Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia



Abstract
:1. Introduction
2. Pathology of Parkinson’s Disease and Dopa-Responsive Dystonia
3. Physiology of Tyrosine Hydroxylase Phosphorylation
4. Linkage of Tyrosine Hydroxylase Phosphorylation to Dopaminergic Pathology
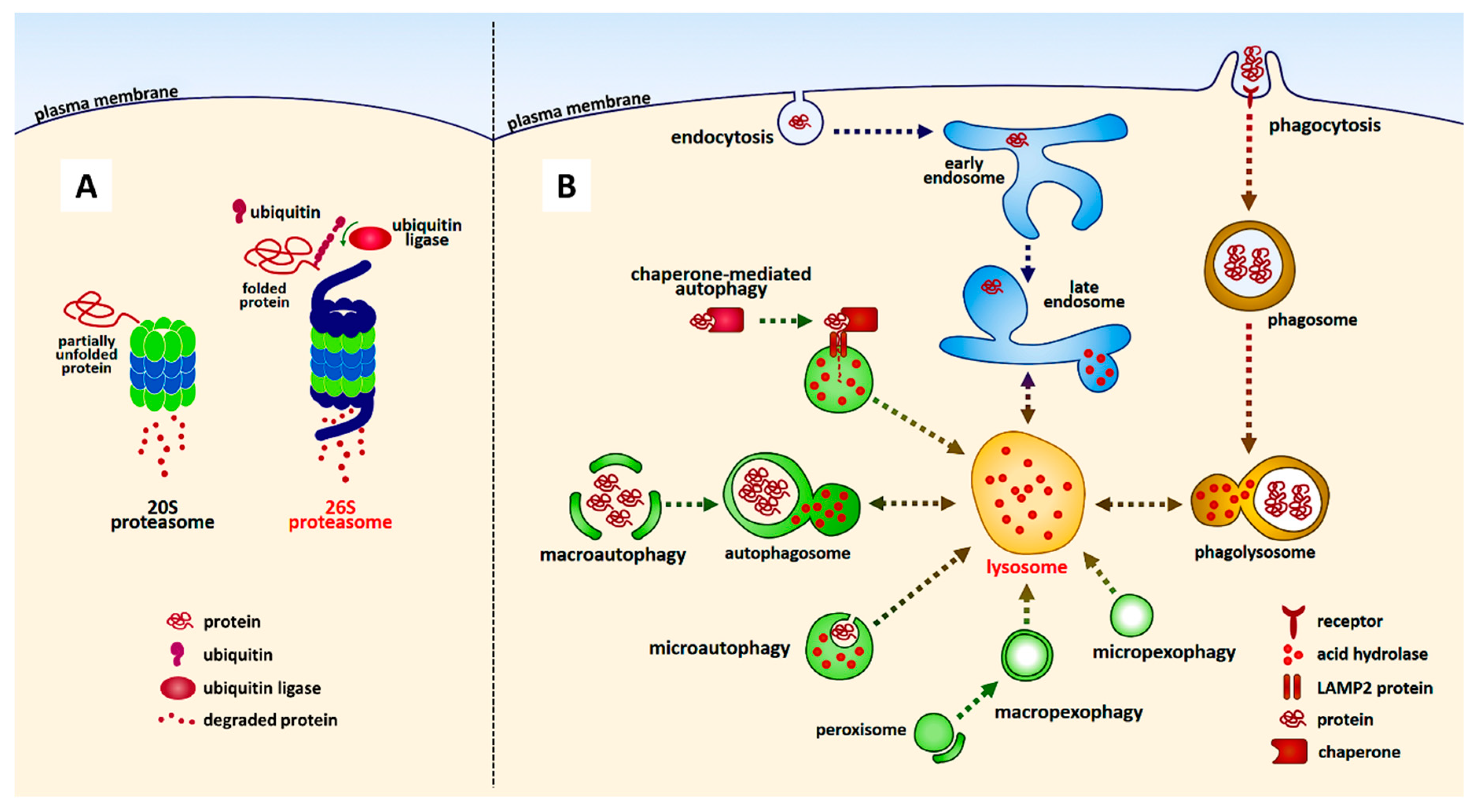
5. Protein Degradation Pathways: Lysosome and Proteasome
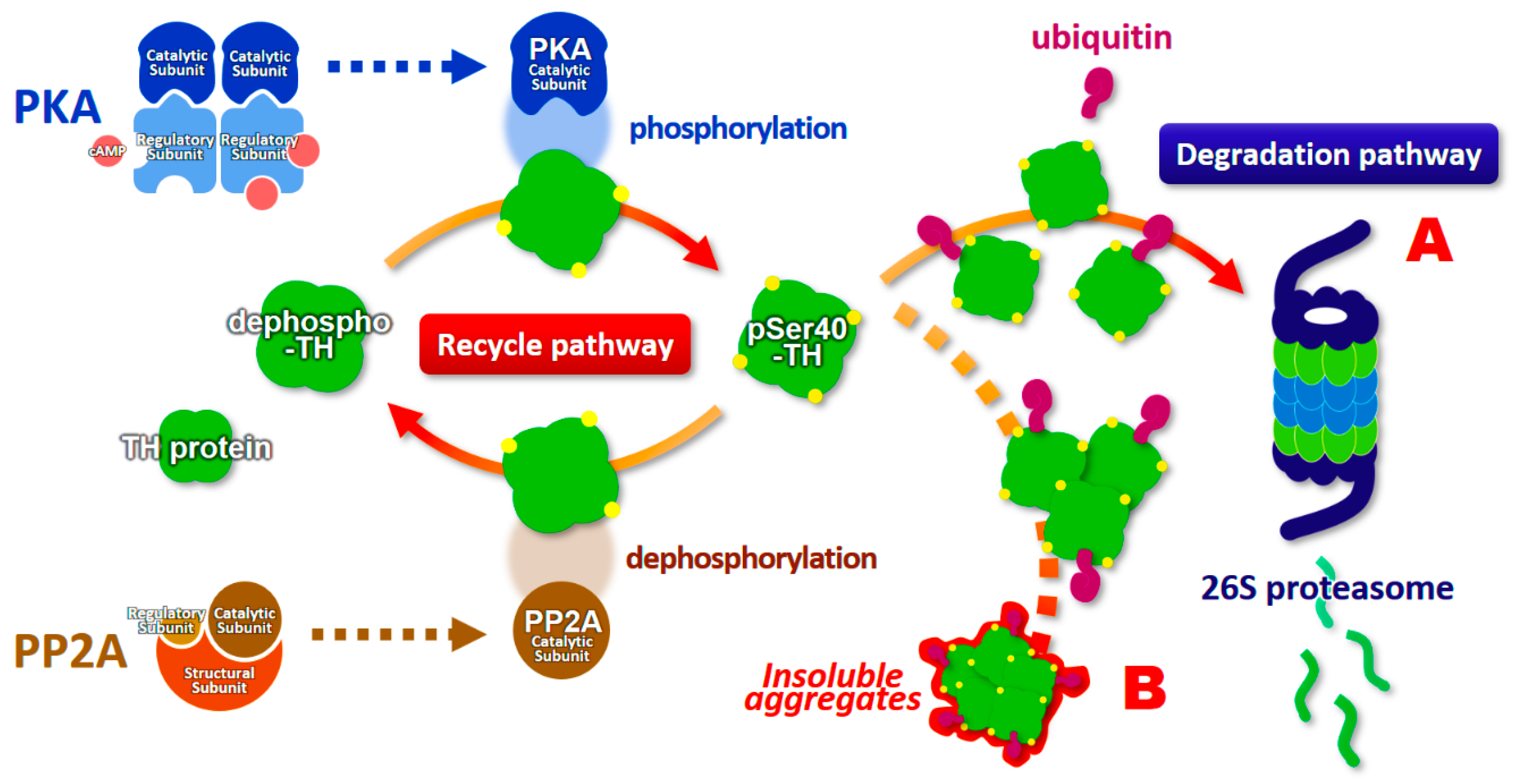
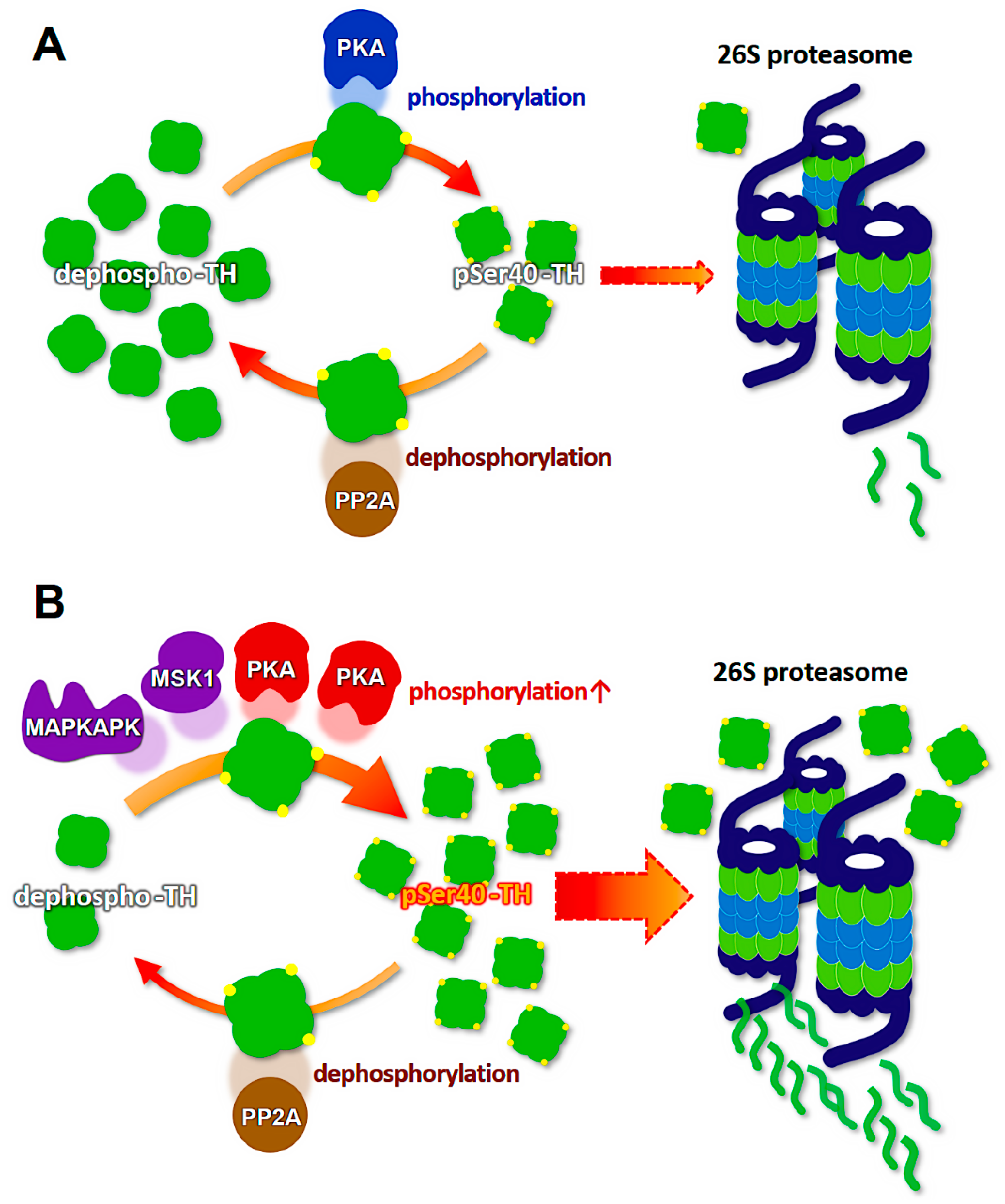
6. Ubiquitination and Proteasomal Degradation of Phosphorylated Tyrosine Hydroxylase
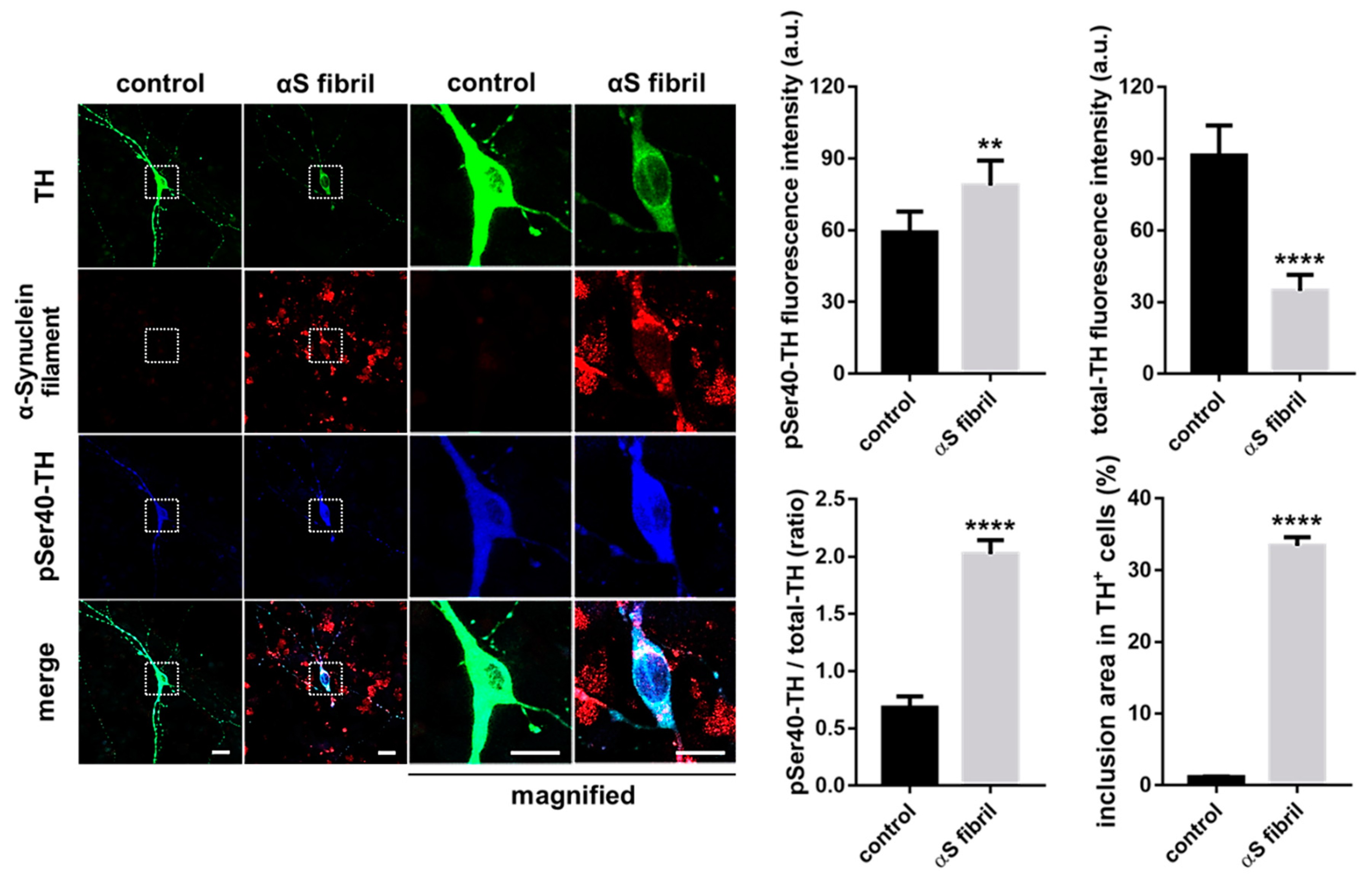
7. Modification of Tyrosine Hydroxylase Phosphorylation by α-Synuclein
8. Novel Therapeutic Targets for α-Synuclein Propagation
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AP-1 | Activator protein 1 |
| CaMKII | Calcium/calmodulin-dependent protein kinase II |
| cAMP | Cyclic adenosine monophosphate |
| CMA | Chaperone-mediated autophagy |
| CSF | Cerebrospinal fluid |
| DLB | Dementia with Lewy bodies |
| DRD | Dopa-responsive dystonia (Segawa disease) |
| ERK | Extracellular signal-regulated kinase |
| FABP | Fatty acid-binding protein |
| GTP | guanosine triphosphate |
| GCH1 | GTP cyclohydrolase 1 |
| Hsc70 | Heat shock cognate protein of 70 kDa |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| LAMP | Lysosome-associated membrane protein |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAPK | mitogen-activated protein kinase |
| MAPKAPK | Mitogen-activated protein kinase activated protein kinase |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSK1 | Mitogen- and stress-activated kinase 1 |
| NGF | Nerve growth factor |
| Nurr1 | Nuclear receptor related-1 |
| PD | Parkinson’s disease |
| PP2A | Protein phosphatase 2a |
| pSer40-TH | Tyrosine hydroxylase phosphorylated at Ser40 |
| SNpc | Substantia nigra pars compacta |
| SRF | Serum-responsive factor |
| TH | Tyrosine hydroxylase |
| VTA | Ventral tegmental area |
References
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Segawa, M. Dopa-responsive dystonia. Handb. Clin. Neurol. 2011, 100, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Nygaard, T.G.; Gutlich, M.; Rajput, A.H.; Pifl, C.; DiStefano, L.; Chang, L.J.; Price, K.; Shimadzu, M.; Hornykiewicz, O.; et al. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology 1999, 53, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kapatos, G.; Haycock, J.W.; Worsley, J.; Wong, H.; Kish, S.J.; Nygaard, T.G. Brain biopterin and tyrosine hydroxylase in asymptomatic dopa-responsive dystonia. Ann. Neurol. 2002, 51, 637–641. [Google Scholar] [CrossRef]
- Leckman, J.F.; Bloch, M.H.; Smith, M.E.; Larabi, D.; Hampson, M. Neurobiological substrates of Tourette’s disorder. J. Child Adolesc. Psychopharmacol. 2010, 20, 237–247. [Google Scholar] [CrossRef]
- Carlsson, M.; Svensson, A. Interfering with glutamatergic neurotransmission by means of NMDA antagonist administration discloses the locomotor stimulatory potential of other transmitter systems. Pharmacol. Biochem. Behav. 1990, 36, 45–50. [Google Scholar] [CrossRef]
- David, C.; Ewert, M.; Seeburg, P.H.; Fuchs, S. Antipeptide antibodies differentiate between long and short isoforms of the D2 dopamine receptor. Biochem. Biophys. Res. Commun. 1991, 179, 824–829. [Google Scholar] [CrossRef]
- Cooper, O.; Greenman, Y. Dopamine Agonists for Pituitary Adenomas. Front. Endocrinol. (Lausanne) 2018, 9, 469. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A. Extrapyramidal system, motor Ganglia and movement disorders. Rev. Neurosci. 1990, 2, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.A. Synergistic interactions of D1- and D2-selective dopamine agonists in animal models for Parkinson’s disease: Sites of action and implications for the pathogenesis of dyskinesias. Can. J. Neurol. Sci. 1992, 19, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haavik, J.; Toska, K. Tyrosine hydroxylase and Parkinson’s disease. Mol. Neurobiol. 1998, 16, 285–309. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Hoefer, P.F.A. Action Potentials of Muscles in Rigidity and Tremor. Arch. Neurol. Psychiatry 1940, 43, 704–725. [Google Scholar] [CrossRef]
