Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin

 , ,
, ,
Abstract
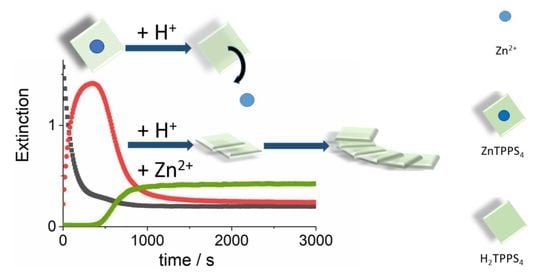
:
1. Introduction
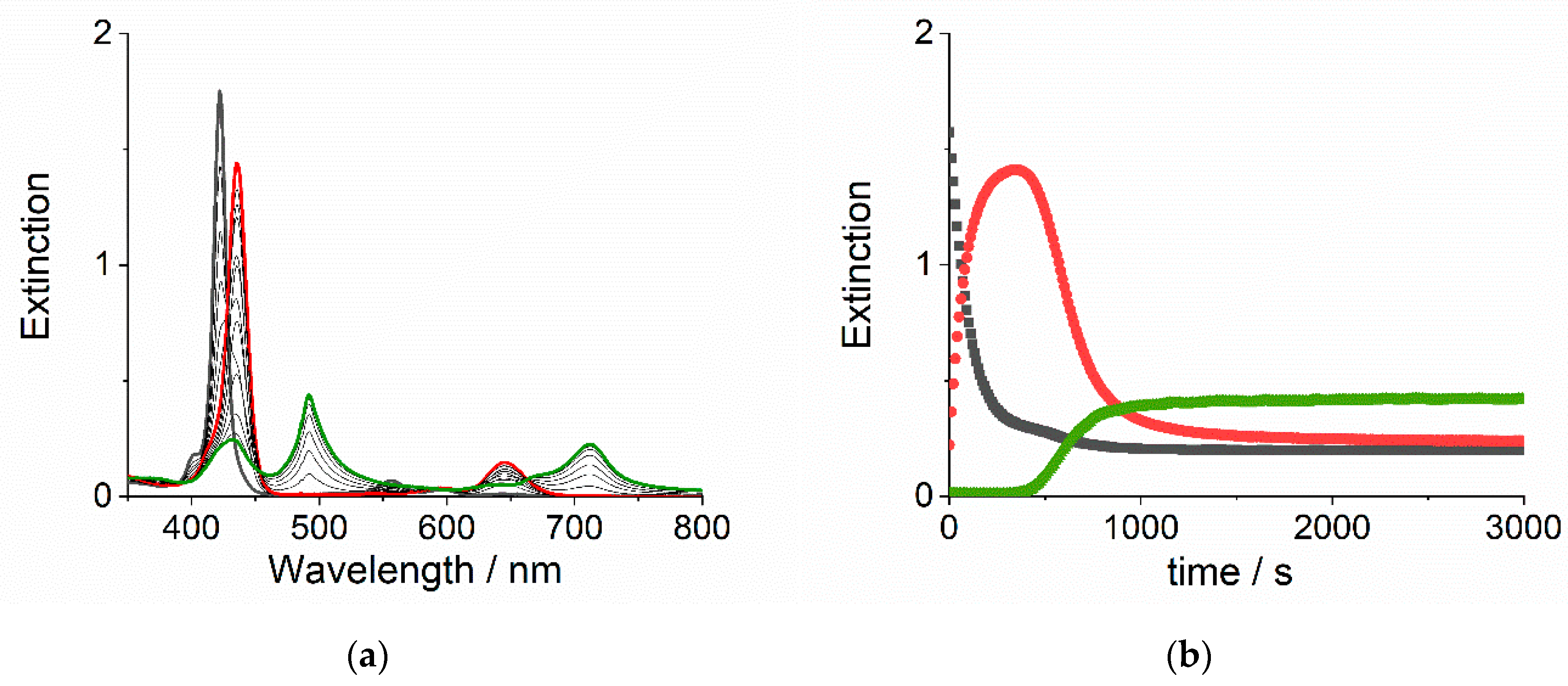
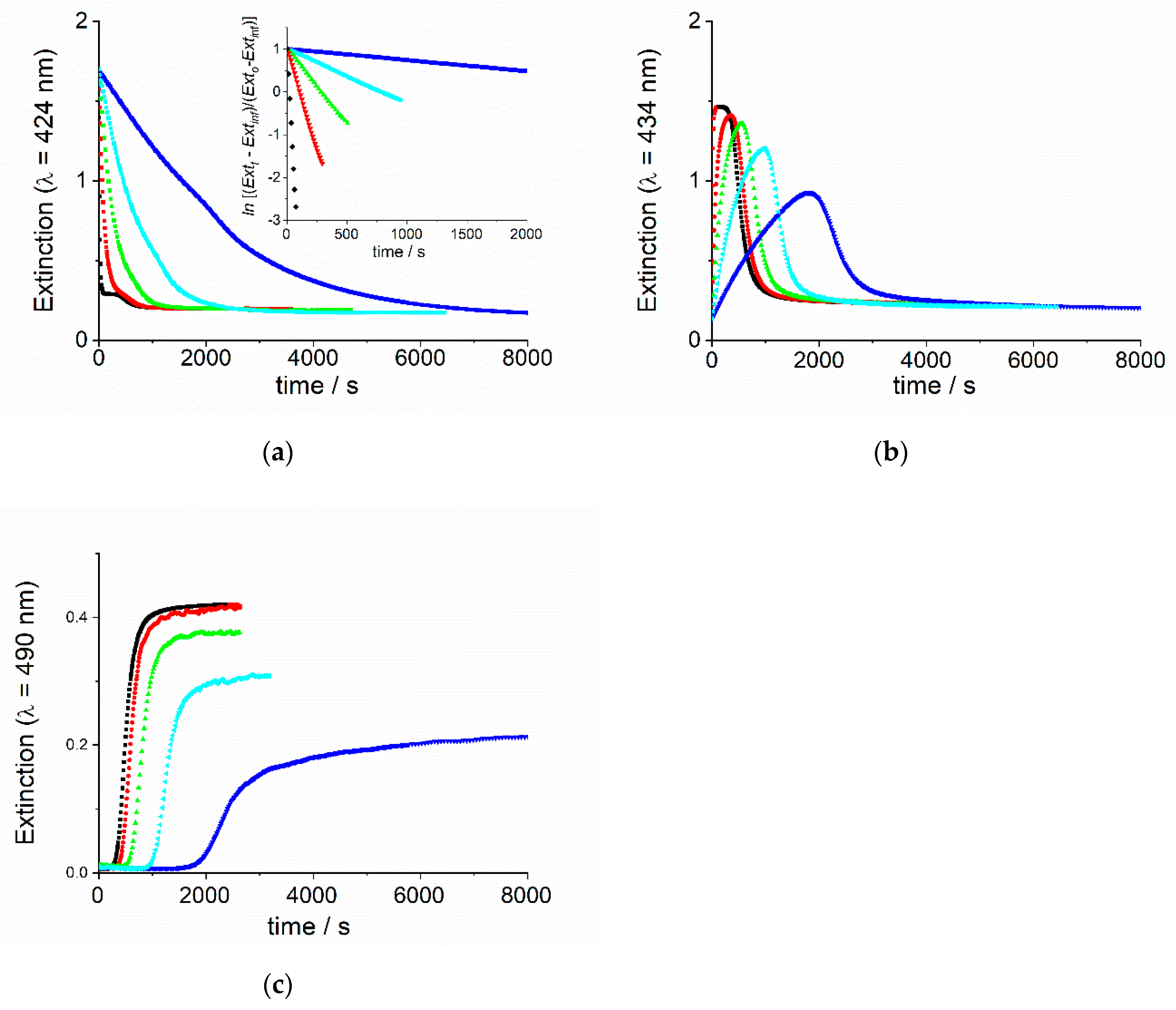
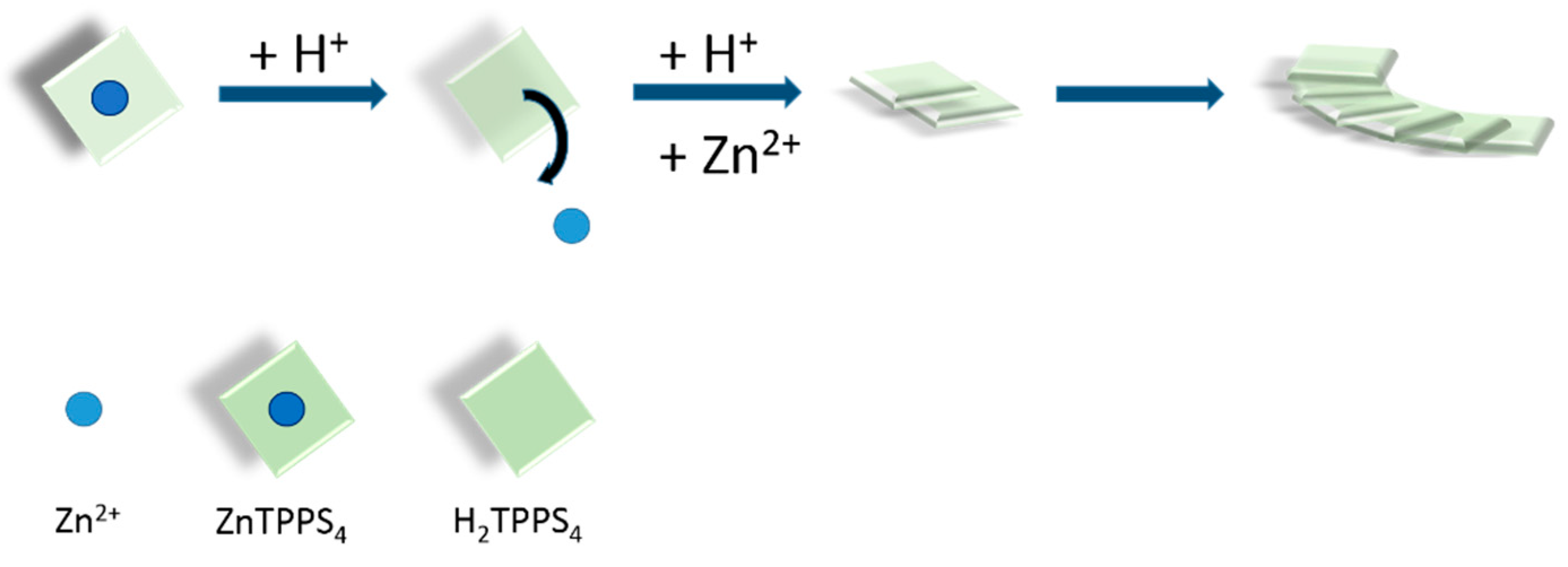
2. Results and Discussion
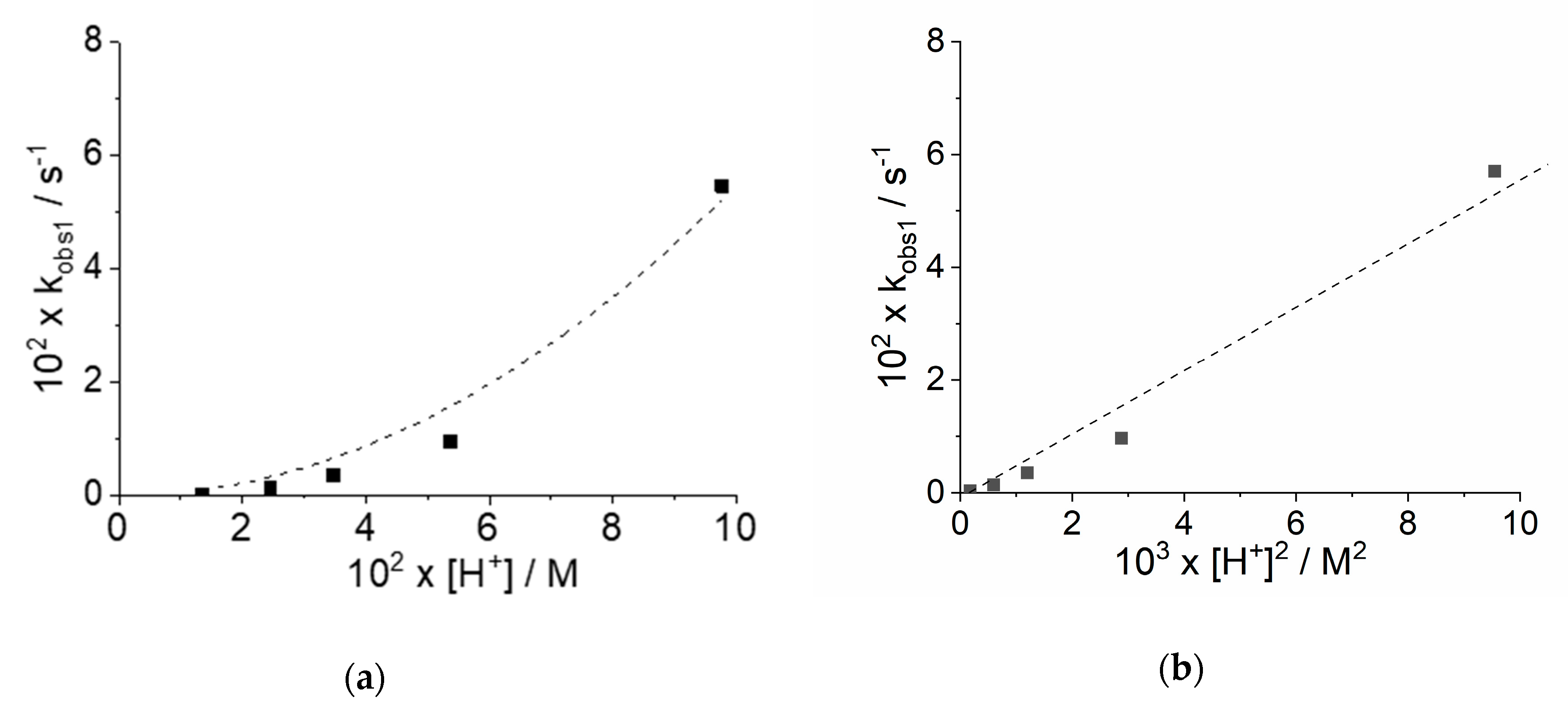
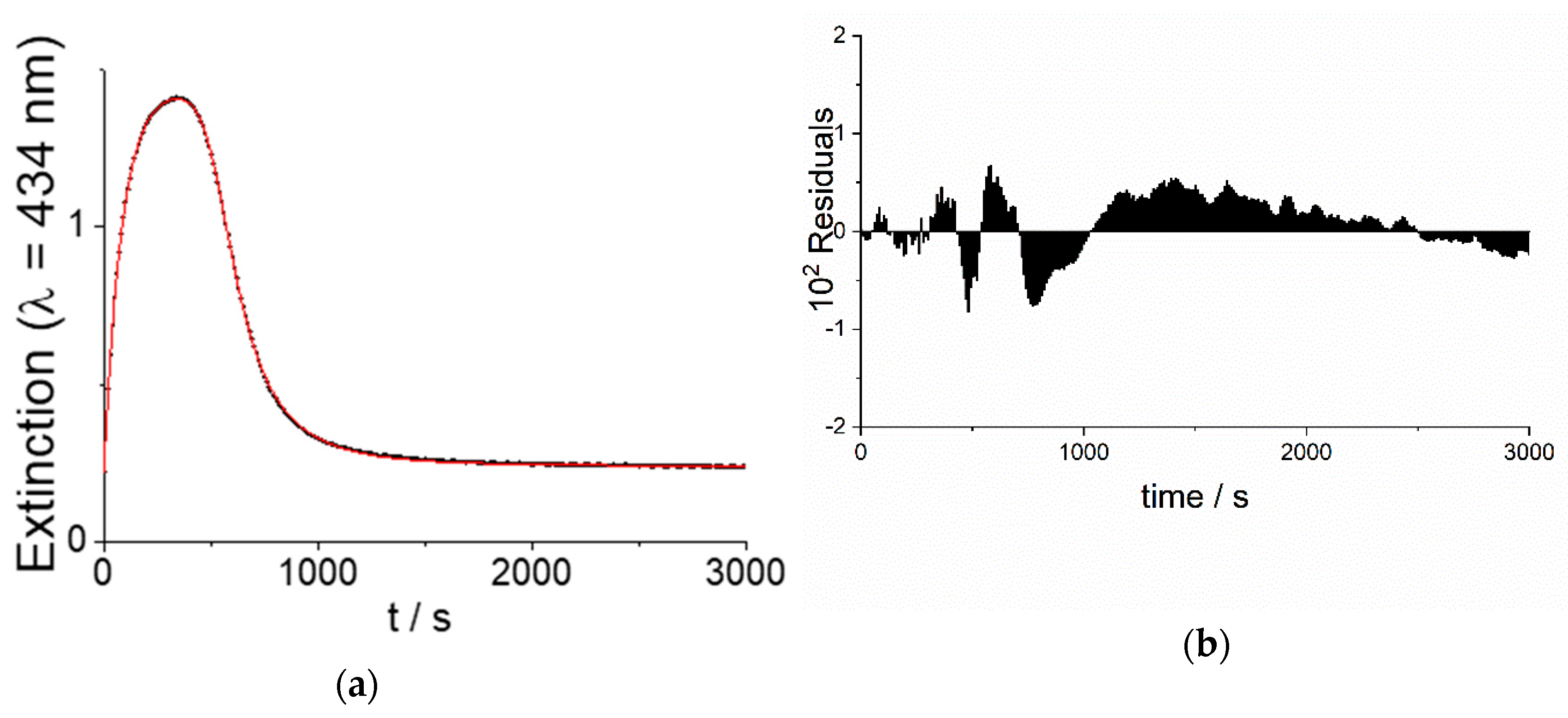
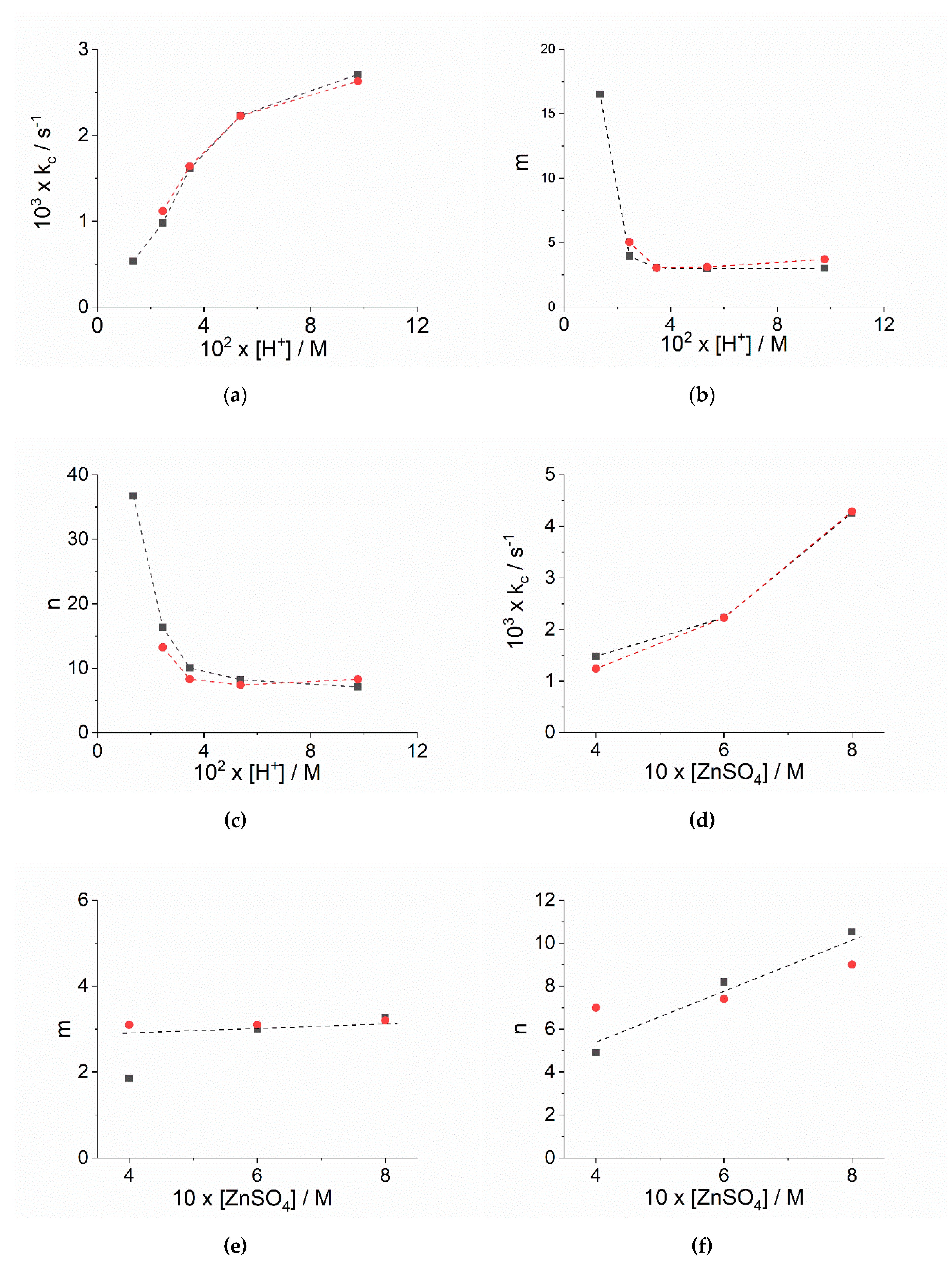
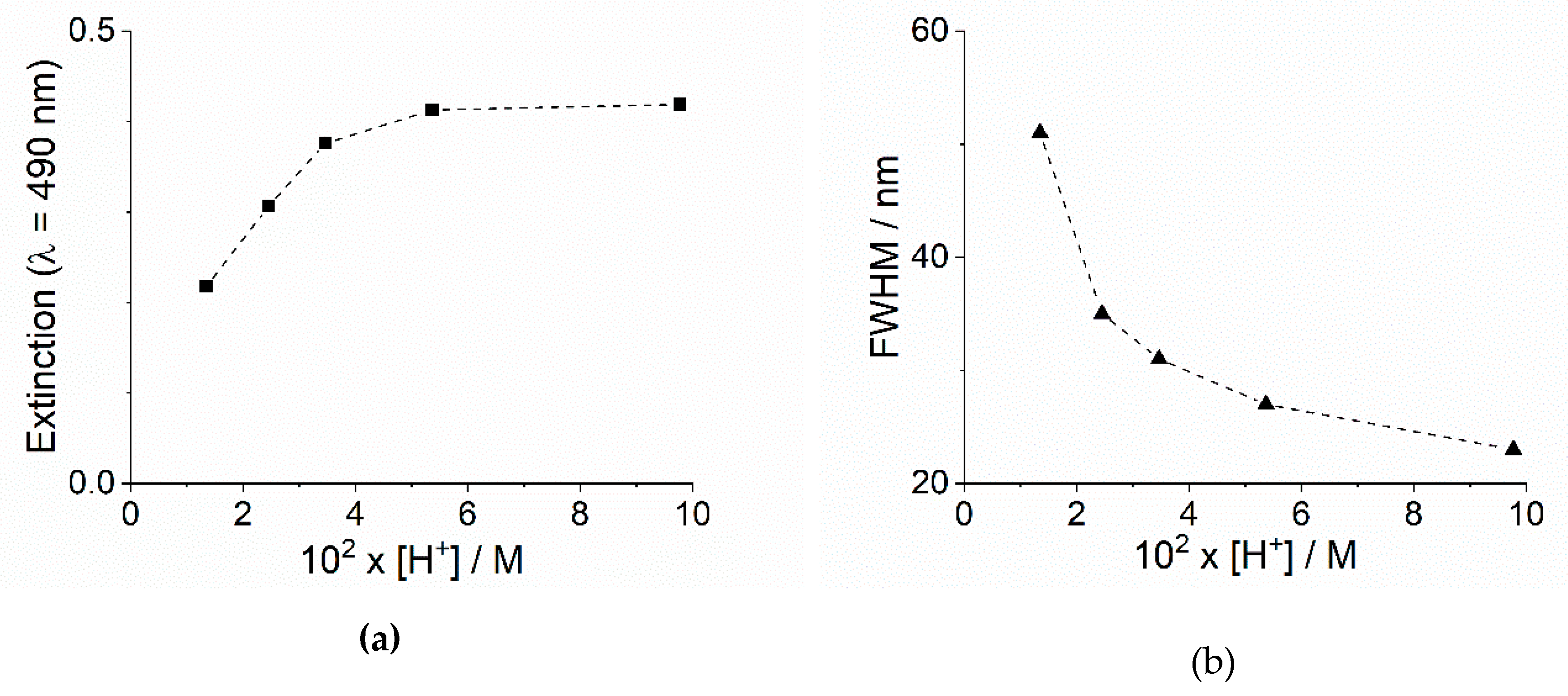
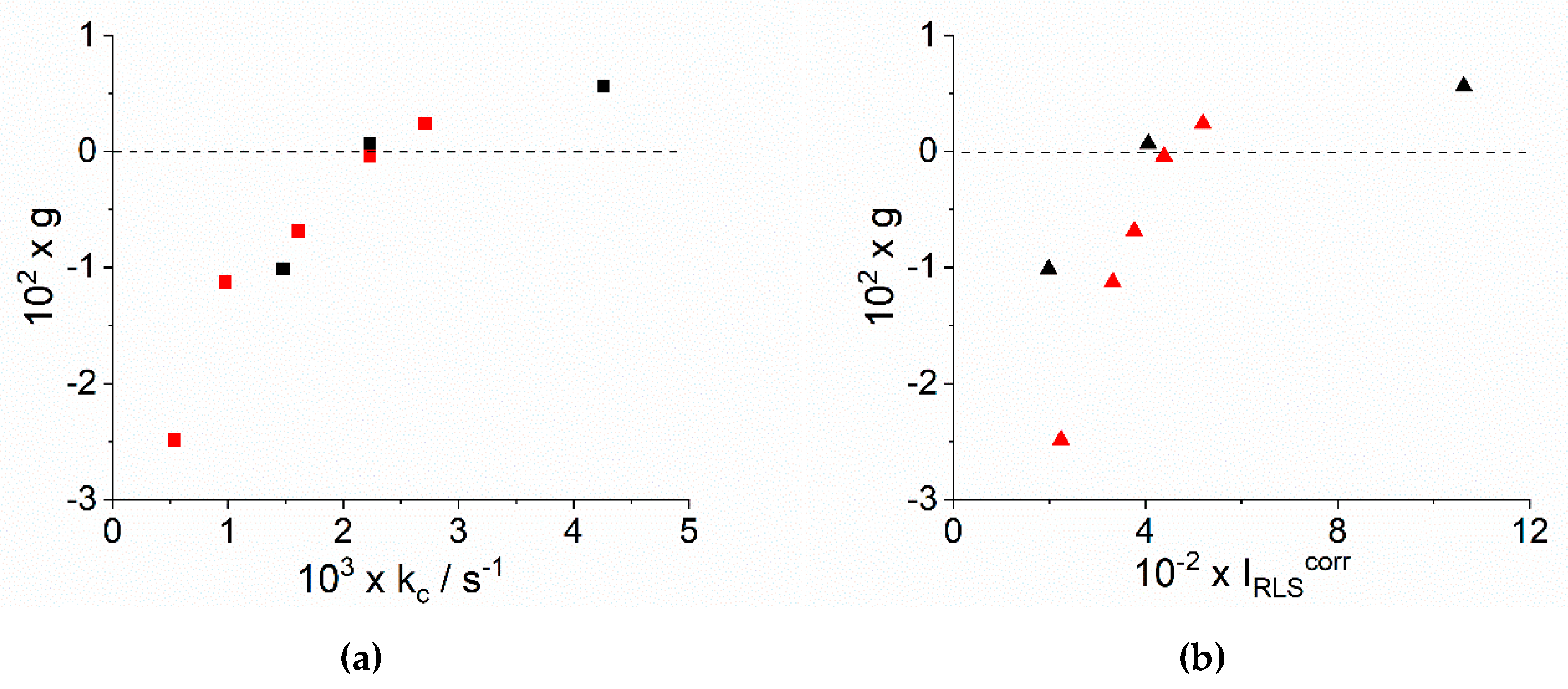
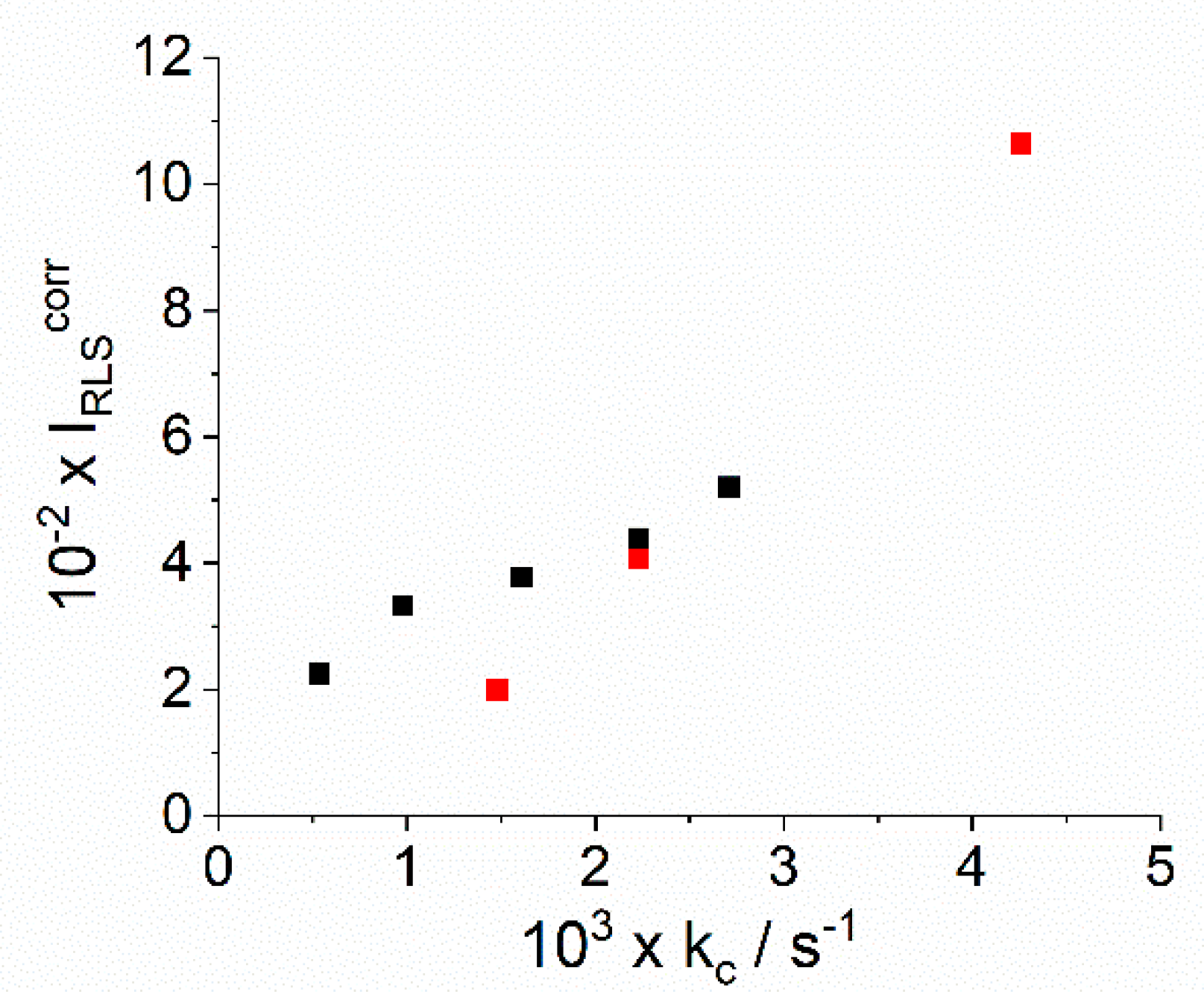
2.1. Kinetic Analysis
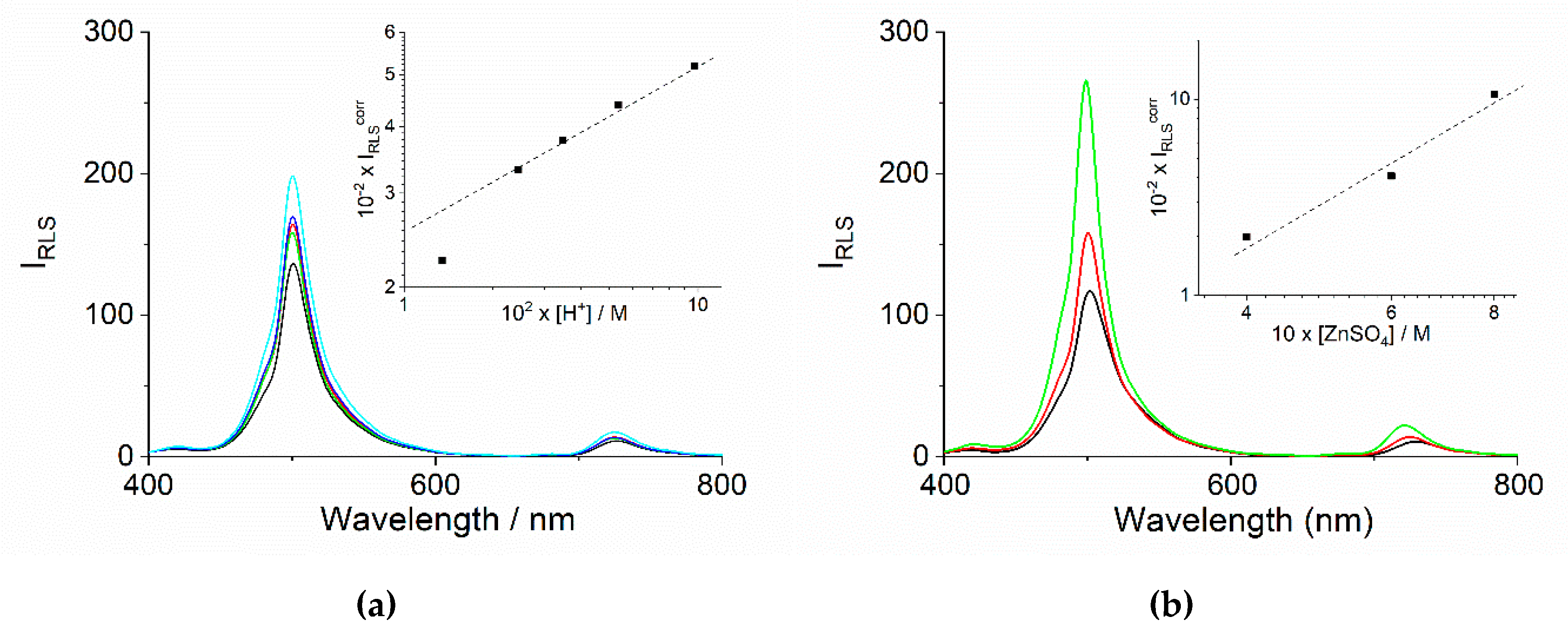
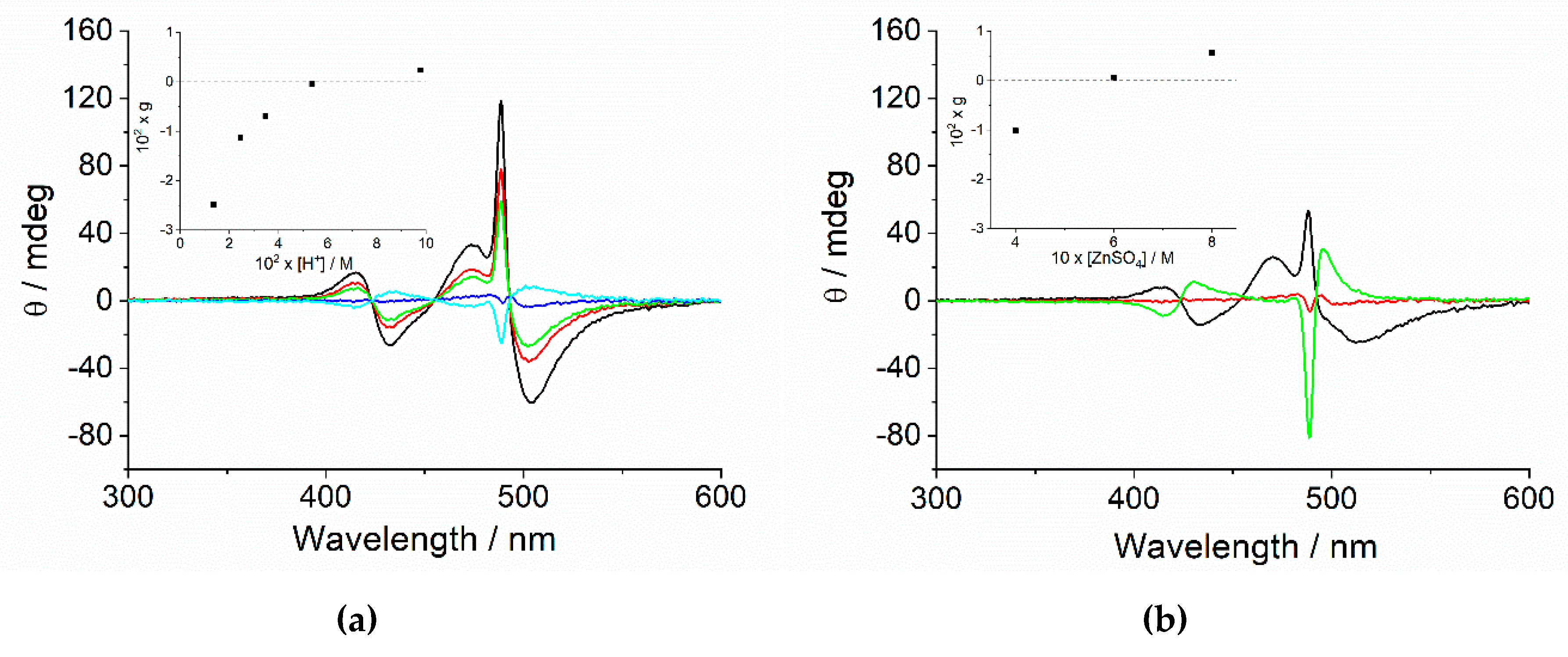
2.2. Chiroptical Properties
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Takayoshi, K. J-aggregates; World Scientific Publishing Company: Tokyo, Japan, 1996. [Google Scholar]
- Kobayashi, T. J-aggregates; World Scientific: Tokyo, Japan, 2012. [Google Scholar]
- Castriciano, M.A.; Donato, M.G.; Villari, V.; Micali, N.; Romeo, A.; Scolaro, L.M. Surfactant-like Behavior of Short-Chain Alcohols in Porphyrin Aggregation. J. Phys. Chem. 2009, 113, 11173–11178. [Google Scholar] [CrossRef] [PubMed]
- Micali, N.; Villari, V.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. From fractal to nanorod porphyrin J-aggregates. Concentration-induced tuning of the aggregate size. J. Phys. Chem. 2006, 110, 8289–8295. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.; Romeo, A.; Villari, V.; Micali, N.; Scolaro, L.M. Structural rearrangements in 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin J-aggregates under strongly acidic conditions. J. Phys. Chem. 2003, 107, 8765–8771. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Aggregation of meso-tetrakis(4-sulfonatophenyl)porphyrin on polyethyleneimine in aqueous solutions and on a glass surface. J. Porphyr. Phthalocyanines 2002, 6, 431–438. [Google Scholar] [CrossRef]
- Micali, N.; Mallamace, F.; Romeo, A.; Purrello, R.; Scolaro, L.M. Mesoscopic structure of meso-tetrakis(4-sulfonatophenyl)porphine J-aggregates. J. Phys. Chem. B 2000, 104, 5897–5904. [Google Scholar] [CrossRef]
- Koti, A.S.R.; Taneja, J.; Periasamy, N. Control of coherence length and aggregate size in the J-aggregate of porphyrin. Chem. Phys. Lett. 2003, 375, 171–176. [Google Scholar] [CrossRef]
- Rotomskis, R.; Augulis, R.; Snitka, V.; Valiokas, R.; Liedberg, B. Hierarchical Structure of TPPS4 J-Aggregates on Substrate Revealed by Atomic Force Microscopy. J. Phys. Chem. B 2004, 108, 2833–2838. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Leone, N.; Cardiano, P.; Manickam, S.; Scolaro, L.M.; Lo Schiavo, S. A new supramolecular polyhedral oligomeric silsesquioxanes (POSS)-porphyrin nanohybrid: Synthesis and spectroscopic characterization. J. Mater. Chem. 2013, 1, 4746–4753. [Google Scholar] [CrossRef]
