Expression Changes and Impact of the Extracellular Matrix on Etoposide Resistant Human Retinoblastoma Cell Lines

 , and
, and
Abstract
:1. Introduction
2. Results
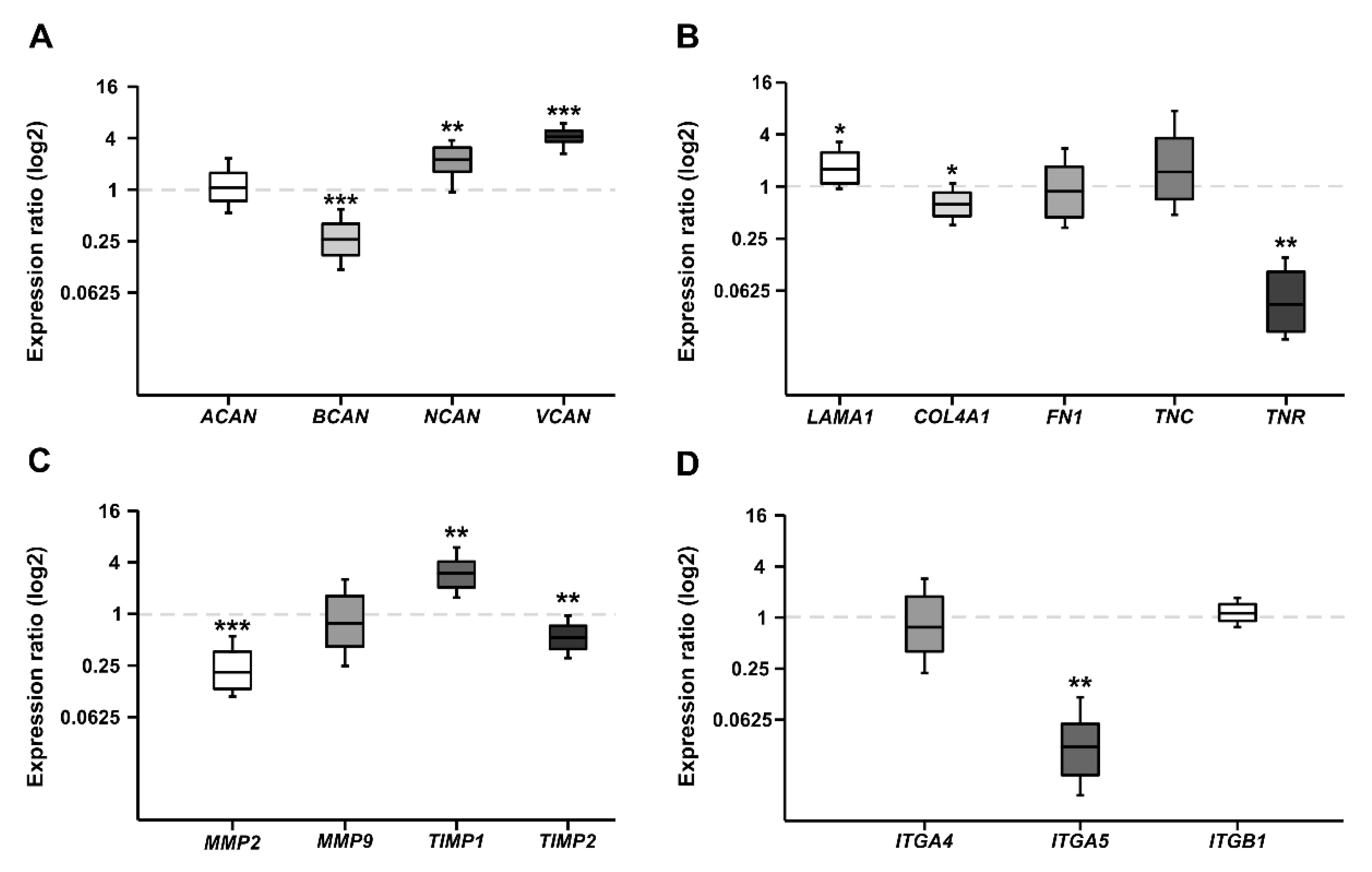
2.1. CSPG mRNA Expression in WERI-RB1 and WERI-ETOR
2.2. Expression of ECM Glycoproteins in WERI-RB1 and WERI-ETOR
2.3. Expression of MMPs and TIMPs in WERI-RB1 and WERI-ETOR
2.4. Expression of Integrin Receptor Subunits in WERI-RB1 and WERI-ETOR
2.5. Expression of CSPGs, ECM Glycoproteins, MMPs, TIMPs, and Integrin Receptor Subunits in Etoposide Sensitive and Resistant Y79 RB cells
2.6. Expression of RPTPβ/ζ Isoforms in WERI-RB1 and WERI-ETOR
2.7. Impact of the ECM on WERI-RB1 and WERI-ETOR Cell Survival
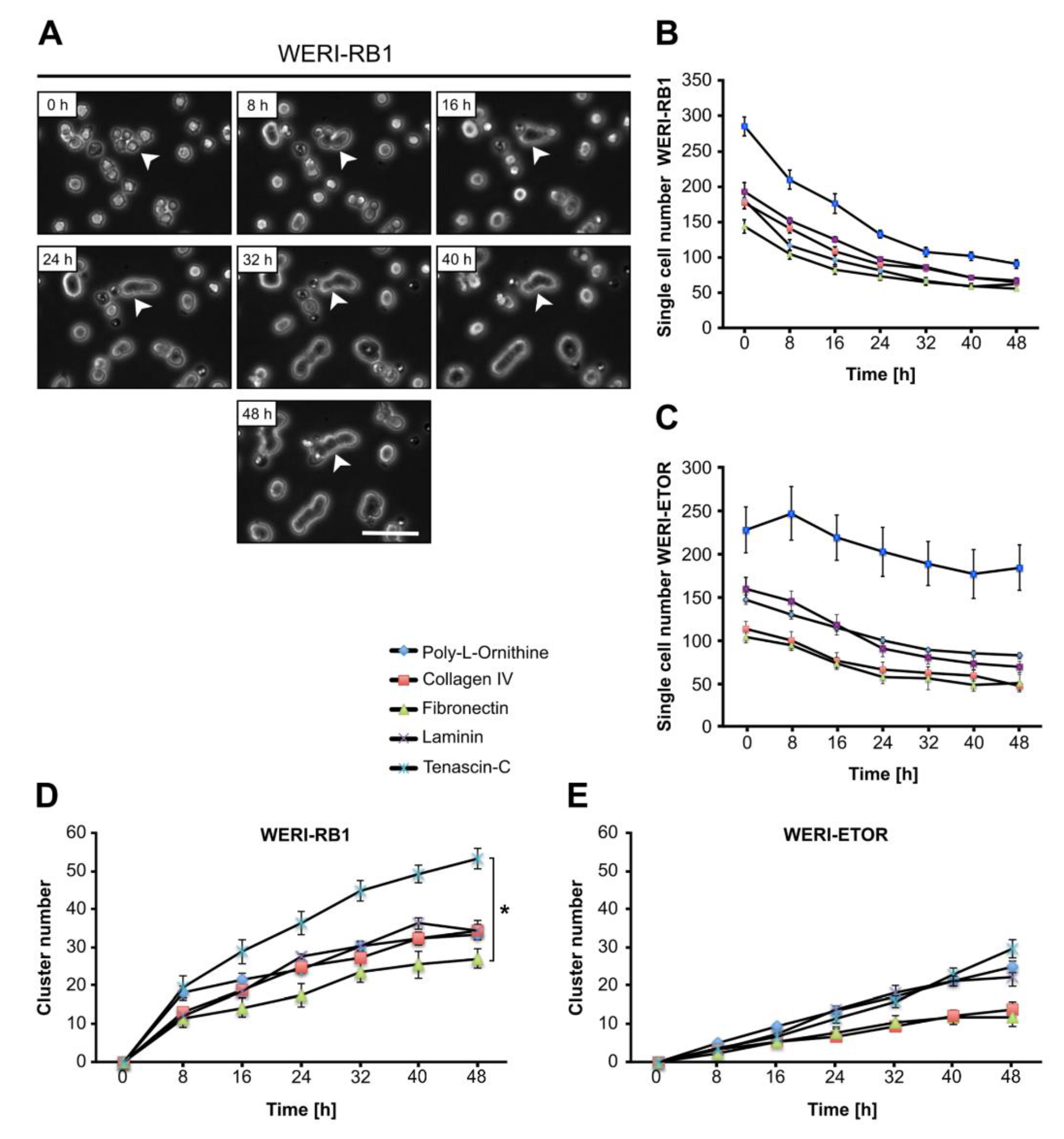
2.8. Influence of the ECM on WERI-RB1 and WERI-ETOR Cell Proliferation
2.9. Cluster Formation Capacity of WERI-RB1 and WERI-ETOR Cells on Different ECM Substrates
3. Discussion
3.1. Expression of CSPGs in RB Cells
3.2. Expression of Glycoproteins and Collagen IV in RB Cells
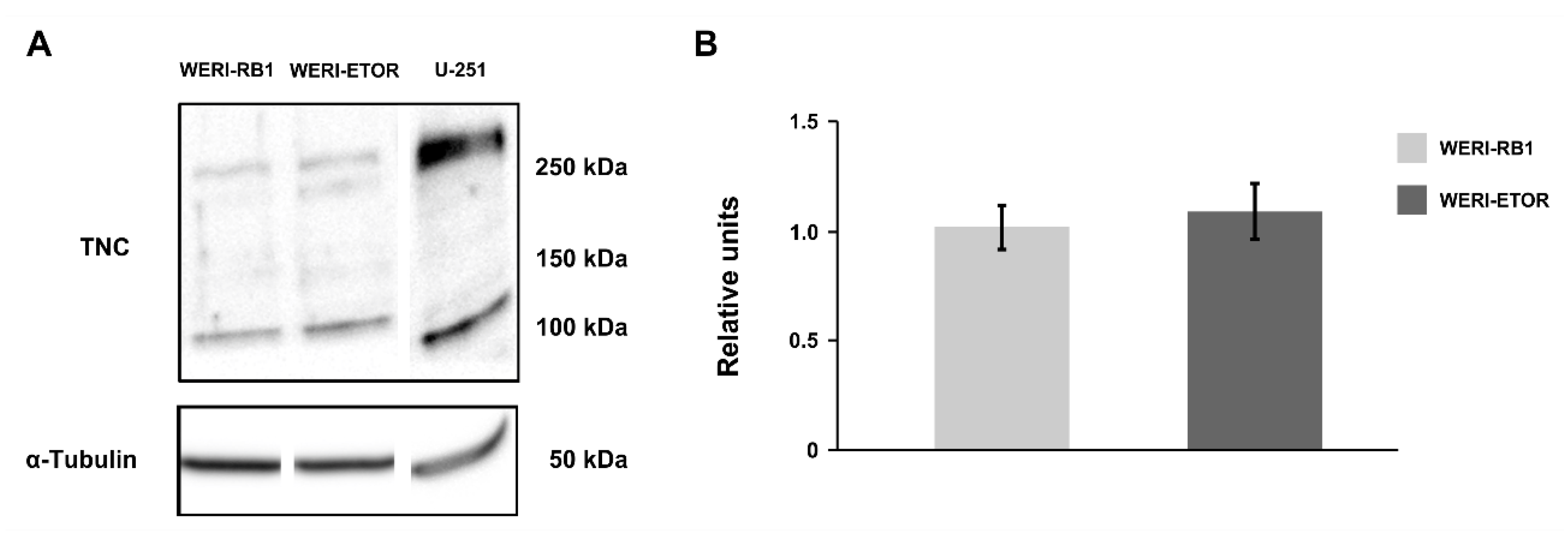
3.3. Expression of Tenascin-C and Tenascin-R in RB Cells
3.4. Expression of MMPs and TIMPs in RB Cells
3.5. Integrin Expression in RB Cells
3.6. Expression of RPTPβ/ζ in RB Cells
3.7. ECM Influence on the Apoptotic Rate of RB Cells
3.8. ECM Influence on the Proliferation Rate of RB Cells
3.9. ECM Influence on Cluster Formation of RB Cells
3.10. Limitations and Future Perspectives of the Study
4. Materials and Methods
4.1. Cultivation and Authentication Verification of Human RB Cell Lines
4.2. Cultivation of the Human Glioblastoma Cell Line U-251-MG and Purification of Human Tenascin-C
4.3. Cell Proliferation and Cluster Formation Analyses Via Time-Lapse Video Microscopy
4.4. RNA Isolation and cDNA Synthesis
4.5. Quantitative Real-Time PCR Analyses (RT-qPCR)
4.6. Semi-Quantitative Reverse-Transcription PCR Analyses (RT-PCR)
4.7. Immunocytochemistry
4.8. Protein Lysis and Western Blot
4.9. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, I.D.; Onadim, Z.; Karaa, E.; Duncan, C.; Chowdhury, T.; Scheimberg, I.; Ohnuma, S.I.; Reddy, M.A.; Sagoo, M.S. The management of retinoblastoma. Oncogene 2018, 37, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Kimani, K.; Dimba, E.A.; Gronsdahl, P.; White, A.; Chan, H.S.; Gallie, B.L. Retinoblastoma. Lancet 2012, 379, 1436–1446. [Google Scholar] [CrossRef] [Green Version]
- Kivela, T. The epidemiological challenge of the most frequent eye cancer: Retinoblastoma, an issue of birth and death. Br. J. Ophthalmol. 2009, 93, 1129–1131. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rojanaporn, D.; Chawla, B.; Sundar, G.; Gopal, L.; Khetan, V. Retinoblastoma in Asia. Eye (Lond.) 2019, 33, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houston, S.K.; Murray, T.G.; Wolfe, S.Q.; Fernandes, C.E. Current update on retinoblastoma. Int. Ophthalmol. Clin. 2011, 51, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.M.; Draper, G.J.; Kingston, J.E. Retinoblastoma in Great Britain 1969-80: Incidence, treatment, and survival. Br. J. Ophthalmol. 1988, 72, 576–583. [Google Scholar] [CrossRef] [Green Version]
- Abramson, D.H.; Schefler, A.C. Update on retinoblastoma. Retina 2004, 24, 828–848. [Google Scholar] [CrossRef]
- Schuler, A.O.; Bornfeld, N. Current therapy aspects of intraocular tumors. Ophthalmologe 2000, 97, 207–222. [Google Scholar]
- Shields, C.L.; De Potter, P.; Himelstein, B.P.; Shields, J.A.; Meadows, A.T.; Maris, J.M. Chemoreduction in the initial management of intraocular retinoblastoma. Arch. Ophthalmol. 1996, 114, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Himelstein, B.; Shields, C.L.; Shields, J.A.; Needle, M.; Miller, D.; Bunin, G.R.; Meadows, A.T. Chemoreduction and local ophthalmic therapy for intraocular retinoblastoma. J. Clin. Oncol. 2000, 18, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Toma, N.M.; Hungerford, J.L.; Plowman, P.N.; Kingston, J.E.; Doughty, D. External beam radiotherapy for retinoblastoma: II. Lens sparing technique. Br. J. Ophthalmol. 1995, 79, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hungerford, J.L.; Toma, N.M.; Plowman, P.N.; Doughty, D.; Kingston, J.E. Whole-eye versus lens-sparing megavoltage therapy for retinoblastoma. Front. Radiat. Oncol. 1997, 30, 81–87. [Google Scholar]
