Dual Targeting of Stromal Cell Support and Leukemic Cell Growth by a Peptidic PKC Inhibitor Shows Effectiveness against B-ALL

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
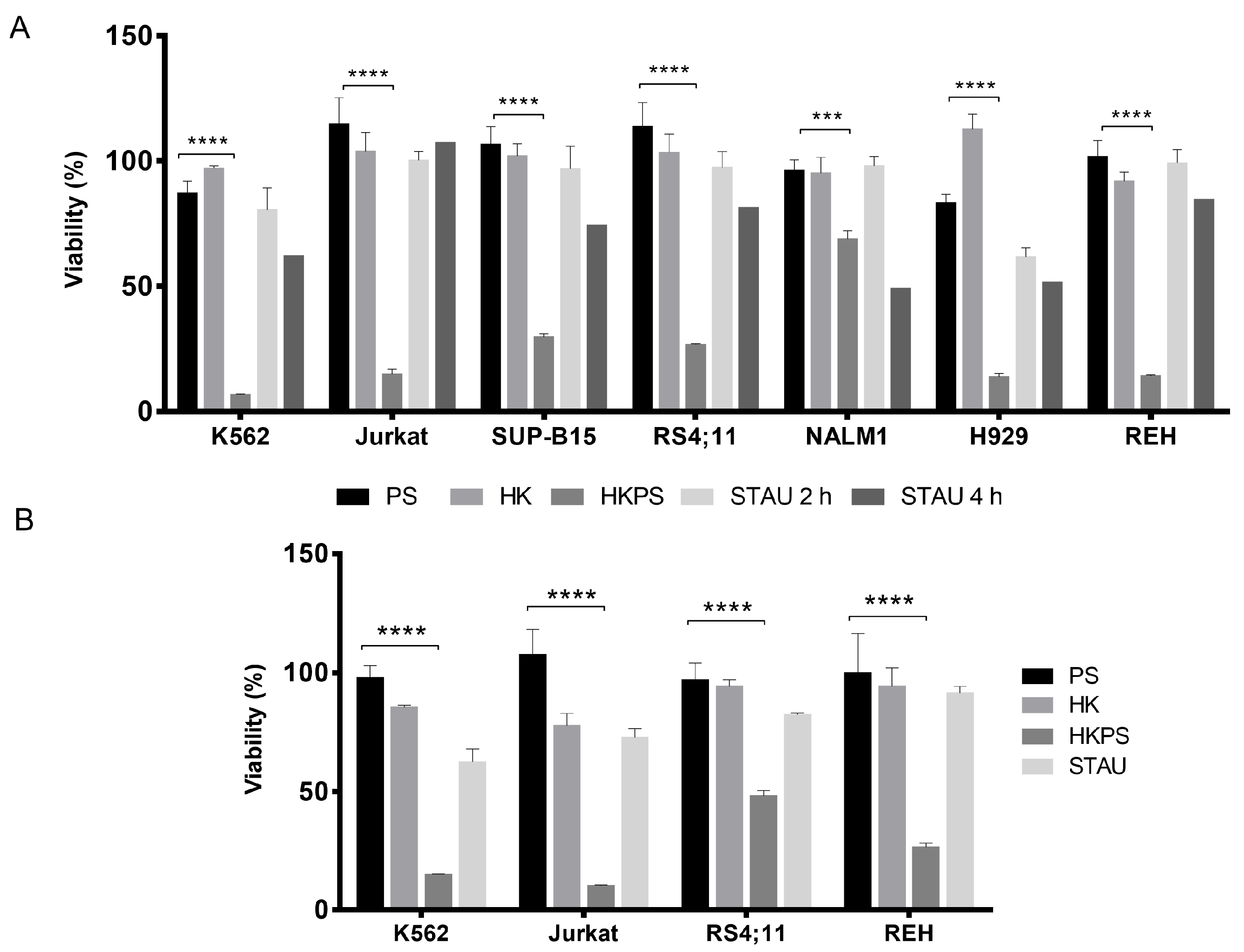
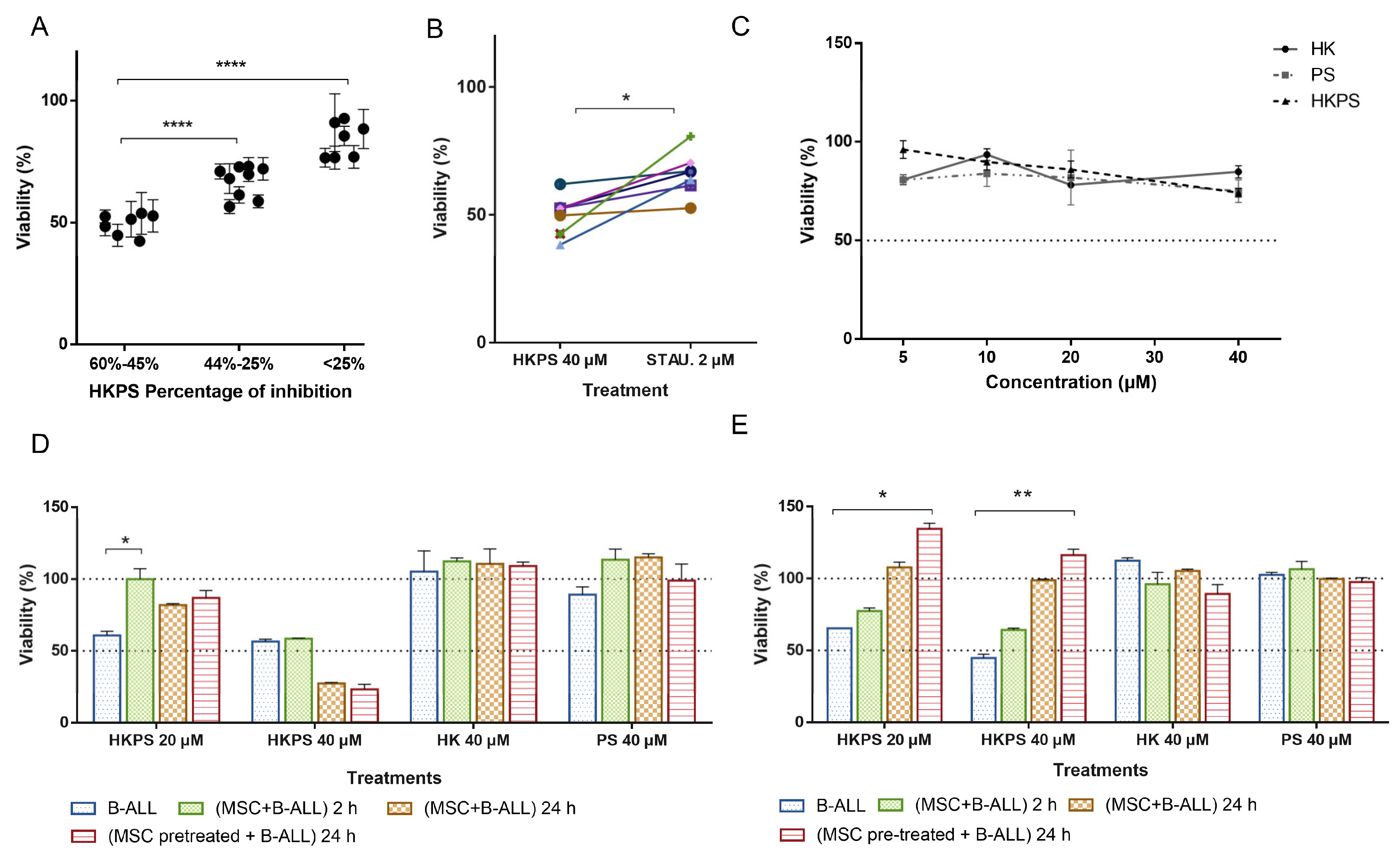
2.1. Cell Growth Inhibition of Leukemic Cell Lines by HKPS
2.2. Cell Growth Inhibition of Leukemic Cells from B-ALL Patients by HKPS
2.3. Cell Growth Inhibition of B-ALL Cells by HKPS in a Co-Culture System with MSC
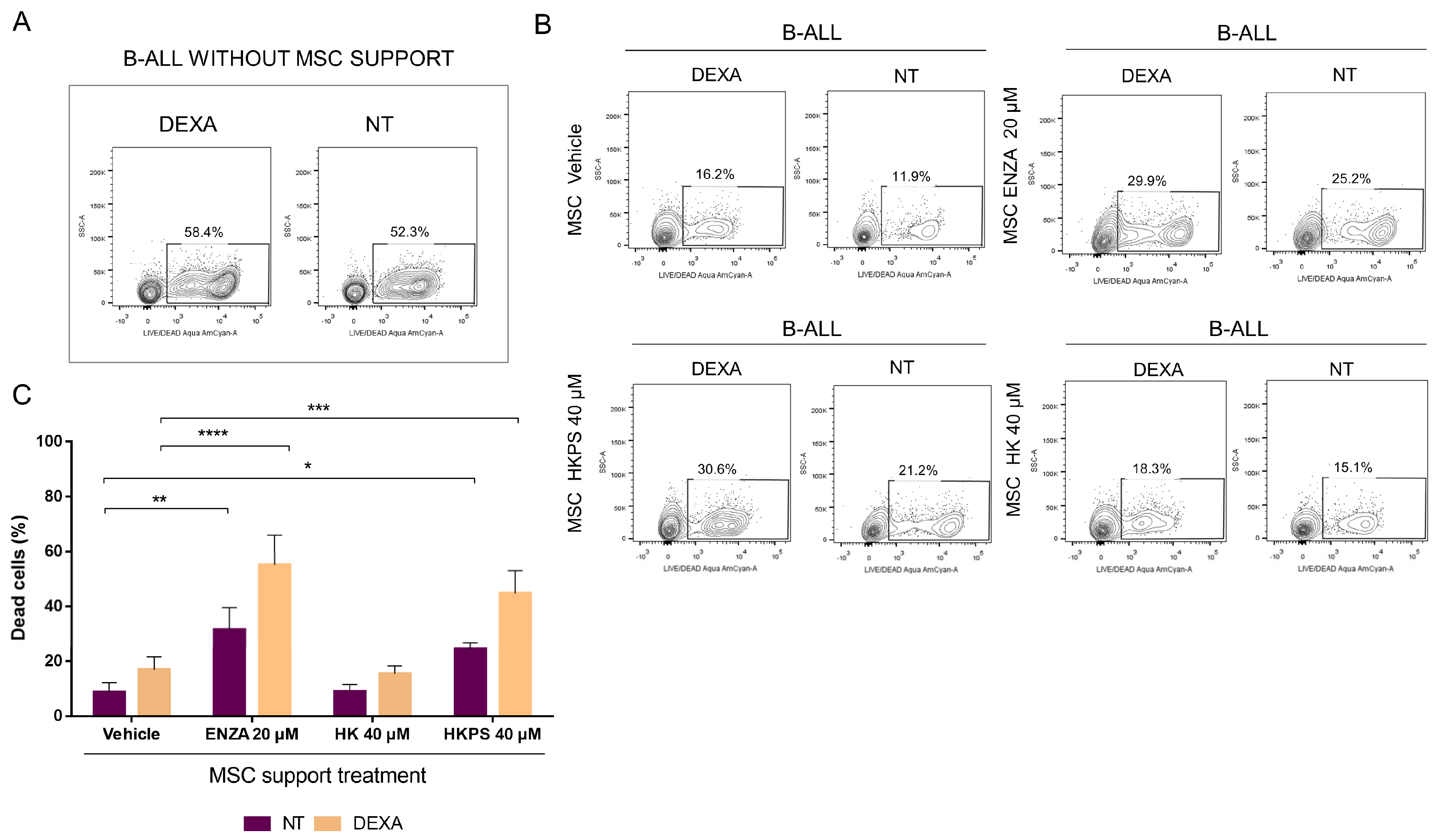
2.4. Differential Cytotoxicity Evaluation of HKPS in MSC and Leukemic Cells
2.5. Role of PKC Inhibition in Cell Adhesion between MSC and B-ALL Cells
2.6. Role of Soluble Factors and Cell–Cell Interactions in MSC Protection
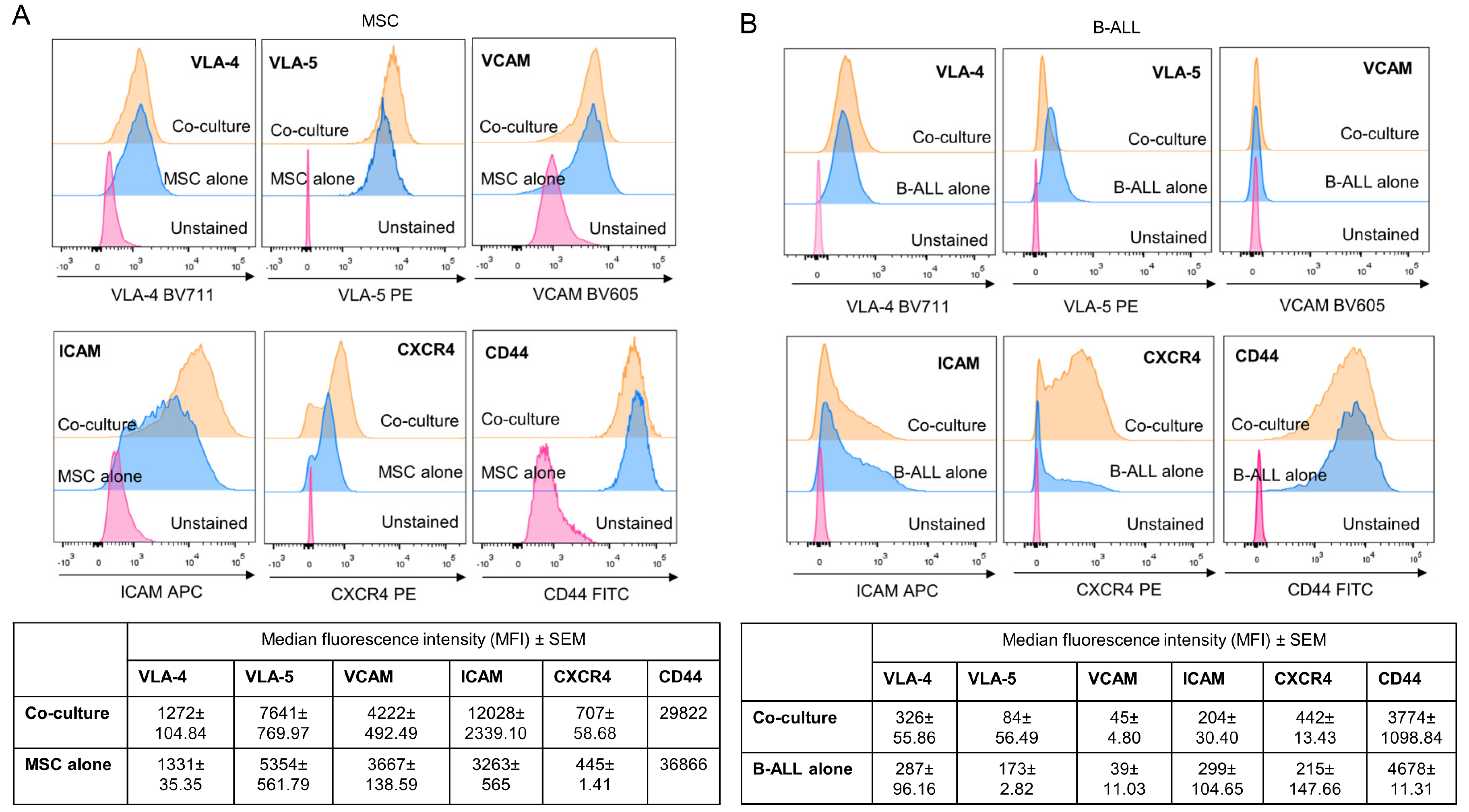
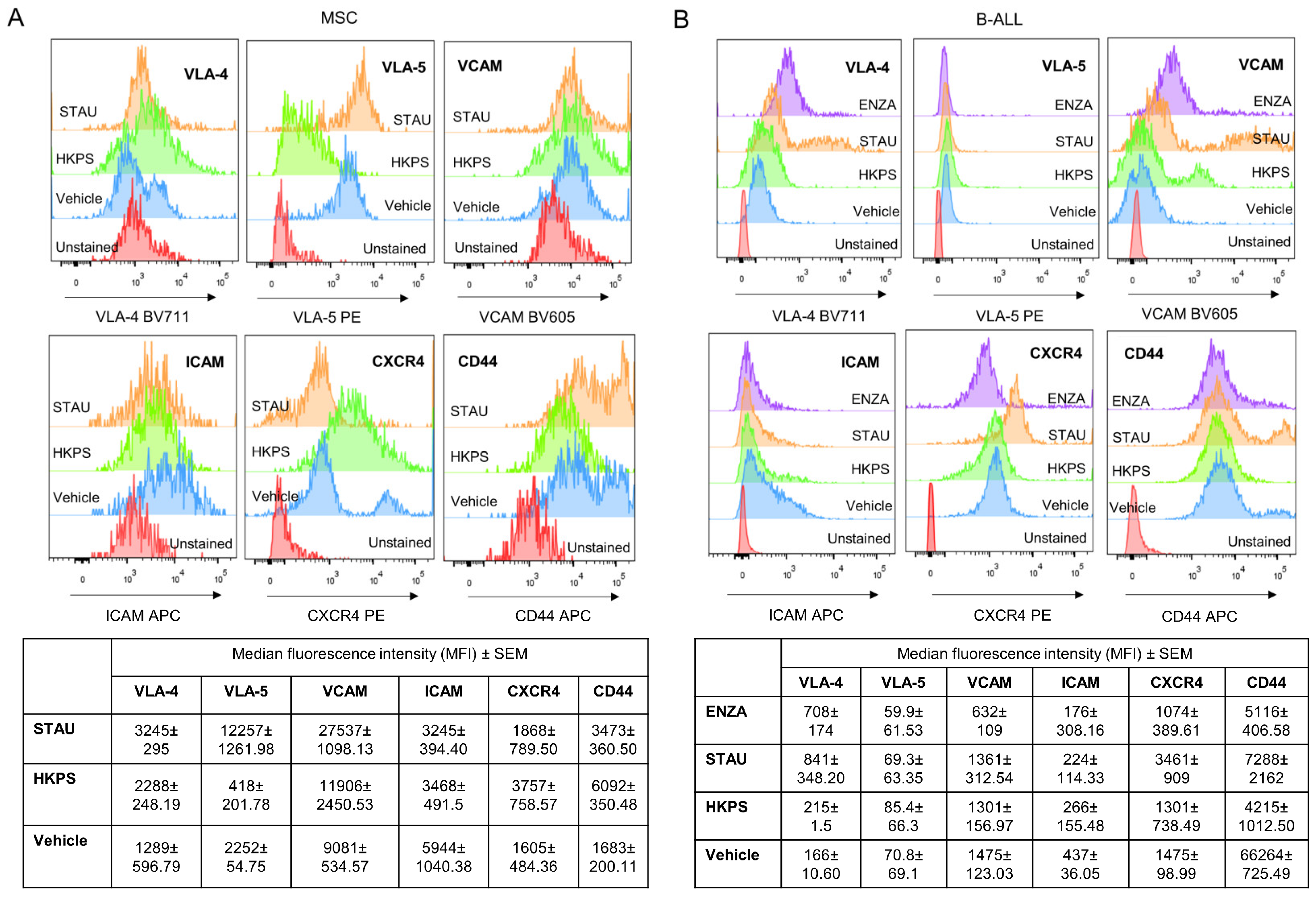
2.7. Adhesion Molecules Involved in MSC and Leukemic Cell Interactions
2.8. HKPS Sensitization of MSC to Dexamethasone Treatment
3. Discussion
4. Materials and Methods
4.1. BM-MSC Isolation (Stromal Cell Support)
4.2. Immunophenotypic and Multipotent Differentiation Capacity in BM-MSC
4.3. Immortalized Leukemic Cell Lines
4.4. Patient B-ALL Cells Isolation and Characterization
4.5. Establishment of the Co-Cultures of Leukemic Cells with MSC
4.6. Peptide Synthesis and Characterization
4.7. Cytotoxicity of Leukemic Cells Induced by PKC Inhibitors
4.8. Differential Cytotoxicity in MSC and Leukemic Cells Determined by Flow Cytometry
4.9. Functional Cell Adhesion Assay of Leukemic Cells
4.10. Contribution of Soluble Factors vs. Direct Cell Contact to the MSC Support
4.10.1. Conditioned Media Evaluation
4.10.2. TW Assay Evaluation
4.11. Expression of Adhesion Molecules in MSC and B-ALL Cells
4.12. Treatment with Cytotoxic Agents
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MSC | Mesenchymal stem cells |
| PKC | Protein kinase C |
| B-ALL | B-cell acute lymphoblastic leukaemia |
| CLL | Chronic lymphoid leukaemia |
| AML | Acute myeloid leukaemia |
| BM | Bone Marrow |
| MNC | Mononuclear cells |
| FBS | Fetal bovine serum |