- Liu, M.H.; Zhang, L.; Wang, T.Y. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Magna, G.; Monti, D.; Di Natale, C.; Paolesse, R.; Stefanelli, M. The Assembly of Porphyrin Systems in Well-Defined Nanostructures: An Update. Molecules 2019, 24, 4307. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, J.J.L.M.; Rowan, A.E.; Nolte, R.J.M.; Sommerdijk, N.A.J.M. Chiral Architectures from Macromolecular Building Blocks. Chem. Rev. 2001, 101, 4039–4070. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, F.J.M.; Jonkheijm, P.; Meijer, E.W.; Schenning, A. About supramolecular assemblies of pi-conjugated systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef] [PubMed]
- Palmans, A.R.A.; Meijer, E.W. Amplification of chirality in dynamic supramolecular aggregates. Angew. Chemie-Int. Ed. 2007, 46, 8948–8968. [Google Scholar] [CrossRef] [PubMed]
- Amabilino, D.B. Chiral nanoscale systems: Preparation, structure, properties and function. Chem. Soc. Rev. 2009, 38, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Iavicoli, P.; Xu, H.; Feldborg, L.N.; Linares, M.; Paradinas, M.; Stafstrom, S.; Ocal, C.; Nieto-Ortega, B.L.; Casado, J.; Navarrete, J.T.L.; et al. Tuning the Supramolecular Chirality of One- and Two-Dimensional Aggregates with the Number of Stereogenic Centers in the Component Porphyrins. J. Am. Chem. Soc. 2010, 132, 9350–9362. [Google Scholar] [CrossRef]
- Oliveras-Gonzalez, C.; Di Meo, F.; Gonzalez-Campo, A.; Beljonne, D.; Norman, P.; Simon-Sorbed, M.; Linares, M.; Amabilino, D.B. Bottom-Up Hierarchical Self-Assembly of Chiral Porphyrins through Coordination and Hydrogen Bonds. J. Am. Chem. Soc. 2015, 137, 15795–15808. [Google Scholar] [CrossRef]
- Monti, D.; Venanzi, M.; Mancini, G.; Di Natale, C.; Paolesse, R. Supramolecular chirality control by solvent changes. Solvodichroic effect on chiral porphyrin aggregation. Chem. Commun. 2005, 2471–2473. [Google Scholar] [CrossRef]
- Monti, D.; Venanzi, M.; Stefanelli, M.; Sorrenti, A.; Mancini, G.; Di Natale, C.; Paolesse, R. Chiral amplification of chiral porphyrin derivatives by templated heteroaggregation. J. Am. Chem. Soc. 2007, 129, 6688. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Zagami, R.; Trapani, M.; Romeo, A.; Patane, S.; Scolaro, L.M. Investigation of the Aggregation Properties of a Chiral Porphyrin Bearing Citronellal Meso Substituent Groups. Chirality 2015, 27, 900–906. [Google Scholar] [CrossRef]