- Muller, K.; Homberg, V.; Lenard, H.G. Motor control in childhood onset dopa-responsive dystonia (Segawa syndrome). Neuropediatrics 1989, 20, 185–191. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Parkinsons Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Matsumine, H.; Asakawa, S.; Kitada, T.; Yoshino, H.; Elibol, B.; Brookes, A.J.; Yamamura, Y.; Kobayashi, T.; Wang, M.; et al. Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene. Biochem. Biophys. Res. Commun. 1998, 249, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Ohta, E.; Hasegawa, K.; Gasser, T.; Obata, F. Independent occurrence of I2020T mutation in the kinase domain of the leucine rich repeat kinase 2 gene in Japanese and German Parkinson’s disease families. Neurosci. Lett. 2007, 417, 21–23. [Google Scholar] [CrossRef]
- Funayama, M.; Hasegawa, K.; Ohta, E.; Kawashima, N.; Komiyama, M.; Kowa, H.; Tsuji, S.; Obata, F. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann. Neurol. 2005, 57, 918–921. [Google Scholar] [CrossRef]
- Balint, B.; Mencacci, N.E.; Valente, E.M.; Pisani, A.; Rothwell, J.; Jankovic, J.; Vidailhet, M.; Bhatia, K.P. Dystonia. Nat. Rev. Dis. Primers 2018, 4, 25. [Google Scholar] [CrossRef]
- Camargo, C.H.F.; Camargos, S.T.; Cardoso, F.E.C.; Teive, H.A.G. The genetics of the dystonias a review based on the new classification of the dystonias. Arq. Neuropsiquiatr. 2015, 73, 350–358. [Google Scholar] [CrossRef]
- Ichinose, H.; Ohye, T.; Takahashi, E.; Seki, N.; Hori, T.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S.; et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994, 8, 236–242. [Google Scholar] [CrossRef]
- Ceravolo, R.; Nicoletti, V.; Garavaglia, B.; Reale, C.; Kiferle, L.; Bonuccelli, U. Expanding the clinical phenotype of DYT5 mutations: Is multiple system atrophy a possible one? Neurology 2013, 81, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Pacheco, O.; Oyama, G.; Briton, A.; Singleton, A.B.; Fernandez, H.H.; Rodriguez, R.L.; Malaty, I.A.; Okun, M.S. A Novel DYT-5 Mutation with Phenotypic Variability within a Colombian Family. Tremor Other Hyperkinet Mov. (N Y) 2013, 3. [Google Scholar] [CrossRef]
- Shetty, A.S.; Bhatia, K.P.; Lang, A.E. Dystonia and Parkinson’s disease: What is the relationship? Neurobiol. Dis. 2019, 132, 104462. [Google Scholar] [CrossRef]
- Montague, P.R.; Hyman, S.E.; Cohen, J.D. Computational roles for dopamine in behavioural control. Nature 2004, 431, 760–767. [Google Scholar] [CrossRef]
- Schultz, W. Behavioral theories and the neurophysiology of reward. Annu. Rev. Psychol. 2006, 57, 87–115. [Google Scholar] [CrossRef] [Green Version]
- Berridge, K.C. From prediction error to incentive salience: Mesolimbic computation of reward motivation. Eur. J. Neurosci. 2012, 35, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Stern, G. The effects of lesions in the substantia nigra. Brain 1966, 89, 449–478. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine Hydroxylase. The initial step in norepinephrine biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar] [PubMed]
- Kaneda, N.; Kobayashi, K.; Ichinose, H.; Kishi, F.; Nakazawa, A.; Kurosawa, Y.; Fujita, K.; Nagatsu, T. Isolation of a novel cDNA clone for human tyrosine hydroxylase: Alternative RNA splicing produces four kinds of mRNA from a single gene. Biochem. Biophys. Res. Commun. 1987, 146, 971–975. [Google Scholar] [CrossRef]
- Grima, B.; Lamouroux, A.; Boni, C.; Julien, J.F.; Javoy-Agid, F.; Mallet, J. A single human gene encoding multiple tyrosine hydroxylases with different predicted functional characteristics. Nature 1987, 326, 707–711. [Google Scholar] [CrossRef]
- Ichinose, H.; Ohye, T.; Fujita, K.; Yoshida, M.; Ueda, S.; Nagatsu, T. Increased heterogeneity of tyrosine hydroxylase in humans. Biochem. Biophys. Res. Commun. 1993, 195, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W. Species differences in the expression of multiple tyrosine hydroxylase protein isoforms. J. Neurochem. 2002, 81, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, C.; Joh, T.H. Limited proteolysis of rat brain tyrosine hydroxylase defines an N-terminal region required for regulation of cofactor binding and directing substrate specificity. J. Mol. Neurosci. 1991, 2, 203–215. [Google Scholar] [PubMed]