- Abramson, D.H.; Marr, B.P.; Dunkel, I.J.; Brodie, S.; Zabor, E.C.; Driscoll, S.J.; Gobin, Y.P. Intra-arterial chemotherapy for retinoblastoma in eyes with vitreous and/or subretinal seeding: 2-year results. Br. J. Ophthalmol. 2012, 96, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Shields, C.L.; Manjandavida, F.P.; Lally, S.E.; Pieretti, G.; Arepalli, S.A.; Caywood, E.H.; Jabbour, P.; Shields, J.A. Intra-arterial chemotherapy for retinoblastoma in 70 eyes: Outcomes based on the international classification of retinoblastoma. Ophthalmology 2014, 121, 1453–1460. [Google Scholar] [CrossRef]
- Munier, F.L.; Beck-Popovic, M.; Balmer, A.; Gaillard, M.C.; Bovey, E.; Binaghi, S. Occurrence of sectoral choroidal occlusive vasculopathy and retinal arteriolar embolization after superselective ophthalmic artery chemotherapy for advanced intraocular retinoblastoma. Retina 2011, 31, 566–573. [Google Scholar] [CrossRef]
- Jia, M.; Wei, Z.; Liu, P.; Zhao, X. Silencing of ABCG2 by MicroRNA-3163 Inhibits Multidrug Resistance in Retinoblastoma Cancer Stem Cells. J. Korean Med. Sci. 2016, 31, 836–842. [Google Scholar] [CrossRef]
- Hoye, A.M.; Erler, J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016, 310, C955–C967. [Google Scholar] [CrossRef] [Green Version]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef] [Green Version]
- Van Obberghen-Schilling, E.; Tucker, R.P.; Saupe, F.; Gasser, I.; Cseh, B.; Orend, G. Fibronectin and tenascin-C: Accomplices in vascular morphogenesis during development and tumor growth. Int. J. Dev. Biol. 2011, 55, 511–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperigkou, Z.; Mohr, B.; Karamanos, N.; Gotte, M. Shed proteoglycans in tumor stroma. Cell Tissue Res. 2016, 365, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Wei, H.; Lu, T. Targeting microenvironment in cancer therapeutics. Oncotarget 2016, 7, 52575–52583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate Functions of Matrix Metalloproteinases in Physiological and Pathological Conditions. J. Cell Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef]
- Kapoor, C.; Vaidya, S.; Hitesh, V.W.; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. 2016, 12, 28–35. [Google Scholar]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Hodkinson, P.S.; Mackinnon, A.C.; Sethi, T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Int. J. Radiat. Biol. 2007, 83, 733–741. [Google Scholar] [CrossRef]
- Reinhard, J.; Brosicke, N.; Theocharidis, U.; Faissner, A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int. J. Biochem. Cell Biol. 2016, 81, 174–183. [Google Scholar] [CrossRef]
- Seigel, G.M.; Campbell, L.M.; Narayan, M.; Gonzalez-Fernandez, F. Cancer stem cell characteristics in retinoblastoma. Mol. Vis. 2005, 11, 729–737. [Google Scholar]
- Seigel, G.M.; Hackam, A.S.; Ganguly, A.; Mandell, L.M.; Gonzalez-Fernandez, F. Human embryonic and neuronal stem cell markers in retinoblastoma. Mol. Vis. 2007, 13, 823–832. [Google Scholar]
- Balla, M.M.; Vemuganti, G.K.; Kannabiran, C.; Honavar, S.G.; Murthy, R. Phenotypic characterization of retinoblastoma for the presence of putative cancer stem-like cell markers by flow cytometry. Invest. Ophthalmol. Vis. Sci. 2009, 50, 1506–15014. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Li, Y.; Peng, F.; Huang, B.; Lin, J.; Zhang, W.; Zheng, J.; Jiang, R.; Song, G.; Ge, J. Identification of tumorigenic retinal stem-like cells in human solid retinoblastomas. Int. J. Cancer 2007, 121, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Srivastava, A.; Kumar, S.; Singh, U.; Goswami, S.; Chawla, B.; Bajaj, M.S.; Kashyap, S.; Kaur, J. Expression of multidrug resistance proteins in retinoblastoma. Int. J. Ophthalmol. 2017, 10, 1655–1661. [Google Scholar] [PubMed]