- Van Hameren, R.; van Buul, A.M.; Castriciano, M.A.; Villari, V.; Micali, N.; Schon, P.; Speller, S.; Scolaro, L.M.; Rowan, A.E.; Elemans, J.A.A.W.; et al. Supramolecular porphyrin polymers in solution and at the solid-liquid interface. Nano Lett. 2008, 8, 253–259. [Google Scholar] [CrossRef]
- Lettieri, R.; Cardova, L.; Gatto, E.; Mazzuca, C.; Monti, D.; Palleschi, A.; Placidi, E.; Drasar, P.; Venanzi, M. Hierarchical transfer of chiral information from the molecular to the mesoscopic scale by Langmuir-Blodgett deposition of tetrasteroid-porphyrins. New J. Chem. 2017, 41, 639–649. [Google Scholar] [CrossRef]
- Stefanelli, M.; Magna, G.; Zurlo, F.; Caso, F.M.; Di Bartolomeo, E.; Antonaroliit, S.; Venanzi, M.; Paolesse, R.; Di Natale, C.; Monti, D. Chiral Selectivity of Porphyrin-ZnO Nanoparticle Conjugates. Acs Appl. Mater. Interfaces 2019, 11, 12077–12087. [Google Scholar] [CrossRef] [PubMed]
- Zelenka, K.; Trnka, T.; Tislerova, I.; Monti, D.; Cinti, S.; Naitana, M.L.; Schiaffino, L.; Venanzi, M.; Laguzzi, G.; Luvidi, L.; et al. Spectroscopic, Morphological, and Mechanistic Investigation of the Solvent-Promoted Aggregation of Porphyrins Modified in meso-Positions by Glucosylated Steroids. Chemistry-A Eur. J. 2011, 17, 13743–13753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, D.; Cantonetti, V.; Venanzi, M.; Ceccacci, F.; Bombelli, C.; Mancini, G. Interaction of a chirally functionalised porphyrin derivative with chiral micellar aggregates. Chem. Commun. 2004, 972–973. [Google Scholar] [CrossRef]
- Bellacchio, E.; Lauceri, R.; Gurrieri, S.; Scolaro, L.M.; Romeo, A.; Purrello, R. Template-imprinted chiral porphyrin aggregates. J. Am. Chem. Soc. 1998, 120, 12353–12354. [Google Scholar] [CrossRef]
- Dordevic, L.; Arcudi, F.; D'Urso, A.; Cacioppo, M.; Micali, N.; Burgi, T.; Purrello, R.; Prato, M. Design principles of chiral carbon nanodots help convey chirality from molecular to nanoscale level. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Gaeta, M.; Oliveri, I.P.; Fragala, M.E.; Failla, S.; D'Urso, A.; Di Bella, S.; Purrello, R. Chirality of self-assembled achiral porphyrins induced by chiral Zn(II) Schiff-base complexes and maintained after spontaneous dissociation of the templates: A new case of chiral memory. Chem. Commun. 2016, 52, 8518–8521. [Google Scholar] [CrossRef]
- Gaeta, M.; Raciti, D.; Randazzo, R.; Gangemi, C.M.A.; Raudino, A.; D’Urso, A.; Fragala, M.E.; Purrello, R. Chirality Enhancement of Porphyrin Supramolecular Assembly Driven by a Template Preorganization Effect. Angew. Chemie-Int. Ed. 2018, 57, 10656–10660. [Google Scholar] [CrossRef]