- Campbell, D.G.; Hardie, D.G.; Vulliet, P.R. Identification of four phosphorylation sites in the N-terminal region of tyrosine hydroxylase. J. Biol. Chem. 1986, 261, 10489–10492. [Google Scholar]
- Goodwill, K.E.; Sabatier, C.; Marks, C.; Raag, R.; Fitzpatrick, P.F.; Stevens, R.C. Crystal structure of tyrosine hydroxylase at 2.3 A and its implications for inherited neurodegenerative diseases. Nat. Struct. Biol. 1997, 4, 578–585. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Hardie, D.G.; Vulliet, P.R. Site-specific phosphorylation of tyrosine hydroxylase after KCl depolarization and nerve growth factor treatment of PC12 cells. J. Biol. Chem. 1990, 265, 22358–22364. [Google Scholar]
- Kumer, S.C.; Vrana, K.E. Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 1996, 67, 443–462. [Google Scholar] [CrossRef]
- Chae, H.D.; Suh, B.C.; Joh, T.H.; Kim, K.T. AP1-mediated transcriptional enhancement of the rat tyrosine hydroxylase gene by muscarinic stimulation. J. Neurochem. 1996, 66, 1264–1272. [Google Scholar] [CrossRef]
- Guo, Z.; Du, X.; Iacovitti, L. Regulation of tyrosine hydroxylase gene expression during transdifferentiation of striatal neurons: Changes in transcription factors binding the AP-1 site. J. Neurosci. 1998, 18, 8163–8174. [Google Scholar] [CrossRef] [Green Version]
- Kawahata, I.; Lai, Y.; Morita, J.; Kato, S.; Ohtaku, S.; Tomioka, Y.; Tabuchi, A.; Tsuda, M.; Sumi-Ichinose, C.; Kondo, K.; et al. V-1/CP complex formation is required for genetic co-regulation of adult nigrostriatal dopaminergic function via the RHO/MAL/SRF pathway in vitro and in vivo. J. Neurol. Sci. 2017, 381, 359–360. [Google Scholar] [CrossRef]
- Kadkhodaei, B.; Ito, T.; Joodmardi, E.; Mattsson, B.; Rouillard, C.; Carta, M.; Muramatsu, S.; Sumi-Ichinose, C.; Nomura, T.; Metzger, D.; et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci. 2009, 29, 15923–15932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkley, P.R.; Bobrovskaya, L.; Graham, M.E.; Von Nagy-Felsobuki, E.I.; Dickson, P.W. Tyrosine hydroxylase phosphorylation: Regulation and consequences. J. Neurochem. 2004, 91, 1025–1043. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, P.R.; Dickson, P.W. Tyrosine hydroxylase phosphorylation in vivo. J. Neurochem. 2019, 149, 706–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, K.; Rich, D.P.; Soderling, T.R. Generation of the Ca2(+)-independent form of Ca2+/calmodulin-dependent protein kinase II in cerebellar granule cells. J. Biol. Chem. 1989, 264, 21830–21836. [Google Scholar]
- Fukunaga, K.; Miyamoto, E.; Soderling, T.R. Regulation of Ca2+/calmodulin-dependent protein kinase II by brain gangliosides. J. Neurochem. 1990, 54, 103–109. [Google Scholar] [CrossRef]
- Soderling, T.R.; Fukunaga, K.; Rich, D.P.; Fong, Y.L.; Smith, K.; Colbran, R.J. Regulation of brain Ca2+/calmodulin-dependent protein kinase II. Adv. Second Messenger Phosphoprot. Res. 1990, 24, 206–211. [Google Scholar]
- Haycock, J.W. Phosphorylation of tyrosine hydroxylase in situ at serine 8, 19, 31, and 40. J. Biol. Chem. 1990, 265, 11682–11691. [Google Scholar]
- Haycock, J.W.; Haycock, D.A. Tyrosine hydroxylase in rat brain dopaminergic nerve terminals. Multiple-site phosphorylation in vivo and in synaptosomes. J. Biol. Chem. 1991, 266, 5650–5657. [Google Scholar]
- Bobrovskaya, L.; Dunkley, P.R.; Dickson, P.W. Phosphorylation of Ser19 increases both Ser40 phosphorylation and enzyme activity of tyrosine hydroxylase in intact cells. J. Neurochem. 2004, 90, 857–864. [Google Scholar] [CrossRef]
- Daubner, S.C.; Lauriano, C.; Haycock, J.W.; Fitzpatrick, P.F. Site-directed mutagenesis of serine 40 of rat tyrosine hydroxylase. Effects of dopamine and cAMP-dependent phosphorylation on enzyme activity. J. Biol. Chem. 1992, 267, 12639–12646. [Google Scholar]
- Okuno, S.; Fujisawa, H. A new mechanism for regulation of tyrosine 3-monooxygenase by end product and cyclic AMP-dependent protein kinase. J. Biol. Chem. 1985, 260, 2633–2635. [Google Scholar] [PubMed]