| STAU | Staurosporine |
| ENZA | Enzastaurine |
| DEXA | Dexamethasone |
| NT | Non treated |
| TW | Transwell |
| MRD | Minimal residual disease |
| VCAM-1 | Vascular cell-adhesion molecule-1 |
| VLA | Very late antigen |
References
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabe, Y.; Konopleva, M. Advances in understanding the leukaemia microenvironment. Br. J. Haematol. 2014, 164, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, I.-H.; Jeong, S.-Y.; Kim, J.-A. Normal and leukemic stem cell niche interactions. Curr. Opin. Hematol. 2019, 26, 249–257. [Google Scholar] [CrossRef]
- Vernot, J.-P.; Bonilla, X.; Rodriguez-Pardo, V.; Vanegas, N.-D. Phenotypic and Functional Alterations of Hematopoietic Stem and Progenitor Cells in an In Vitro Leukemia-Induced Microenvironment. Int. J. Mol. Sci. 2017, 18, 199. [Google Scholar] [CrossRef] [Green Version]
- Makrynikola, V.; Bradstock, K.F. Adhesion of precursor-B acute lymphoblastic leukaemia cells to bone marrow stromal proteins. Leukemia 1993, 7, 86–92. [Google Scholar]
- Manabe, A.; Yi, T.; Kumagai, M.A.; Campana, D. Use of stroma-supported cultures of leukemic cells to assess antileukemic drugs. I. Cytotoxicity of interferon alpha in acute lymphoblastic leukemia. Leukemia 1993, 7, 1990–1995. [Google Scholar]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B- lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar] [CrossRef] [Green Version]
- Bendall, L.J.; Daniel, A.; Kortlepel, K.; Gottlieb, D.J. Bone marrow adherent layers inhibit apoptosis of acute myeloid leukemia cells. Exp. Hematol. 1994, 22, 1252–1260. [Google Scholar]
- Panayiotidis, P.; Jones, D.; Ganeshaguru, K.; Foroni, L.; Hoffbrand, A.V. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br. J. Haematol. 1996, 92, 97–103. [Google Scholar] [CrossRef]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Jacamo, R.; Chen, Y.; Wang, Z.; Wencai, M.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, D.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filshie, R.; Gottlieb, D.; Bradstock, K. VLA-4 is involved in the engraftment of the human pre-B acute lymphoblastic leukaemia cell line NALM-6 in SCID mice. Br. J. Haematol. 1998, 102, 1292–1300. [Google Scholar] [CrossRef]