- McFall, R.C.; Sery, T.W.; Makadon, M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res. 1977, 37, 1003–1010. [Google Scholar] [PubMed]
- Stephan, H.; Boeloeni, R.; Eggert, A.; Bornfeld, N.; Schueler, A. Photodynamic therapy in retinoblastoma: Effects of verteporfin on retinoblastoma cell lines. Invest. Ophthalmol. Vis. Sci. 2008, 49, 3158–3163. [Google Scholar] [CrossRef] [Green Version]
- Mergler, S.; Cheng, Y.; Skosyrski, S.; Garreis, F.; Pietrzak, P.; Kociok, N.; Dwarakanath, A.; Reinach, P.S.; Kakkassery, V. Altered calcium regulation by thermosensitive transient receptor potential channels in etoposide-resistant WERI-Rb1 retinoblastoma cells. Exp. Eye Res. 2012, 94, 157–173. [Google Scholar] [CrossRef]
- Kakkassery, V.; Skosyrski, S.; Luth, A.; Kleuser, B.; van der Giet, M.; Tate, R.; Reinhard, J.; Faissner, A.; Joachim, S.C.; Kociok, N. Etoposide Upregulates Survival Favoring Sphingosine-1-Phosphate in Etoposide-Resistant Retinoblastoma Cells. Pathol. Oncol. Res. 2019, 25, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Reid, T.W.; Albert, D.M.; Rabson, A.S.; Russell, P.; Craft, J.; Chu, E.W.; Tralka, T.S.; Wilcox, J.L. Characteristics of an established cell line of retinoblastoma. J. Natl. Cancer Inst. 1974, 53, 347–360. [Google Scholar] [CrossRef]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Balcioglu, H.E.; Danen, E.H. Integrin signaling in control of tumor growth and progression. Int. J. Biochem. Cell Biol. 2013, 45, 1012–1015. [Google Scholar] [CrossRef]
- Missan, D.S.; DiPersio, M. Integrin control of tumor invasion. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 309–324. [Google Scholar] [CrossRef]
- Barnea, G.; Grumet, M.; Milev, P.; Silvennoinen, O.; Levy, J.B.; Sap, J.; Schlessinger, J. Receptor tyrosine phosphatase beta is expressed in the form of proteoglycan and binds to the extracellular matrix protein tenascin. J. Biol. Chem. 1994, 269, 14349–14352. [Google Scholar]
- Grumet, M.; Milev, P.; Sakurai, T.; Karthikeyan, L.; Bourdon, M.; Margolis, R.K.; Margolis, R.U. Interactions with tenascin and differential effects on cell adhesion of neurocan and phosphacan, two major chondroitin sulfate proteoglycans of nervous tissue. J. Biol. Chem. 1994, 269, 12142–12146. [Google Scholar]
- Norman, S.A.; Golfinos, J.G.; Scheck, A.C. Expression of a receptor protein tyrosine phosphatase in human glial tumors. J. Neurooncol. 1998, 36, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanos, N.K. Special issue: Translating extracellular matrix: From cancer progression to therapeutics. Semin. Cancer Biol. 2020, 62, iii–v. [Google Scholar] [CrossRef] [PubMed]
- Rauch, U. Extracellular matrix components associated with remodeling processes in brain. Cell Mol. Life Sci. 2004, 61, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Varga, I.; Hutoczki, G.; Szemcsak, C.D.; Zahuczky, G.; Toth, J.; Adamecz, Z.; Kenyeres, A.; Bognar, L.; Hanzely, Z.; Klekner, A. Brevican, neurocan, tenascin-C and versican are mainly responsible for the invasiveness of low-grade astrocytoma. Pathol. Oncol. Res. 2012, 18, 413–420. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Dwyer, C.A.; Bi, W.L.; Viapiano, M.S.; Matthews, R.T. Brevican knockdown reduces late-stage glioma tumor aggressiveness. J. Neurooncol. 2014, 120, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimakopoulou, A.P.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. The biological role of chondroitin sulfate in cancer and chondroitin-based anticancer agents. Vivo 2008, 22, 385–389. [Google Scholar]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isogai, Z.; Shinomura, T.; Yamakawa, N.; Takeuchi, J.; Tsuji, T.; Heinegard, D.; Kimata, K. 2B1 antigen characteristically expressed on extracellular matrices of human malignant tumors is a large chondroitin sulfate proteoglycan, PG-M/versican. Cancer Res. 1996, 56, 3902–3908. [Google Scholar] [PubMed]