- Fujisawa, H.; Okuno, S. Regulatory mechanism of tyrosine hydroxylase activity. Biochem. Biophys. Res. Commun. 2005, 338, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Royo, M.; Fitzpatrick, P.F.; Daubner, S.C. Mutation of regulatory serines of rat tyrosine hydroxylase to glutamate: Effects on enzyme stability and activity. Arch. Biochem. Biophys. 2005, 434, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Haavik, J.; Schelling, D.L.; Campbell, D.G.; Andersson, K.K.; Flatmark, T.; Cohen, P. Identification of protein phosphatase 2A as the major tyrosine hydroxylase phosphatase in adrenal medulla and corpus striatum: Evidence from the effects of okadaic acid. FEBS Lett. 1989, 251, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, C.A.; Hall, A.; Sim, A.T.; Bunn, S.J.; Marley, P.D.; Cheah, T.B.; Dunkley, P.R. Tyrosine hydroxylase phosphorylation in digitonin-permeabilized bovine adrenal chromaffin cells: The effect of protein kinase and phosphatase inhibitors on Ser19 and Ser40 phosphorylation. J. Neurochem. 1997, 69, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.B.; Sim, A.T.; Goncalves, C.A.; Dunkley, P.R. Tyrosine hydroxylase dephosphorylation by protein phosphatase 2A in bovine adrenal chromaffin cells. Neurochem. Res. 2002, 27, 207–213. [Google Scholar] [CrossRef]
- Haycock, J.W. Peptide substrates for ERK1/2: Structure-function studies of serine 31 in tyrosine hydroxylase. J. Neurosci. Methods 2002, 116, 29–34. [Google Scholar] [CrossRef]
- Ichinose, H.; Ohye, T.; Fujita, K.; Pantucek, F.; Lange, K.; Riederer, P.; Nagatsu, T. Quantification of mRNA of tyrosine hydroxylase and aromatic L-amino acid decarboxylase in the substantia nigra in Parkinson’s disease and schizophrenia. J. Neural Transm. Park. Dis. Dement. Sect. 1994, 8, 149–158. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kiuchi, K.; Kojima, K.; Kondo, T.; Narabayashi, H.; Rausch, D.; Riederer, P.; Jellinger, K.; Nagatsu, T. Homospecific activity (activity per enzyme protein) of tyrosine hydroxylase increases in parkinsonian brain. J. Neural Transm. 1988, 72, 77–82. [Google Scholar] [CrossRef]
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural Transm. 2009, 116, 1571–1578. [Google Scholar] [CrossRef]
- Kawahata, I.; Yagishita, S.; Hasegawa, K.; Nagatsu, I.; Nagatsu, T.; Ichinose, H. Immunohistochemical analyses of the postmortem human brains from patients with Parkinson’s disease with anti-tyrosine hydroxylase antibodies. Biog. Amines 2009, 23, 1–7. [Google Scholar]
- Baumann, A.; Jorge-Finnigan, A.; Jung-Kc, K.; Sauter, A.; Horvath, I.; Morozova-Roche, L.A.; Martinez, A. Tyrosine Hydroxylase Binding to Phospholipid Membranes Prompts Its Amyloid Aggregation and Compromises Bilayer Integrity. Sci. Rep. 2016, 6, 39488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahata, I.; Ichinose, H. Long-term up-regulation of the level of phosphorylated tyrosine hydroxylase by depletion of dopamine or biopterin. Neurosci. Res. 2009, 65, S66. [Google Scholar] [CrossRef]
- Kawahata, I.; Ohtaku, S.; Tomioka, Y.; Ichinose, H.; Yamakuni, T. Dopamine or biopterin deficiency potentiates phosphorylation at (40)Ser and ubiquitination of tyrosine hydroxylase to be degraded by the ubiquitin proteasome system. Biochem. Biophys. Res. Commun. 2015, 465, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sumi-Ichinose, C.; Urano, F.; Kuroda, R.; Ohye, T.; Kojima, M.; Tazawa, M.; Shiraishi, H.; Hagino, Y.; Nagatsu, T.; Nomura, T.; et al. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. A study from 6-pyruvoyltetrahydropterin synthase knockout mice. J. Biol. Chem. 2001, 276, 41150–41160. [Google Scholar] [CrossRef] [Green Version]
- Takazawa, C.; Fujimoto, K.; Homma, D.; Sumi-Ichinose, C.; Nomura, T.; Ichinose, H.; Katoh, S. A brain-specific decrease of the tyrosine hydroxylase protein in sepiapterin reductase-null mice--as a mouse model for Parkinson’s disease. Biochem. Biophys. Res. Commun. 2008, 367, 787–792. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Stern, S.T.; Adiseshaiah, P.P.; Crist, R.M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 2012, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Cohignac, V.; Landry, M.J.; Boczkowski, J.; Lanone, S. Autophagy as a Possible Underlying Mechanism of Nanomaterial Toxicity. Nanomaterials 2014, 4, 548–582. [Google Scholar] [CrossRef] [Green Version]