- Liu, C.-C.; Leclair, P.; Yap, S.Q.; Lim, C.J. The Membrane-Proximal KXGFFKR Motif of -Integrin Mediates Chemoresistance. Mol. Cell. Biol. 2013, 33, 4334–4345. [Google Scholar] [CrossRef] [Green Version]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Dürig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein Kinase C-β-Dependent Activation of NF-κB in Stromal Cells Is Indispensable for the Survival of Chronic Lymphocytic Leukemia B Cells In Vivo. Cancer Cell 2013, 23, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Altman, A.; Kong, K.-F. Protein Kinase C Enzymes in the Hematopoietic and Immune Systems. Annu. Rev. Immunol. 2016, 34, 511–538. [Google Scholar] [CrossRef]
- Komada, F.; Nishikawa, M.; Uemura, Y.; Morita, K.; Hidaka, H.; Shirakawa, S. Expression of Three Major Protein Kinase C Isozymes in Various Types of Human Leukemic Cells. Cancer Res. 1991, 51, 4271–4278. [Google Scholar]
- Jiffar, T.; Kurinna, S.; Suck, G.; Carlson-Bremer, D.; Ricciardi, M.R.; Konopleva, M.; Andreeff, M.; Ruvolo, P.P. PKC α mediates chemoresistance in acute lymphoblastic leukemia through effects on Bcl2 phosphorylation. Leukemia 2004, 18, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Li, Q.; Gao, Y.; Zhao, L.; Liu, Y. Increased PKCα activity by Rack1 overexpression is responsible for chemotherapy resistance in T-cell acute lymphoblastic leukemia-derived cell line. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Abrams, S.T.; Lakum, T.; Lin, K.; Jones, G.M.; Treweeke, A.T.; Farahani, M.; Hughes, M.; Zuzel, M.; Slupsky, J.R. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase CβII. Blood 2007, 109, 1193–1201. [Google Scholar] [CrossRef] [Green Version]