- Pirinen, R.; Leinonen, T.; Bohm, J.; Johansson, R.; Ropponen, K.; Kumpulainen, E.; Kosma, V.M. Versican in nonsmall cell lung cancer: Relation to hyaluronan, clinicopathologic factors, and prognosis. Hum. Pathol. 2005, 36, 44–50. [Google Scholar] [CrossRef]
- Quirico-Santos, T.; Fonseca, C.O.; Lagrota-Candido, J. Brain sweet brain: Importance of sugars for the cerebral microenvironment and tumor development. Arq. Neuropsiquiatr. 2010, 68, 799–803. [Google Scholar] [CrossRef] [Green Version]
- Abaskharoun, M.; Bellemare, M.; Lau, E.; Margolis, R.U. Expression of hyaluronan and the hyaluronan-binding proteoglycans neurocan, aggrecan, and versican by neural stem cells and neural cells derived from embryonic stem cells. Brain Res. 2010, 1327, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Klausmeyer, A.; Garwood, J.; Faissner, A. Differential expression of phosphacan/RPTPbeta isoforms in the developing mouse visual system. J. Comp. Neurol. 2007, 504, 659–679. [Google Scholar] [CrossRef]
- Popp, S.; Andersen, J.S.; Maurel, P.; Margolis, R.U. Localization of aggrecan and versican in the developing rat central nervous system. Dev. Dyn. 2003, 227, 143–149. [Google Scholar] [CrossRef]
- Felemban, M.; Dorgau, B.; Hunt, N.C.; Hallam, D.; Zerti, D.; Bauer, R.; Ding, Y.; Collin, J.; Steel, D.; Krasnogor, N.; et al. Extracellular matrix component expression in human pluripotent stem cell-derived retinal organoids recapitulates retinogenesis in vivo and reveals an important role for IMPG1 and CD44 in the development of photoreceptors and interphotoreceptor matrix. Acta Biomater. 2018, 74, 207–221. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlund, D.; Franklin, O.; Lundberg, E.; Lundin, C.; Sund, M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Burnier, J.V.; Wang, N.; Michel, R.P.; Hassanain, M.; Li, S.; Lu, Y.; Metrakos, P.; Antecka, E.; Burnier, M.N.; Ponton, A.; et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene 2011, 30, 3766–3783. [Google Scholar] [CrossRef] [Green Version]
- Campochiaro, P.A.; Jerdon, J.A.; Glaser, B.M. The extracellular matrix of human retinal pigment epithelial cells in vivo and its synthesis in vitro. Invest. Ophthalmol. Vis. Sci. 1986, 27, 1615–1621. [Google Scholar] [PubMed]
- Pina, Y.; Boutrid, H.; Schefler, A.; Dubovy, S.; Feuer, W.; Jockovich, M.E.; Murray, T.G. Blood vessel maturation in retinoblastoma tumors: Spatial distribution of neovessels and mature vessels and its impact on ocular treatment. Invest. Ophthalmol. Vis. Sci. 2009, 50, 1020–1024. [Google Scholar] [CrossRef]
- Jockovich, M.E.; Pina, Y.; Alegret, A.; Cebulla, C.; Feuer, W.; Murray, T.G. Heterogeneous tumor vasculature in retinoblastoma: Implications for vessel targeting therapy. Retina 2008, 28 (Suppl. 3), S81–S86. [Google Scholar] [CrossRef]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour. Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [Green Version]
- Skubitz, A.P.; Grossman, M.D.; McCarthy, J.B.; Wayner, E.A.; Cameron, J.D. The decreased adhesion of Y79 retinoblastoma cells to extracellular matrix proteins is due to a deficit of integrin receptors. Invest. Ophthalmol. Vis. Sci. 1994, 35, 2820–2833. [Google Scholar]
- Nowak, M.; Freudenberg, U.; Tsurkan, M.V.; Werner, C.; Levental, K.R. Modular GAG-matrices to promote mammary epithelial morphogenesis in vitro. Biomaterials 2017, 112, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Amin, J.D.; Kirschmann, D.A.; Schiemann, W.P. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-beta signaling in breast cancer cells. Neoplasia 2011, 13, 406–418. [Google Scholar] [CrossRef] [Green Version]