- Lauceri, R.; Fasciglione, G.F.; D’Urso, A.; Marini, S.; Purrello, R.; Coletta, M. Kinetic investigation of porphyrin interaction with chiral templates reveals unexpected features of the induction and self-propagation mechanism of chiral memory. J. Am. Chem. Soc. 2008, 130, 10476–10477. [Google Scholar] [CrossRef] [Green Version]
- Lauceri, R.; Raudino, A.; Scolaro, L.M.; Micali, N.; Purrello, R. From achiral porphyrins to template-imprinted chiral aggregates and further. Self-replication of chiral memory from scratch. J. Am. Chem. Soc. 2002, 124, 894–895. [Google Scholar] [CrossRef]
- Randazzo, R.; Lauceri, R.; Mammana, A.; D'Urso, A.; Purrello, R. Interactions of Lambda and Delta Enantiomers of Ruthenium(II) Cationic Complexes with Achiral Anionic Porphyrins. Chirality 2009, 21, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, R.; Mammana, A.; D’Urso, A.; Lauceri, R.; Purrello, R. Reversible “Chiral Memory” in Ruthenium Tris(phenanthroline)-Anionic Porphyrin Complexes. Angew. Chemie-Int. Ed. 2008, 47, 9879–9882. [Google Scholar] [CrossRef] [PubMed]
- Rosaria, L.; D’Urso, A.; Mammana, A.; Purrello, R. Chiral memory: Induction, amplification, and switching in porphyrin assemblies. Chirality 2008, 20, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Romeo, A.; Zagami, R.; Micali, N.; Scolaro, L.M. Kinetic effects of tartaric acid on the growth of chiral J-aggregates of tetrakis(4-sulfonatophenyl)porphyrin. Chem. Commun. 2012, 48, 4872–4874. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Romeo, A.; De Luca, G.; Villari, V.; Scolaro, L.M.; Micali, N. Scaling the Chirality in Porphyrin J-Nanoaggregates. J. Am. Chem. Soc. 2011, 133, 765–767. [Google Scholar] [CrossRef]
- Purrello, R.; Scolaro, L.M.; Bellacchio, E.; Gurrieri, S.; Romeo, A. Chiral H- and J-Type Aggregates of meso-Tetrakis(4-sulfonatophenyl)porphine on a-Helical Polyglutamic Acid Induced by Cationic Porphyrins. Inorg. Chem. 1998, 37, 3647–3648. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Liu, M. Fabrication of Chiral Langmuir-Schaefer Films from Achiral TPPS and Amphiphiles through the Adsorption at the Air/Water Interface. J. Phys. Chem. B 2003, 107, 2565–2569. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, J.; Liu, M. Supramolecular Chirality of Achiral TPPS Complexed with Chiral Molecular Films. J. Phys. Chem. B 2003, 107, 12768–12773. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, M. Aggregation and Induced Chirality of an Anionic meso-Tetraphenylsulfonato Porphyrin (TPPS) on a Layer-by-Layer Assembled DNA/PAH Matrix. J. Phys. Chem. B 2004, 108, 2880–2884. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.H. Supramolecular Chirality and Chiral Inversion of Tetraphenylsulfonato Porphyrin Assemblies on Optically Active Polylysine. J. Phys. Chem. B 2009, 113, 14015–14020. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, X.; Li, Y.; Ma, R.; An, Y.; Shi, L. Chiral Micelles of Achiral TPPS and Diblock Copolymer Induced by Amino Acids. Macromolecules 2009, 42, 6253–6260. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Escudero, C.; Acosta-Reyes, F.; Casas, M.T.; Altoe, V.; Aloni, S.; Oncins, G.; Sorrenti, A.; Crusats, J.; Campos, J.L.; et al. Structure vs. properties-chirality, optics and shapes - in amphiphilic porphyrin J-aggregates. J. Mater. Chem. C 2013, 1, 3337–3346. [Google Scholar] [CrossRef] [Green Version]
- Randazzo, R.; Gaeta, M.; Gangemi, C.M.A.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Chiral Recognition of L- and D- Amino Acid by Porphyrin Supramolecular Aggregates. Molecules 2018, 24, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, M.; Mazzaglia, A.; Piperno, A.; Cordaro, A.; Zagami, R.; Castriciano, M.A.; Romeo, A.; Monsù Scolaro, L. Novel Nanohybrids Based on Supramolecular Assemblies of Meso-tetrakis-(4-sulfonatophenyl) Porphyrin J-aggregates and Amine-Functionalized Carbon Nanotubes. Nanomaterials 2020, 10, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, M.; Castriciano, M.A.; Romeo, A.; De Luca, G.; Machado, N.; Howes, B.D.; Smulevich, G.; Scolaro, L.M. Nanohybrid Assemblies of Porphyrin and Au-10 Cluster Nanoparticles. Nanomaterials 2019, 9, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hachemi, Z.; Balaban, T.S.; Campos, J.L.; Cespedes, S.; Crusats, J.; Escudero, C.; Kamma-Lorger, C.S.; Llorens, J.; Malfois, M.; Mitchell, G.R.; et al. Effect of Hydrodynamic Forces on meso-(4-Sulfonatophenyl)-Substituted Porphyrin J-Aggregate Nanoparticles: Elasticity, Plasticity and Breaking. Chem. Eur. J. 2016, 22, 9740–9749. [Google Scholar] [CrossRef]
- Escudero, C.; Crusat, J.; Diez-Perez, I.; El-Hachemi, Z.; Ribo, J.M. Folding and hydrodynamic forces in J-aggregates of 5-phenyl-10,15,20-tris-(4-sulfo-phenyl)porphyrin. Angew. Chem. Int. Ed. 2006, 45, 8032–8035. [Google Scholar] [CrossRef]
- Ribo, J.M.; Crusats, J.; Sagues, F.; Claret, J.; Rubires, R. Chiral Sign Induction by Vortices During the Formation of Mesophases in Stirred Solutions. Science 2001, 292, 2063–2066. [Google Scholar] [CrossRef]
- D’Urso, A.; Randazzo, R.; Lo Faro, L.; Purrello, R. Vortexes and Nanoscale Chirality. Angew. Chem. Int. Edn Engl. 2010, 49, 108–112. [Google Scholar] [CrossRef]
