H. Traumatic Brain Injury Pathophysiology and Treatments: Early, Intermediate, and Late Phases Post-Injury. Int. J. Mol. Sci. 2014, 15, 309–341. [Google Scholar] [CrossRef]
- Weber, J.T. Altered Calcium Signaling Following Traumatic Brain Injury. Front. Pharmacol. 2012, 3, 60. [Google Scholar] [CrossRef]
- Weber, J.T.; Rzigalinski, B.A.; Ellis, E.A. Traumatic Injury of Cortical Neurons Causes Changes in Intracellular Calcium Stores and Capacitative Calcium Influx. J. Biol. Chem. 2001, 276, 1800–1807. [Google Scholar] [CrossRef]
- Cucchiaroni, M.L.; Viscomi, M.T.; Bernardi, G.; Molinari, M.; Guatteo, E.; Mercuri, N.B. Metabotropic glutamate receptor 1 mediates the electrophysiological and toxic actions of the cycad derivative beta-N-Methylamino-L-alanine on substantia nigra pars compacta DAergic neurons. J. Neurosci. 2010, 30, 5176–5188. [Google Scholar] [CrossRef]
- Chen, T.; Fei, F.; Jiang, X.F.; Zhang, L.; Qu, Y.; Huo, K.; Fei, Z. Down-regulation of Homer1b/c attenuates glutamate-mediated excitotoxicity through endoplasmic reticulum and mitochondria pathways in rat cortical neurons. Free Radic Biol. Med. 2012, 52, 208–217. [Google Scholar] [CrossRef]
- Hou, P.F.; Liu, Z.H.; Li, N.; Cheng, W.J.; Guo, S.W. Knockdown of STIM1 Improves Neuronal Survival After Traumatic Neuronal Injury Through Regulating mGluR1-Dependent Ca2+ Signaling in Mouse Cortical Neurons. Cell. Mol. Neurobiol. 2015, 35, 283–292. [Google Scholar] [CrossRef]
- Fan, X.X.; Hao, Y.Y.; Guo, S.W.; Zhao, X.P.; Xiang, Y.; Feng, F.X.; Liang, G.T.; Dong, Y.W. Knockdown of RTN1-C attenuates traumatic neuronal injury through regulating intracellular Ca2+ homeostasis. Neurochem. Int. 2018, 121, 19–25. [Google Scholar] [CrossRef]
- Rao, W.; Zhang, L.; Peng, C.; Hui, H.; Wang, K.; Su, N.; Wang, L.; Dai, S.; Yang, Y.; Chen, T.; et al. Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim. Biophys. Acta 2015, 1852, 2402–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Tobe, B.; Jain, P.A.; Niles, W.; Winquist, A.; Mastrangelo, L.; Snyder, E.Y. The Application and Future of Neural Stem Cells in Regenerative Medicine. In Translational Regenerative Medicine, 1st ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 403–413. [Google Scholar]
- Zipfel, G.J.; Lee, J.M.; Choi, D.W. Reducting calcium overload in the ischemic brain. N. Engl. J. Med. 1999, 341, 1543–1544. [Google Scholar] [CrossRef]
- Zhang, M.; Song, J.N.; Wub, Y.; Zhao, Y.L.; Pang, H.G.; Fu, Z.F.; Zhang, B.F.; Ma, X.D. Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats. Neuroreport 2014, 25, 507–513. [Google Scholar] [CrossRef]
- Varga-Szabo, D.; Braun, A.; Kleinschnitz, C.; Bender, M.; Pleines, I.; Pham, M.; Renné, T.; Stoll, G.; Nieswandt, B. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J. Exp. Med. 2008, 205, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Chakroborty, S.; Stutzmann, G.E. Calcium channelopathies and Alzheimer’s disease: Insight into therapeutic success and failures. Eur. J. Pharmacol. 2014, 739, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Konnerth, A. Impairments of neural circuit function in Alzheimer’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150429. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, X.; Li, G.; Zhang, Y.; Wu, Y.; Song, W. Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef]
- Sinnen, B.L.; Bowen, A.B.; Gibson, E.S.; Kennedy, M.J. Local and Use-Dependent Effects of β-Amyloid Oligomers on NMDA Receptor Function Revealed by Optical Quantal Analysis. J. Neurosci. 2016, 45, 11532–11543. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed]
- Gazda, K.; Kuznicki, J.; Wegierski, T. Knockdown of amyloid precursor protein increases calcium levels in the endoplasmic reticulum. Sci. Rep. 2017, 7, 14512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, S.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflügers Arch. Eur. J. Physiol. 2010, 459, 441–449. [Google Scholar] [CrossRef]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging Pathways Driving Early Synaptic Pathology in Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, Y. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Speshilova, A.; Alexandrov, S.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 2015, 10, 37. [Google Scholar] [CrossRef]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal 2016, 9, ra89. [Google Scholar] [CrossRef]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuznicki, J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1050–1057. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, E.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Fon Tacer, K.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Wu, J.; Ryskamp, D.A.; Liang, X.; Egorova, P.; Zakharova, O.; Hung, G.; Bezprozvanny, I. Enhanced Store-Operated Calcium Entry Leads to Striatal Synaptic Loss in a Huntington’s Disease Mouse Model. J. Neurosci. 2016, 36, 125–141. [Google Scholar] [CrossRef]