- Rossow, L.; Veitl, S.; Vorlova, S.; Wax, J.K.; Kuhn, A.E.; Maltzahn, V.; Upcin, B.; Karl, F.; Hoffmann, H.; Gatzner, S.; et al. LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene 2018, 37, 4921–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.M.; Bird, D.; Welti, J.C.; Gourlaouen, M.; Lang, G.; Murray, G.I.; Reynolds, A.R.; Cox, T.R.; Erler, J.T. Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res. 2013, 73, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.M.; Bird, D.; Lang, G.; Cox, T.R.; Erler, J.T. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 2013, 32, 1863–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Deng, L.; Zhu, J.; Rychahou, P.G.; Xu, R. Prolyl-4-hydroxylase alpha subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, G.; Stewart, R.L.; Chen, J.; Gao, T.; Scott, T.L.; Samayoa, L.M.; O’Connor, K.; Lane, A.N.; Xu, R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat. Commun. 2018, 9, 4456. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, S.K.; Olden, K.; Yamada, K.M. Fibronectin and integrins in invasion and metastasis. Cancer Metastasis. Rev. 1995, 14, 173–189. [Google Scholar] [CrossRef]
- Bae, Y.K.; Kim, A.; Kim, M.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum. Pathol. 2013, 44, 2028–2037. [Google Scholar] [CrossRef]
- Li, Y.; Miao, L.; Yu, M.; Shi, M.; Wang, Y.; Yang, J.; Xiao, Y.; Cai, H. alpha1-antitrypsin promotes lung adenocarcinoma metastasis through upregulating fibronectin expression. Int. J. Oncol. 2017, 50, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.J.; Yan, Q.; Zhang, G.L.; Wie, Y.H.; Yuan, W.B.; Xu, W.; Huang, L.F. Expression and Prognostic Role of FHIT, Fibronectin, and PTEN in Hepatocellular Carcinoma. Clin. Lab. 2016, 62, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J. Cancer. 2017, 8, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Banerji, A.; Frei, E.; Chatterjee, A. Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci. 2008, 82, 467–476. [Google Scholar] [CrossRef]
- Reinhard, J.; Roll, L.; Faissner, A. Tenascins in Retinal and Optic Nerve Neurodegeneration. Front. Integr. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, J.; Joachim, S.C.; Faissner, A. Extracellular matrix remodeling during retinal development. Exp. Eye Res. 2015, 133, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Brosicke, N.; van Landeghem, F.K.; Scheffler, B.; Faissner, A. Tenascin-C is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013, 354, 409–430. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Tucker, R.P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. 2011, 3, a004960. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Bak, K.H.; Kim, Y.S.; Kim, J.M.; Ko, Y.; Oh, S.J.; Kim, K.M.; Hong, E.K. Expression of tenascin-C in astrocytic tumors: Its relevance to proliferation and angiogenesis. Surg. Neurol. 2000, 54, 235–240. [Google Scholar] [CrossRef]
- Gong, X.G.; Lv, Y.F.; Li, X.Q.; Xu, F.G.; Ma, Q.Y. Gemcitabine resistance induced by interaction between alternatively spliced segment of tenascin-C and annexin A2 in pancreatic cancer cells. Biol. Pharm. Bull. 2010, 33, 1261–1267. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.A.; Chen, C.H.; Sun, H.S.; Cheng, C.P.; Tseng, V.S.; Hsu, H.S.; Su, W.C.; Lai, W.W.; Wang, Y.C. Global Oct4 target gene analysis reveals novel downstream PTEN and TNC genes required for drug-resistance and metastasis in lung cancer. Nucleic Acids Res. 2015, 43, 1593–1608. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; He, X.; Wei, W.; Wang, J.; Zhang, T.; Shen, X. Tenascin-C induces resistance to apoptosis in pancreatic cancer cell through activation of ERK/NF-kappaB pathway. Apoptosis 2015, 20, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Al-Yahyaee, A.; Abdullah, N.; Sam, J.E.; Al-Zeheimi, N.; Yaish, M.W.; Adham, S.A. Neuropilin-1 promotes the oncogenic Tenascin-C/integrin beta3 pathway and modulates chemoresistance in breast cancer cells. BMC Cancer 2018, 18, 533. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, I.; Hirakoshi, J.; Kusakabe, M.; Masunaga, A.; Itoyama, S.; Sakakura, T. Relationships among tenascin expression, DNA ploidy patterns, and multidrug resistance gene product (P-glycoprotein) in human colon carcinoma. Jpn. J. Cancer Res. 1993, 84, 703–707. [Google Scholar] [CrossRef]
- Brosicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adh. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Pesheva, P.; Probstmeier, R.; Skubitz, A.P.; McCarthy, J.B.; Furcht, L.T.; Schachner, M. Tenascin-R (J1 160/180 inhibits fibronectin-mediated cell adhesion--functional relatedness to tenascin-C. J. Cell Sci. 1994, 107, 2323–2333. [Google Scholar] [PubMed]
- Long, H.; Zhou, B.; Jiang, F.G. Expression of MMP-2 and MMP-9 in retinoblastoma and their significance. Int. J. Ophthalmol. 2011, 4, 489–491. [Google Scholar] [PubMed]
- Adithi, M.; Nalini, V.; Kandalam, M.; Krishnakumar, S. Expression of matrix metalloproteinases and their inhibitors in retinoblastoma. J. Pediatr. Hematol. Oncol. 2007, 29, 399–405. [Google Scholar] [CrossRef]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef]
- Jeleniewicz, W.; Cybulski, M.; Nowakowski, A.; Stenzel-Bembenek, A.; Guz, M.; Marzec-Kotarska, B.; Kotarski, J.; Stepulak, A. MMP-2 mRNA Expression in Ovarian Cancer Tissues Predicts Patients’ Response to Platinum-Taxane Chemotherapy. Anticancer. Res. 2019, 39, 1821–1827. [Google Scholar] [CrossRef]
- Nalluri, S.; Ghoshal-Gupta, S.; Kutiyanawalla, A.; Gayatri, S.; Lee, B.R.; Jiwani, S.; Rojiani, A.M.; Rojiani, M.V. TIMP-1 Inhibits Apoptosis in Lung Adenocarcinoma Cells via Interaction with Bcl-2. PLoS ONE 2015, 10, e0137673. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.W.; Zhu, H.Y.; Wang, J.Z.; Fu, B.; Lu, Y.; Hong, Q.; Xie, Y.S.; Chen, X.M. Tissue inhibitor of metalloproteinase-1 counteracts glucolipotoxicity in the pancreatic beta-cell line INS-1. Chin. Med. J. Engl. 2011, 124, 258–261. [Google Scholar] [PubMed]
- Li, Y.; Yang, X.; Du, X.; Lei, Y.; He, Q.; Hong, X.; Tang, X.; Wen, X.; Zhang, P.; Sun, Y.; et al. RAB37 Hypermethylation Regulates Metastasis and Resistance to Docetaxel-Based Induction Chemotherapy in Nasopharyngeal Carcinoma. Clin. Cancer Res. 2018, 24, 6495–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, H.; Nishikawa, M.; Tei, H.; Furukawa, J.; Harada, K.; Fujisawa, M. Significance of circulating matrix metalloproteinase-9 to tissue inhibitor of metalloproteinases-2 ratio as a predictor of disease progression in patients with metastatic renal cell carcinoma receiving sunitinib. Urol. Oncol. 2014, 32, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Halon, A.; Nowak-Markwitz, E.; Donizy, P.; Matkowski, R.; Maciejczyk, A.; Gansukh, T.; Gyorffy, B.; Spaczynski, M.; Zabel, M.; Lage, H.; et al. Enhanced immunoreactivity of TIMP-2 in the stromal compartment of tumor as a marker of favorable prognosis in ovarian cancer patients. J. Histochem. Cytochem. 2012, 60, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waleh, N.S.; Murphy, B.J.; Zaveri, N.T. Increase in tissue inhibitor of metalloproteinase-2 (TIMP-2) levels and inhibition of MMP-2 activity in a metastatic breast cancer cell line by an anti-invasive small molecule SR13179. Cancer Lett. 2010, 289, 111–118. [Google Scholar] [CrossRef]
- Wang, Y.; Shenouda, S.; Baranwal, S.; Rathinam, R.; Jain, P.; Bao, L.; Hazari, S.; Dash, S.; Alahari, S.K. Integrin subunits alpha5 and alpha6 regulate cell cycle by modulating the chk1 and Rb/E2F pathways to affect breast cancer metastasis. Mol. Cancer 2011, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Zheng, Y.; Liu, S.; Peng, Z. Fibronectin Regulates the Dynamic Formation of Ovarian Cancer Multicellular Aggregates and the Expression of Integrin Receptors. Asian Pac. J. Cancer Prev. 2018, 19, 2493–2498. [Google Scholar]
- Muller-Tidow, C.; Ji, P.; Diederichs, S.; Potratz, J.; Baumer, N.; Kohler, G.; Cauvet, T.; Choudary, C.; van der Meer, T.; Chan, W.Y.; et al. The cyclin A1-CDK2 complex regulates DNA double-strand break repair. Mol. Cell Biol. 2004, 24, 8917–8928. [Google Scholar] [CrossRef] [Green Version]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin alpha5beta1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef] [Green Version]