- Takami, M.; Katayama, K.; Noguchi, K.; Sugimoto, Y. Protein kinase C alpha-mediated phosphorylation of PIM-1L promotes the survival and proliferation of acute myeloid leukemia cells. Biochem. Biophys. Res. Commun. 2018, 503, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, R.; Vukovic, M.; Tarafdar, A.; Cosimo, E.; Dunn, K.; McCaig, A.M.; Holroyd, A.; McClanahan, F.; Ramsay, A.G.; Gribben, J.G.; et al. Generation of a poor prognostic chronic lymphocytic leukemia-like disease model: PKCα subversion induces up-regulation of PKCβII expression in B lymphocytes. Haematologica 2015, 100, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, R.; Soh, J.W.; Michie, A.M. Subversion of protein kinase Cα signaling in hematopoietic progenitor cells results in the generation of a B-cell chronic lymphocytic leukemia-like population in vivo. Cancer Res. 2006, 66, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redig, A.J.; Platanias, L.C. Protein kinase C signalling in leukemia. Leuk. Lymphoma 2008, 49, 1255–1262. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [Green Version]
- Tarafdar, A.; Michie, A.M. Protein kinase C in cellular transformation: A valid target for therapy? Biochem. Soc. Trans. 2014, 42, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, M.; Yao, S.; Lint, Y.Z. Controlling epidermal growth factor (EGF)-stimulated Ras activation in intact cells by a cell-permeable peptide mimicking phosphorylated EGF receptor. J. Biol. Chem. 1996, 271, 27456–27461. [Google Scholar] [CrossRef] [Green Version]
- Perdomo-Arciniegas, A.M.; Patarroyo, M.E.; Vernot, J.-P. Novel Chimeric Peptide Inhibits Protein Kinase C and Induces Apoptosis in Human Immune Cells. Int. J. Pept. Res. Ther. 2008, 14, 64–74. [Google Scholar] [CrossRef]
- Faul, M.M.; Gillig, J.R.; Jirousek, M.R.; Ballas, L.M.; Schotten, T.; Kahl, A.; Mohr, M. Acyclic N-(azacycloalkyl)bisindolylmaleimides: Isozyme selective inhibitors of PKCβ. Bioorganic Med. Chem. Lett. 2003, 13, 1857–1859. [Google Scholar] [CrossRef]