- Kenney, D.L.; Benarroch, E.E. The autophagy-lysosomal pathway: General concepts and clinical implications. Neurology 2015, 85, 634–645. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B. Autophagy and lysosomal pathways in nervous system disorders. Mol Cell Neurosci 2018, 91, 167–208. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F.; Terlecky, S.R.; Chiang, H.L.; Olson, T.S.; Isenman, L.D.; Short-Russell, S.R.; Freundlieb, S.; Terlecky, L.J. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin. Cell Biol. 1990, 1, 449–455. [Google Scholar] [PubMed]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef]
- Hershko, A.; Leshinsky, E.; Ganoth, D.; Heller, H. ATP-dependent degradation of ubiquitin-protein conjugates. Proc. Natl. Acad. Sci. USA 1984, 81, 1619–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [PubMed]
- Ciechanover, A.; Elias, S.; Heller, H.; Hershko, A. “Covalent affinity” purification of ubiquitin-activating enzyme. J. Biol. Chem. 1982, 257, 2537–2542. [Google Scholar]
- Hershko, A.; Ciechanover, A.; Heller, H.; Haas, A.L.; Rose, I.A. Proposed role of ATP in protein breakdown: Conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. USA 1980, 77, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- DeMartino, G.N.; Slaughter, C.A. The proteasome, a novel protease regulated by multiple mechanisms. J. Biol. Chem. 1999, 274, 22123–22126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickart, C.M. Ubiquitin biology: An old dog learns an old trick. Nat. Cell Biol. 2000, 2, E139–E141. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Ubiquitin in chains. Trends Biochem. Sci. 2000, 25, 544–548. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Olanow, C.W. Proteolytic stress: A unifying concept for the etiopathogenesis of Parkinson’s disease. Ann. Neurol. 2003, 53, S73–S84. [Google Scholar] [CrossRef]
- Lazar, M.A.; Truscott, R.J.; Raese, J.D.; Barchas, J.D. Thermal denaturation of native striatal tyrosine hydroxylase: Increased thermolability of the phosphorylated form of the enzyme. J. Neurochem. 1981, 36, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Døskeland, A.P.; Flatmark, T. Ubiquitination of soluble and membrane-bound tyrosine hydroxylase and degradation of the soluble form. Eur. J. Biochem. 2002, 269, 1561–1569. [Google Scholar] [CrossRef]
- Urano, F.; Hayashi, N.; Arisaka, F.; Kurita, H.; Murata, S.; Ichinose, H. Molecular mechanism for pterin-mediated inactivation of tyrosine hydroxylase: Formation of insoluble aggregates of tyrosine hydroxylase. J. Biochem. 2006, 139, 625–635. [Google Scholar] [CrossRef]
- Nakashima, A.; Mori, K.; Kaneko, Y.S.; Hayashi, N.; Nagatsu, T.; Ota, A. Phosphorylation of the N-terminal portion of tyrosine hydroxylase triggers proteasomal digestion of the enzyme. Biochem. Biophys. Res. Commun. 2011, 407, 343–347. [Google Scholar] [CrossRef]
- Nakashima, A.; Ohnuma, S.; Kodani, Y.; Kaneko, Y.S.; Nagasaki, H.; Nagatsu, T.; Ota, A. Inhibition of deubiquitinating activity of USP14 decreases tyrosine hydroxylase phosphorylated at Ser19 in PC12D cells. Biochem. Biophys. Res. Commun. 2016, 472, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Carbajosa, N.A.; Corradi, G.; Verrilli, M.A.; Guil, M.J.; Vatta, M.S.; Gironacci, M.M. Tyrosine hydroxylase is short-term regulated by the ubiquitin-proteasome system in PC12 cells and hypothalamic and brainstem neurons from spontaneously hypertensive rats: Possible implications in hypertension. PLoS ONE 2015, 10, e0116597. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.F.; Calipari, E.S.; Jones, S.R. Regulation of Tyrosine Hydroxylase Expression and Phosphorylation in Dopamine Transporter-Deficient Mice. ACS Chem. Neurosci. 2016, 7, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Bennett, M.C.; Bishop, J.F.; Leng, Y.; Chock, P.B.; Chase, T.N.; Mouradian, M.M. Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 1999, 274, 33855–33858. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Lee, V.M. Are ubiquitination pathways central to Parkinson’s disease? Cell 2003, 114, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Tehranian, R.; Dietrich, P.; Stefanis, L.; Perez, R.G. Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 2005, 118, 3523–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, H.; Montoya, S.E.; Alerte, T.N.; Wang, J.; Wu, J.; Peng, X.; Hong, C.S.; Friedrich, E.E.; Mader, S.A.; Pedersen, C.J.; et al. Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 2010, 285, 17648–17661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Vila, M.; Lincoln, S.; McCormack, A.; Picciano, M.; LaFrancois, J.; Yu, X.; Dickson, D.; Langston, W.J.; McGowan, E.; et al. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol. Dis. 2001, 8, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Rathke-Hartlieb, S.; Kahle, P.J.; Neumann, M.; Ozmen, L.; Haid, S.; Okochi, M.; Haass, C.; Schulz, J.B. Sensitivity to MPTP is not increased in Parkinson’s disease-associated mutant alpha-synuclein transgenic mice. J. Neurochem. 2001, 77, 1181–1184. [Google Scholar] [CrossRef]
- Richfield, E.K.; Thiruchelvam, M.J.; Cory-Slechta, D.A.; Wuertzer, C.; Gainetdinov, R.R.; Caron, M.G.; Di Monte, D.A.; Federoff, H.J. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 2002, 175, 35–48. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Ng, A.S.L.; Tan, Y.J.; Lu, Z.; Ng, E.Y.L.; Ng, S.Y.E.; Chia, N.S.Y.; Setiawan, F.; Xu, Z.; Tay, K.Y.; Prakash, K.M.; et al. Plasma alpha-synuclein detected by single molecule array is increased in PD. Ann. Clin. Transl. Neurol. 2019, 6, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Hastings, T.G. Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J. Neurochem. 2004, 89, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Alerte, T.N.; Akinfolarin, A.A.; Friedrich, E.E.; Mader, S.A.; Hong, C.S.; Perez, R.G. Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: Lessons from viral transduction of knockout mice. Neurosci. Lett. 2008, 435, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toska, K.; Kleppe, R.; Armstrong, C.G.; Morrice, N.A.; Cohen, P.; Haavik, J. Regulation of tyrosine hydroxylase by stress-activated protein kinases. J. Neurochem. 2002, 83, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef]
- Collier, T.J.; Lipton, J.; Daley, B.F.; Palfi, S.; Chu, Y.; Sortwell, C.; Bakay, R.A.; Sladek, J.R., Jr.; Kordower, J.H. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: Diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol. Dis. 2007, 26, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef]
- Anisimova, A.S.; Alexandrov, A.I.; Makarova, N.E.; Gladyshev, V.N.; Dmitriev, S.E. Protein synthesis and quality control in aging. Aging (Albany NY) 2018, 10, 4269–4288. [Google Scholar] [CrossRef]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s disease as a result of aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism—chronic treatment with L-dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef]
- Cotzias, G.C.; Miller, S.T.; Tang, L.C.; Papavasiliou, P.S. Levodopa, fertility, and longevity. Science 1977, 196, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Papavasiliou, P.S.; Cotzias, G.C.; Rosal, V.L.; Miller, S.T. Treatment of parkinsonism with N-n-propyl norapomorphine and levodopa (with or without carbidopa). Arch. Neurol. 1978, 35, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Srivanitchapoom, P. Levodopa-induced Dyskinesia: Clinical Features, Pathophysiology, and Medical Management. Ann. Indian Acad. Neurol. 2017, 20, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recasens, A.; Ulusoy, A.; Kahle, P.J.; Di Monte, D.A.; Dehay, B. In vivo models of alpha-synuclein transmission and propagation. Cell Tissue Res. 2018, 373, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut-brain axis and the spread of alpha-synuclein pathology: Vagal highway or dead end? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Lena, C.; et al. alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.; Marano, M.M.; Tandon, A. Import and Export of Misfolded alpha-Synuclein. Front. Neurosci. 