- Jacquemet, G.; Green, D.M.; Bridgewater, R.E.; von Kriegsheim, A.; Humphries, M.J.; Norman, J.C.; Caswell, P.T. RCP-driven alpha5beta1 recycling suppresses Rac and promotes RhoA activity via the RacGAP1-IQGAP1 complex. J. Cell Biol. 2013, 202, 917–935. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Koutsioumpa, M.; Poimenidi, E.; Pantazaka, E.; Theodoropoulou, C.; Skoura, A.; Megalooikonomou, V.; Kieffer, N.; Courty, J.; Mizumoto, S.; Sugahara, K.; et al. Receptor protein tyrosine phosphatase beta/zeta is a functional binding partner for vascular endothelial growth factor. Mol. Cancer 2015, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamantopoulou, Z.; Kitsou, P.; Menashi, S.; Courty, J.; Katsoris, P. Loss of receptor protein tyrosine phosphatase beta/zeta (RPTPbeta/zeta) promotes prostate cancer metastasis. J. Biol. Chem. 2012, 287, 40339–40349. [Google Scholar] [CrossRef] [Green Version]
- Rauch, U.; Clement, A.; Retzler, C.; Frohlich, L.; Fassler, R.; Gohring, W.; Faissner, A. Mapping of a defined neurocan binding site to distinct domains of tenascin-C. J. Biol. Chem. 1997, 272, 26905–26912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokosaki, Y.; Matsuura, N.; Higashiyama, S.; Murakami, I.; Obara, M.; Yamakido, M.; Shigeto, N.; Chen, J.; Sheppard, D. Identification of the ligand binding site for the integrin alpha9 beta1 in the third fibronectin type III repeat of tenascin-C. J. Biol. Chem. 1998, 273, 11423–11428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siri, A.; Knauper, V.; Veirana, N.; Caocci, F.; Murphy, G.; Zardi, L. Different susceptibility of small and large human tenascin-C isoforms to degradation by matrix metalloproteinases. J. Biol. Chem. 1995, 270, 8650–8654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.H.; Chung, C.P. Tenascin-C promotes cell survival by activation of Akt in human chondrosarcoma cell. Cancer Lett. 2005, 229, 101–105. [Google Scholar] [CrossRef]
- Ni, W.D.; Yang, Z.T.; Cui, C.A.; Cui, Y.; Fang, L.Y.; Xuan, Y.H. Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 486, 607–612. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef]
- Morgan, M.; Saba, S.; Gower, W. Fibronectin influences cellular proliferation and apoptosis similarly in LNCaP and PC-3 prostate cancer cell lines. Urol. Oncol. 2000, 5, 155–159. [Google Scholar] [CrossRef]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar] [PubMed]
- De Jong-Hesse, Y.; Kampmeier, J.; Lang, G.K.; Lang, G.E. Effect of extracellular matrix on proliferation and differentiation of porcine lens epithelial cells. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 695–700. [Google Scholar] [CrossRef]
- Illario, M.; Cavallo, A.L.; Monaco, S.; Di Vito, E.; Mueller, F.; Marzano, L.A.; Troncone, G.; Fenzi, G.; Rossi, G.; Vitale, M. Fibronectin-induced proliferation in thyroid cells is mediated by alphavbeta3 integrin through Ras/Raf-1/MEK/ERK and calcium/CaMKII signals. J. Clin. Endocrinol.Metab. 2005, 90, 2865–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orend, G.; Huang, W.; Olayioye, M.A.; Hynes, N.E.; Chiquet-Ehrismann, R. Tenascin-C blocks cell-cycle progression of anchorage-dependent fibroblasts on fibronectin through inhibition of syndecan-4. Oncogene 2003, 22, 3917–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, B.; Ladanyi, A.; Tanko, L.; Sreter, L.; Tulassay, Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin. Cancer Res. 2001, 7, 4080–4085. [Google Scholar] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Radwanska, A.; Grall, D.; Schaub, S.; Divonne, S.B.F.; Ciais, D.; Rekima, S.; Rupp, T.; Sudaka, A.; Orend, G.; Van Obberghen-Schilling, E. Counterbalancing anti-adhesive effects of Tenascin-C through fibronectin expression in endothelial cells. Sci. Rep. 2017, 7, 12762. [Google Scholar] [CrossRef] [Green Version]
- Faissner, A.; Kruse, J. J1/tenascin is a repulsive substrate for central nervous system neurons. Neuron 1990, 5, 627–637. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Gene | Primer Sequence | Accession Number | Product Size (bp) |