- Crusats, J.; El-Hachemi, Z.; Ribo, J.M. Hydrodynamic Effects on Chiral Induction. Chem. Soc. Rev. 2010, 39, 569–577. [Google Scholar] [CrossRef]
- Micali, N.; Engelkamp, H.; van Rhee, P.G.; Christianen, P.C.M.; Scolaro, L.M.; Maan, J.C. Selection of supramolecular chirality by application of rotational and magnetic forces. Nat. Chem. 2012, 4, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Canillas, A.; Purrello, R.; Ribo, J.M. Evidence of induced chirality in stirred solutions of supramolecular nanofibers. Opt. Lett. 2009, 34, 2177–2179. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.S.; Li, Y.K.; Yan, F.S.; Liu, C.; Sang, Y.T.; Tian, F.; Feng, Q.; Duan, P.F.; Zhang, L.; Shi, X.H.; et al. Control over the emerging chirality in supramolecular gels and solutions by chiral microvortices in milliseconds. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Romeo, A.; Castriciano, M.A.; Occhiuto, I.; Zagami, R.; Pasternack, R.F.; Scolaro, L.M. Kinetic Control of Chirality in Porphyrin J-Aggregates. J. Am. Chem. Soc. 2014, 136, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Monsu Scolaro, L.; Castriciano, M.; Romeo, A.; Mazzaglia, A.; Mallamace, F.; Micali, N. Nucleation effects in the aggregation of water-soluble porphyrin aqueous solutions. Phys. A Stat. Mech. Appl. 2002, 304, 158–169. [Google Scholar] [CrossRef]
- Mallamace, F.; Micali, N.; Trusso, S.; Scolaro, L.M.; Romeo, A.; Terracina, A.; Pasternack, R.F. Experimental evidence for self-similar structures in the aggregation of porphyrins in aqueous solutions. Phys. Rev. Lett. 1996, 76, 4741–4744. [Google Scholar] [CrossRef]
- Mallamace, F.; Monsu’ Scolaro, L.; Romeo, A.; Micali, N. Crossover in the Kinetic Growth Process of Porphyrin Aggregation. Phys. Rev. Lett. 1999, 82, 3480–3483. [Google Scholar] [CrossRef]
- Romeo, A.; Castriciano, M.A.; Zagami, R.; Pollicino, G.; Monsu Scolaro, L.; Pasternack, R.F. Effect of zinc cations on the kinetics for supramolecular assembling and the chirality of porphyrin J-aggregates. Chem. Sci. 2017, 8, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Hambright, P. The coordination chemistry of metalloporphyrins. Coord. Chem. Rev. 1971, 6, 247–268. [Google Scholar] [CrossRef]
- Gaeta, M.; Randazzo, R.; Cristaldi, D.A.; D’Urso, A.; Purrello, R.; Fragala, M.E. ZnTPPS demetalation: Role of polyelectrolytes on aggregation after protonation in acid. J. Porphyr. Phthalocyanines 2017, 21, 426–430. [Google Scholar] [CrossRef]