2018, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, J.; Hasegawa, T.; Sugeno, N.; Yoshida, S.; Akiyama, T.; Fujimori, K.; Hatakeyama, H.; Miki, Y.; Tomiyama, A.; Kawata, Y.; et al. Extracellular alpha-synuclein enters dopaminergic cells by modulating flotillin-1-assisted dopamine transporter endocytosis. FASEB J. 2019, 33, 10240–10256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delenclos, M.; Trendafilova, T.; Mahesh, D.; Baine, A.M.; Moussaud, S.; Yan, I.K.; Patel, T.; McLean, P.J. Investigation of Endocytic Pathways for the Internalization of Exosome-Associated Oligomeric Alpha-Synuclein. Front. Neurosci. 2017, 11, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahata, I.; Bousset, L.; Melki, R.; Fukunaga, K. Fatty Acid-Binding Protein 3 is Critical for alpha-Synuclein Uptake and MPP(+)-Induced Mitochondrial Dysfunction in Cultured Dopaminergic Neurons. Int. J. Mol. Sci. 2019, 20, 5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuki, Y.; Matsuo, K.; Kawahata, I.; Fukui, N.; Mizobata, T.; Kawata, Y.; Owada, Y.; Shioda, N.; Fukunaga, K. Fatty Acid Binding Protein 3 Enhances the Spreading and Toxicity of alpha-Synuclein in Mouse Brain. Int. J. Mol. Sci. 2020, 21, 2230. [Google Scholar] [CrossRef] [Green Version]
- Perlmutter, J.S.; Tempel, L.W.; Black, K.J.; Parkinson, D.; Todd, R.D. MPTP induces dystonia and parkinsonism. Clues to the pathophysiology of dystonia. Neurology 1997, 49, 1432–1438. [Google Scholar] [CrossRef]
- Tabbal, S.D.; Mink, J.W.; Antenor, J.A.; Carl, J.L.; Moerlein, S.M.; Perlmutter, J.S. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience 2006, 141, 1281–1287. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Parkinsons Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [Green Version]
- Lai, F.; Jiang, R.; Xie, W.; Liu, X.; Tang, Y.; Xiao, H.; Gao, J.; Jia, Y.; Bai, Q. Intestinal Pathology and Gut Microbiota Alterations in a Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Mouse Model of Parkinson’s Disease. Neurochem. Res. 2018, 43, 1986–1999. [Google Scholar] [CrossRef]
- Matsuo, K.; Cheng, A.; Yabuki, Y.; Takahata, I.; Miyachi, H.; Fukunaga, K. Inhibition of MPTP-induced alpha-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice. Neuropharmacology 2019, 150, 164–174. [Google Scholar] [CrossRef]
- Cheng, A.; Shinoda, Y.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Development of FABP3 ligands that inhibit arachidonic acid-induced alpha-synuclein oligomerization. Brain Res. 2019, 1707, 190–197. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Veerhuis, R.; De Vente, J.; Verhey, F.R.; Vreeling, F.; Van Boxtel, M.P.; Glatz, J.F.; Pelsers, M.A. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 2011, 18, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Kasai, T.; Ohmichi, T.; Kishi, Y.; Kakeya, T.; Waragai, M.; Kondo, M.; Allsop, D.; Tokuda, T. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: Pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 2017, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Yang, T.; Shankar, G.; Smith, I.M.; Shen, Y.; Walsh, D.M.; Selkoe, D.J. A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 2009, 66, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewczuk, P.; Kornhuber, J.; Vanmechelen, E.; Peters, O.; Heuser, I.; Maier, W.; Jessen, F.; Burger, K.; Hampel, H.; Frolich, L.; et al. Amyloid beta peptides in plasma in early diagnosis of Alzheimer’s disease: A multicenter study with multiplexing. Exp. Neurol. 2010, 223, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Van Oijen, M.; Hofman, A.; Soares, H.D.; Koudstaal, P.J.; Breteler, M.M. Plasma Abeta(1-40) and Abeta(1-42) and the risk of dementia: A prospective case-cohort study. Lancet Neurol. 2006, 5, 655–660. [Google Scholar] [CrossRef]
- Graff-Radford, N.R.; Crook, J.E.; Lucas, J.; Boeve, B.F.; Knopman, D.S.; Ivnik, R.J.; Smith, G.E.; Younkin, L.H.; Petersen, R.C.; Younkin, S.G. Association of low plasma Abeta42/Abeta40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2007, 64, 354–362. [Google Scholar] [CrossRef]