- Ebinger, S.; Özdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Alves, C.C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Möricke, A.; Reiter, A.; Zimmermann, M.; Gadner, H.; Stanulla, M.; Dördelmann, M.; Löning, L.; Beier, R.; Ludwig, W.D.; Ratei, R.; et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: Treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 2008, 111, 4477–4489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudry, M.E.; Fortney, J.E.; York, T.; Hall, B.M.; Gibson, L.F. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood 2000, 96, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [Green Version]
- Bailey, L.C.; Lange, B.J.; Rheingold, S.R.; Bunin, N.J. Bone-marrow relapse in paediatric acute lymphoblastic leukaemia. Lancet Oncol. 2008, 9, 873–883. [Google Scholar] [CrossRef]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef]
- Meggio, F.; Deana, A.D.; Ruzzene, M.; Brunati, A.M.; Cesaro, L.; Guerra, B.; Meyer, T.; Mett, H.; Fabbro, D.; Furet, P.; et al. Different Susceptibility of Protein Kinases to Staurosporine Inhibition: Kinetic Studies and Molecular Bases for the Resistance of Protein Kinase CK2. Eur. J. Biochem. 1995, 234, 317–322. [Google Scholar] [CrossRef]
- ŌMura, S.; Sasaki, Y.; Iwai, Y.; Takeshima, H. Staurosporine, a Potentially Important Gift from a Microorganism. J. Antibiot. 1995, 48, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T.; Rennert, P. NF-κB activation by protein kinase C isoforms and B-cell function. EMBO Rep. 2003, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Shishido, S.; Bönig, H.; Kim, Y.M. Role of integrin alpha4 in drug resistance of leukemia. Front. Oncol. 2014, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Slayton, W.B. Integrin VLA-5 and FAK are good targets to improve treatment response in the Philadelphia chromosome positive acute lymphoblastic leukemia. Front. Oncol. 2014, 4, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradstock, K.F.; Gottlieb, D.J. Interaction of acute leukemia cells with the bone marrow microenvironment: Implications for control of minimal residual disease. Leuk. Lymphoma 1995, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bogetto, L.; Gabriele, E.; Cariati, R.; Dolcetti, R.; Spessotto, P.; Doglioni, C.; Boiocchi, M.; Perris, R.; Colombatti, A. Bidirectional