- Li, S.H.; Li, X.J. Huntingtin–protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Nopoulos, P.C. Huntington disease: A single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar] [PubMed]
- Raymond, L.A. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem. Biophys. Res. Commun. 2017, 483, 1051–1062. [Google Scholar] [CrossRef]
- Czeredys, M.; Maciag, F.; Methner, A.; Kuznicki, J. Tetrahydrocarbazoles decrease elevated SOCE in medium spiny neurons from transgenic YAC128 mice, a model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1194–1205. [Google Scholar] [CrossRef]
- Doria, J.G.; Silva, F.R.; Souza, J.M.; Vieira, L.B.; Carvalho, T.G.; Reis, H.J.; Pereira, G.S.; Dobransky, T.; Ribeiro, F.M. Metabotropic glutamate receptor 5 positive allosteric modulators are neuroprotective in a mouse model of Huntington’s disease. Br. J. Pharmacol. 2013, 169, 909–992. [Google Scholar] [CrossRef]
- Wu, J.; Ryskamp, D.; Birnbaumer, L.; Bezprozvanny, I. Inhibition of TRPC1-Dependent Store-Operated Calcium Entry Improves Synaptic Stability and Motor Performance in a Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2018, 7, 35–50. [Google Scholar] [CrossRef]
- Czeredys, M.; Gruszczynska-Biegala, J.; Schacht, T.; Methner, A.; Kuznicki, J. Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington’s disease. Front. Mol. Neurosci. 2013, 6, 42. [Google Scholar] [CrossRef]
- Czeredys, M.; Vigont, V.A.; Boeva, V.A.; Mikoshiba, K.; Kaznacheyeva, E.V.; Kuznicki, J. Huntingtin-Associated Protein 1A Regulates Store-Operated Calcium Entry in Medium Spiny Neurons From Transgenic YAC128 Mice, a Model of Huntington‘s Disease. Front. Cell. Neurosci. 2018, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Vigont, V.A.; Zimina, O.A.; Glushankova, L.N.; Kolobkova, J.A.; Ryazantseva, M.A.; Mozhayeva, G.N.; Kaznacheyeva, E.V. STIM1 Protein Activates Store-Operated Calcium Channels in Cellular Model of Huntington’s Disease. Acta Naturae 2014, 6, 40–47. [Google Scholar]
- Vigont, V.; Kolobkova, Y.; Skopin, A.; Zimina, O.; Zenin, V.; Glushankova, L.; Kaznacheyeva, E.V. Both Orai1 and TRPC1 are involved in excessive store-operated calcium entry in striatal neurons expressing mutant Huntingtin exon 1. Front. Physiol. 2015, 6, 337. [Google Scholar] [CrossRef]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Pathological Mechanism | STIM Expression | Role of STIM | Effect | References |
|---|---|---|---|---|---|
| TBI | Glutamate toxicity mediated by mGluR | STIM1 is overexpressed | - Increasing SOCE-mediated Ca2+ influx - Contributing to Ca2+ overload | toxic (blockade of STIM expression -> protective) | [82,83] |
| STIM2 is overexpressed | [84] | ||||
| Cerebral ischemia | Glutamate toxicity mediated by NMDAR | STIM1 is overexpressed | - Increasing SOCE-mediated Ca2+ influx - Contributing to Ca2+ overload | toxic (blockade of STIM expression -> protective) | [87] |
| STIM2 is overexpressed | [45] | ||||
| AD | Glutamate toxicity mediated by mGluR and NMDAR | STIM1 is downregulated | - Reducing SOCE-mediated Ca2+ influx - Stabilizes dendritic spines | neuroprotective | [101] |
| STIM2 is downregulated | [52,94] | ||||
| HD | Glutamate toxicity mediated by mGluR and NMDAR | STIM1 is overexpressed | - Increasing SOCE-mediated Ca2+ influx - Contributing to Ca2+ overload - Disrupting dendritic spines | toxic (blockade of STIM expression -> protective) | [112,116] |
| STIM2 is overexpressed | [106,112] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serwach, K.; Gruszczynska-Biegala, J. STIM Proteins and Glutamate Receptors in Neurons: Role in Neuronal Physiology and Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092289
Serwach K, Gruszczynska-Biegala J. STIM Proteins and Glutamate Receptors in Neurons: Role in Neuronal Physiology and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(9):2289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092289
Chicago/Turabian StyleSerwach, Karolina, and Joanna Gruszczynska-Biegala. 2019. "STIM Proteins and Glutamate Receptors in Neurons: Role in Neuronal Physiology and Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 9: 2289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092289