- Trapani, M.; Occhiuto, I.G.; Zagami, R.; De Luca, G.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M.; Pasternack, R.F. Mechanism for Copper(II)-Mediated Disaggregation of a Porphyrin J-Aggregate. Acs Omega 2018, 3, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Occhiuto, I.; De Luca, G.; Trapani, M.; Scolaro, L.M.; Pasternack, R.F. Peripheral Stepwise Degradation of a Porphyrin J-Aggregate. Inorg. Chem. 2012, 51, 10074–10076. [Google Scholar] [CrossRef] [PubMed]
- Zagami, R.; Castriciano, M.A.; Romeo, A.; Trapani, M.; Pedicini, R.; Scolaro, L.M. Tuning supramolecular chirality in nano and mesoscopic porphyrin J-aggregates. Dyes Pigment. 2017, 142, 255–261. [Google Scholar] [CrossRef]
- Scolaro, L.M.; Romeo, A.; Castriciano, M.A.; Micali, N. Unusual optical properties of porphyrin fractal J-aggregates. Chem. Commun. 2005, 3018–3020. [Google Scholar] [CrossRef] [PubMed]
- Micali, N.; Villari, V.; Scolaro, L.M.; Romeo, A.; Castriciano, M.A. Light scattering enhancement in an aqueous solution of spermine-induced fractal J-aggregate composite. Phys. Rev. 2005, 72. [Google Scholar] [CrossRef]
- Romeo, A.; Castriciano, M.A.; Scolaro, L.M. Spectroscopic and kinetic investigations on porphyrin J-aggregates induced by polyamines. J. Porphyr. Phthalocyanines 2010, 14, 713–721. [Google Scholar] [CrossRef]
- Cheung, S.K.; Dixon, F.L.; Fleischer, E.B.; Jeter, D.Y.; Krishnamurthy, M. Kinetic studies of the formation, acid-catalyzed solvolysis, and cupric ion displacement of a zinc porphyrin in aqueous solutions. Bioinorg. Chem. 1973, 2, 281–294. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Fleming, C.; Herring, S.; Collings, P.J.; dePaula, J.; DeCastro, G.; Gibbs, E.J. Aggregation kinetics of extended porphyrin and cyanine dye assemblies. Biophys. J. 2000, 79, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Pasternack, R.F.; Gibbs, E.J.; Collings, P.J.; dePaula, J.C.; Turzo, L.C.; Terracina, A. A nonconventional approach to supramolecular formation dynamics. The kinetics of assembly of DNA-bound porphyrins. J. Am. Chem. Soc. 1998, 120, 5873–5878. [Google Scholar] [CrossRef]
- Parkash, J.; Robblee, J.H.; Agnew, J.; Gibbs, E.; Collings, P.; Pasternack, R.F.; de Paula, J.C. Depolarized resonance light scattering by porphyrin and chlorophyll a aggregates. Biophys. J. 1998, 74, 2089–2099. [Google Scholar] [CrossRef] [Green Version]