induction of the cognate receptor-ligand alpha4/VCAM-1 pair defines a novel mechanism of tumor intravasation. Blood 2000, 95, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Moses, B.S.; Slone, W.L.; Thomas, P.; Evans, R.; Piktel, D.; Angel, P.M.; Walsh, C.M.; Cantrell, P.S.; Rellick, S.L.; Martin, K.H.; et al. Bone marrow microenvironment modulation of acute lymphoblastic leukemia phenotype. Exp. Hematol. 2016, 44, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasa, M.; Miura, Y.; Fujishiro, A.; Fujii, S.; Sugino, N.; Yoshioka, S.; Yokota, A.; Hishita, T.; Hirai, H.; Andoh, A.; et al. Bortezomib interferes with adhesion of B cell precursor acute lymphoblastic leukemia cells through SPARC up-regulation in human bone marrow mesenchymal stromal/stem cells. Int. J. Hematol. 2017, 105, 1–11. [Google Scholar] [CrossRef]
- Hart, C.; Drewel, D.; Mueller, G.; Grassinger, J.; Zaiss, M.; Kunz-Schughart, L.A.; Andreesen, R.; Reichle, A.; Holler, E.; Hennemann, B. Expression and Function of Homing-Essential Molecules and Enhanced In Vivo Homing Ability of Human Peripheral Blood-Derived Hematopoietic Progenitor Cells after Stimulation with Stem Cell Factor. Stem Cells 2004, 22, 580–589. [Google Scholar] [CrossRef]
- Barwe, S.P.; Quagliano, A.; Gopalakrishnapillai, A. Eviction from the sanctuary: Development of targeted therapy against cell adhesion molecules in acute lymphoblastic leukemia. Semin. Oncol. 2017, 44, 101–112. [Google Scholar] [CrossRef]
- Liu, Y.F.; Zhang, S.Y.; Chen, Y.Y.; Shi, K.; Zou, B.; Liu, J.; Yang, Q.; Jiang, H.; Wei, L.; Li, C.Z.; et al. ICAM-1 Deficiency in the Bone Marrow Niche Impairs Quiescence and Repopulation of Hematopoietic Stem Cells. Stem Cell Rep. 2018, 11, 258–273. [Google Scholar] [CrossRef]
- Leung, K.T.; Zhang, C.; Chan, K.Y.Y.; Li, K.; Cheung, J.T.K.; Ng, M.H.L.; Zhang, X.B.; Sit, T.; Lee, W.Y.W.; Kang, W.; et al. CD9 blockade suppresses disease progression of high-risk pediatric B-cell precursor acute lymphoblastic leukemia and enhances chemosensitivity. Leukemia 2020, 34, 709–720. [Google Scholar] [CrossRef]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: Development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef]