- Schupf, N.; Patel, B.; Pang, D.; Zigman, W.B.; Silverman, W.; Mehta, P.D.; Mayeux, R. Elevated plasma beta-amyloid peptide Abeta(42) levels, incident dementia, and mortality in Down syndrome. Arch. Neurol. 2007, 64, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Haga, H.; Yamada, R.; Izumi, H.; Shinoda, Y.; Kawahata, I.; Miyachi, H.; Fukunaga, K. Novel fatty acid-binding protein 3 ligand inhibits dopaminergic neuronal death and improves motor and cognitive impairments in Parkinson’s disease model mice. Pharmacol. Biochem. Behav. 2020, 191, 172891. [Google Scholar] [CrossRef]
- Kawahata, I.; Yoshida, M.; Sun, W.; Nakajima, A.; Lai, Y.; Osaka, N.; Matsuzaki, K.; Yokosuka, A.; Mimaki, Y.; Naganuma, A.; et al. Potent activity of nobiletin-rich Citrus reticulata peel extract to facilitate cAMP/PKA/ERK/CREB signaling associated with learning and memory in cultured hippocampal neurons: Identification of the substances responsible for the pharmacological action. J. Neural Transm. 2013, 120, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Suzuki, T.; Rico, E.G.; Kusano, S.; Tamura, H.; Mimaki, Y.; Yamakuni, T. Fermented Citrus reticulata (ponkan) fruit squeezed draff that contains a large amount of 4′-demethylnobiletin prevents MK801-induced memory impairment. J. Nat. Med. 2017, 71, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Xu, H.; Takahashi, M.; Murata, K.; Han, W.; Yamaguchi, Y.; Fujii, A.; Yamaguchi, K.; Yamakuni, T. Royal jelly coordinately enhances hippocampal neuronal expression of somatostatin and neprilysin genes conferring neuronal protection against toxic soluble amyloid-β oligomers implicated in Alzheimer’s disease pathogenesis. J. Funct. Foods 2018, 51, 28–38. [Google Scholar] [CrossRef]
- Kawahata, I. Drug discovery study of fundamental therapy for dopamine related diseases targeting new V-1 / CP complex. Impact 2019, 2019, 49–51. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Choi, H.J.; Jang, Y.J.; Kim, H.J.; Hwang, O. Tetrahydrobiopterin is released from and causes preferential death of catecholaminergic cells by oxidative stress. Mol. Pharmacol. 2000, 58, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Choi, J.H.; Chang, J.W.; Kim, S.W.; Hwang, O. Immobilization stress causes increases in tetrahydrobiopterin, dopamine, and neuromelanin and oxidative damage in the nigrostriatal system. J. Neurochem. 2005, 95, 89–98. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evidence | Year | Reference |
|---|---|---|
| Activated tyrosine hydroxylase purified from bovine striatum decreases its thermal stability | 1981 | [97] |
| Human recombinant TH protein is ubiquitinated and degraded in the reticulocyte lysate system | 2002 | [98] |
| Recombinant human TH forms insoluble aggregates in the presence of tetrahydrobiopterin | 2006 | [99] |
| Proteasomal inhibition accumulates ubiquitinated TH protein phosphorylated at 40Ser to form insoluble aggregates in NGF-differentiated PC12D cells | 2009 | [70] |
| Phosphorylation of the N-terminal domain of tyrosine hydroxylase triggers proteasomal digestion | 2011 | [100] |
| Short-term inhibition of proteasome increases the accumulation of ubiquitinated TH protein in PC12 cell and brainstem neurons | 2015 | [102] |
| Dopamine or biopterin deficiency potentiates phosphorylation at 40Ser and ubiquitination of TH protein to be degraded by the ubiquitin proteasome system | 2015 | [74] |
| Inhibition of USP14 to activate proteasome decreases TH protein phosphorylated at 19Ser | 2016 | [101] |
| Dopamine transporter-deficiency increases TH phosphorylation and decreases TH protein in striatum and nucleus accumbens | 2016 | [103] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawahata, I.; Fukunaga, K. Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia. Int. J. Mol. Sci. 2020, 21, 3779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113779
Kawahata I, Fukunaga K. Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia. International Journal of Molecular Sciences. 2020; 21(11):3779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113779
Chicago/Turabian StyleKawahata, Ichiro, and Kohji Fukunaga. 2020. "Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia" International Journal of Molecular Sciences 21, no. 11: 3779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21113779