- Pasternack, R.F.; Collings, P.J. Resonance Light-Scattering—A New Technique for Studying Chromophore Aggregation. Science 1995, 269, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Short, J.M.; Berriman, J.A.; Kübel, C.; El-Hachemi, Z.; Naubron, J.-V.; Balaban, T.S. Electron Cryo-Microscopy of TPPS4⋅2HCl Tubes Reveals a Helical Organisation Explaining the Origin of their Chirality. ChemPhysChem 2013, 14, 3209–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villari, V.; Mazzaglia, A.; Castriciano, M.A.; Luca, G.d.; Romeo, A.; Scolaro, L.M.; Micali, N. Optical enhancement and structural properties of a hybrid organic-inorganic ternary nanocomposite. J. Phys. Chem. C 2011, 115, 5435–5439. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Zagami, R.; Trapani, M.; Bolzonello, L.; Romeo, A.; Castriciano, M.A.; Collini, E.; Monsu Scolaro, L. The role of counter-anions in the kinetics and chirality of porphyrin J-aggregates. Chem. Commun. 2016, 52, 11520–11523. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 103 [H+]/M | 103 kobs1/s−1 a | 103 kc/s−1 b | mb | nb | 103 kc/s−1 c | mc | mc |
|---|---|---|---|---|---|---|---|
| 13.5 d | 0.261 ± 0.001 | 0.537 ± 0.002 | 16.5 ± 2.7 | 36.7 ± 6.3 | e | e | e |
| 24.6 d | 1.29 ± 0.01 | 0.979 ± 0.001 | 3.9 ± 0.1 | 16.4 ± 0.4 | 1.12 ± 0.38 | 5.0 ± 0.9 | 13.3 ± 5.8 |
| 34.7 d | 3.55 ± 0.02 | 1.61 ± 0.01 | 3.1 ± 0.1 | 10.0 ± 0.1 | 1.64 ± 0.60 | 3.0 ± 0.3 | 8.3 ± 3.6 |
| 53.7 d | 9.76 ± 0.05 | 2.23 ± 0.01 | 3.0 ± 0.1 | 8.2 ± 0.1 | 2.23 ± 0.02 | 3.1 ± 0.1 | 7.4 ± 0.2 |
| 97.7 d | 54.7 ± 0.7 | 2.71 ± 0.01 | 3.0 ± 0.1 | 7.1 ± 0.1 | 2.63 ± 0.20 | 3.7 ± 0.1 | 8.3 ± 0.9 |
| 53.7 f | 12.3 ± 0.1 | 1.48 ± 0.01 | 1.8 ± 0.1 | 4.9 ± 0.1 | 1.24 ± 0.26 | 3.1 ± 0.3 | 7.0 ± 1.9 |
| 53.7 g | 9.59 ± 0.08 | 2.23 ± 0.01 | 3.0 ± 0.1 | 8.2 ± 0.2 | 2.23 ± 0.60 | 3.1 ± 0.3 | 7.4 ± 2.6 |
| 53.7 h | 10.8 ± 0.1 | 4.26 ± 0.01 | 3.3 ± 0.1 | 10.5 ± 0.1 | 4.29 ± 0.99 | 3.2 ± 0.2 | 9.0 ± 2.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Occhiuto, I.G.; Castriciano, M.A.; Trapani, M.; Zagami, R.; Romeo, A.; Pasternack, R.F.; Monsù Scolaro, L. Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin. Int. J. Mol. Sci. 2020, 21, 4001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114001
Occhiuto IG, Castriciano MA, Trapani M, Zagami R, Romeo A, Pasternack RF, Monsù Scolaro L. Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin. International Journal of Molecular Sciences. 2020; 21(11):4001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114001
Chicago/Turabian StyleOcchiuto, Ilaria Giuseppina, Maria Angela Castriciano, Mariachiara Trapani, Roberto Zagami, Andrea Romeo, Robert F. Pasternack, and Luigi Monsù Scolaro. 2020. "Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin" International Journal of Molecular Sciences 21, no. 11: 4001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114001