- Burger, J.A.; Burger, M.; Kipps, T.J. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood 1999, 94, 3658–3667. [Google Scholar] [CrossRef]
- Bonilla, X.; Vanegas, N.D.P.; Vernot, J.P. Acute leukemia induces senescence and impaired osteogenic differentiation in mesenchymal stem cells endowing leukemic cells with functional advantages. Stem Cells Int. 2019, 2019, 3864948. [Google Scholar] [CrossRef]
- Amigo-Jiménez, I.; Bailón, E.; Aguilera-Montilla, N.; Terol, M.J.; García-Marco, J.A.; García-Pardo, A. Bone marrow stroma-induced resistance of chronic lymphocytic leukemia cells to arsenic trioxide involves Mcl-1 upregulation and is overcome by inhibiting the PI3Kδ or PKCβ signaling pathways. Oncotarget 2015, 6, 44832–44848. [Google Scholar] [CrossRef]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2008, 22, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Mraz, M.; Zent, C.S.; Church, A.K.; Jelinek, D.F.; Wu, X.; Pospisilova, S.; Ansell, S.M.; Novak, A.J.; Kay, N.E.; Witzig, T.E.; et al. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin α-4-β-1 (VLA-4) with natalizumab can overcome this resistance. Br. J. Haematol. 2011, 155, 53–64. [Google Scholar] [CrossRef]
- Redondo-Muñoz, J.; García-Pardo, A.; Teixidó, J. Molecular players in hematologic tumor cell trafficking. Front. Immunol. 2019, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmanns, C.; Fan, D.; O’Brian, C.A.; Bucana, C.D.; Fidler, I.J. Orthotopic and ectopic organ environments differentially influence the sensitivity of murine colon carcinoma cells to doxorubicin and 5-fluorouracil. Int. J. Cancer 1992, 52, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Pontiggia, O.; Sampayo, R.; Raffo, D.; Motter, A.; Xu, R.; Bissell, M.J.; De Kier Joffé, E.B.; Simian, M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: A role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res. Treat. 2012, 133, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336. [Google Scholar] [CrossRef] [Green Version]
- Staroselsky, A.N.; Fan, D.; O’Brian, C.A.; Bucana, C.D.; Gupta, K.P.; Fidler, I.J. Site-dependent differences in response of the UV-2237 murine fibrosarcoma to systemic therapy with adriamycin. Cancer Res. 1990, 50, 7775–7780. [Google Scholar] [PubMed]
- World Medical Association declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [CrossRef] [PubMed] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Amblard, M.; Fehrentz, J.A.; Martinez, J.; Subra, G. Methods and protocols of modern solid phase peptide synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]
- Pears, C.J.; Parker, P.J. Domain interactions in protein kinase C. J. Cell Sci. 1991, 100, 683–686. [Google Scholar] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Aparicio, P.F.; Vanegas, N.-D.P.; Uribe, G.I.; Ortiz-Montero, P.; Cadavid-Cortés, C.; Lagos, J.; Flechas-Afanador, J.; Linares-Ballesteros, A.; Vernot, J.-P. Dual Targeting of Stromal Cell Support and Leukemic Cell Growth by a Peptidic PKC Inhibitor Shows Effectiveness against B-ALL. Int. J. Mol. Sci. 2020, 21, 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103705
Ruiz-Aparicio PF, Vanegas N-DP, Uribe GI, Ortiz-Montero P, Cadavid-Cortés C, Lagos J, Flechas-Afanador J, Linares-Ballesteros A, Vernot J-P. Dual Targeting of Stromal Cell Support and Leukemic Cell Growth by a Peptidic PKC Inhibitor Shows Effectiveness against B-ALL. International Journal of Molecular Sciences. 2020; 21(10):3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103705
Chicago/Turabian StyleRuiz-Aparicio, Paola Fernanda, Natalia-Del Pilar Vanegas, Gloria Inés Uribe, Paola Ortiz-Montero, Camila Cadavid-Cortés, Jimmy Lagos, Jessica Flechas-Afanador, Adriana Linares-Ballesteros, and Jean-Paul Vernot. 2020. "Dual Targeting of Stromal Cell Support and Leukemic Cell Growth by a Peptidic PKC Inhibitor Shows Effectiveness against B-ALL" International Journal of Molecular Sciences 21, no. 10: 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103705