|---|---|---|---|---|
| Aggrecan for | ACAN | tcaccagtgaggacctcgt | NM_001135.3 | 84 |
| Aggrecan rev | ggcggtagtggaagacgac | NM_013227.3 | ||
| β-Actin for | ACTB | ccaaccgcgagaagatga | NM_001101.3 | 97 |
| β-Actin rev | ccagaggcgtacagggatag | |||
| Brevican for | BCAN | tggaagctccactccagaa | NM_021948.4 | 76 |
| Brevican rev | cggtaccatggattgtgtttc | |||
| Collagen IV for | COL4A1 | gggagaaaagggtgaagca | NM_001845.5 | 72 |
| Collagen IV rev | ccaaaggtcctgtgcctataa | |||
| Fibronectin for | FN1 | cttccacaggaggcctacac | NM_212482.1 | 114 |
| NM_002026.2 | ||||
| NM_212478.1 | ||||
| Fibronectin rev | cgcaaaatatgctggaacttt | NM_212476.1 | ||
| NM_212474.1 | ||||
| GAPDH for | GAPDH | tccactggcgtcttcacc | NM_001289746.1 | 114 |
| NM_001289745.1 | ||||
| GAPDH rev | ggcagagatgatgaccctttt | NM_001256799.2 | ||
| NM_002046.5 | ||||
| α4-Integrin for | ITGA4 | gatgaaaatgagcctgaaacg | NM_000885.4 | 80 |
| α4-Integrin rev | gccatactattgccagtgttga | |||
| α5-Integrin for | ITGA5 | cccattgaatttgacagcaa | NM_002205.2 | 92 |
| α5-Integrin rev | tgcaaggacttgtactccaca | |||
| β1-Integrin for | ITGB1 | cgatgccatcatgcaagt | NM_002211 | 71 |
| β1-Integrin rev | acaccagcagccgtgtaac | NM_133376 | ||
| α1-Laminin for | LAMA1 | aggatgacctccattctgactt | NM_005559.3 | 60 |
| α1-Laminin rev | ccttacatgggcactgacct | |||
| MMP-2 for | MMP2 | ctacgaccgcgacaagaagt | NM_001302510.1 | 114 |
| NM_001302509.1 | ||||
| NM_001302508.1 | ||||
| MMP-2 rev | agttcccaccaacagtggac | NM_001127891.2 | ||
| NM_004530.5 | ||||
| MMP-7 for | MMP7 | gctgacatcatgattggcttt | NM_002423.3 | 113 |
| MMP-7 rev | tctcctccgagacctgtcc | |||
| MMP-9 for | MMP9 | gtaccacggccaactacgac | NM_004994.2 | 74 |
| MMP-9 rev | gccgtcctgggtgtagagt | |||
| Neurocan for | NCAN | aggggttcggagctatgg | NM_004386.2 | 95 |
| Neurocan rev | ggcccacgtagaagacctc | |||
| S18 for | RPS18 | cttccacaggaggcctacac | NM_022551.2 | 82 |
| S18 rev | cgcaaaatatgctggaacttt | |||
| TIMP-1 for | TIMP1 | agtggcactcattgcttgtg | NM_003254.2 | 104 |
| TIMP-1 rev | ggactggaagcccttttca | |||
| TIMP-2 for | TIMP2 | gaagagcctgaaccacaggt | NM_003255.4 | 85 |
| TIMP-2 rev | cggggaggagatgtagcac | |||
| Tenascin-C for | TNC | gtctccagcctgccacag | NM_002160.3 | 82 |
| Tenascin-C rev | cgcctttccagccaggttc | |||
| Tenascin-R for | TNR | agtgacctctgagcccattg | NM_003285.2 | 61 |
| Tenascin-R rev | gatgtcttttgggggatcaa | |||
| Versican for | VCAN | tcccaggaaacttacgatg | NM_004385.4 | 70 |
| Versican rev | ggggacagtgaggtgggaa |
| Primer Name | Gene | Primer Sequence | Accession Number | Amplicon Size (bp) |
|---|---|---|---|---|
| β-Actin for | ACTB | tgacggggtcacccacactgtgcccatcta | NM_001101.3 | 661 |
| β-Actin rev | ctagaagcatttgcggtggacgatggaggg | |||
| CP RPTPβ/ζ for | PTPRZ1 | agtgtgcaagtgcttgcctat | XM_005250519.1 | 555 |
| CP RPTPβ/ζ rev | tgcagaatagtcactctgctg | XM_017012477.1 | ||
| EC RPTPβ/ζ for | tgcctacttcccaactgag | NM_002851.2 | 509 | |
| EC RPTPβ/ζ rev | cagcatgagtagtggac | |||
| TEC RPTPβ/ζ for | tgcctacttcccaactgag | NM_001206839.1 | 369 | |
| TEC RPTPβ/ζ rev | attgctccgacatcatctg | 2949 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinhard, J.; Wagner, N.; Krämer, M.M.; Jarocki, M.; Joachim, S.C.; Dick, H.B.; Faissner, A.; Kakkassery, V. Expression Changes and Impact of the Extracellular Matrix on Etoposide Resistant Human Retinoblastoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 4322. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124322
Reinhard J, Wagner N, Krämer MM, Jarocki M, Joachim SC, Dick HB, Faissner A, Kakkassery V. Expression Changes and Impact of the Extracellular Matrix on Etoposide Resistant Human Retinoblastoma Cell Lines. International Journal of Molecular Sciences. 2020; 21(12):4322. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124322
Chicago/Turabian StyleReinhard, Jacqueline, Natalie Wagner, Miriam M. Krämer, Marvin Jarocki, Stephanie C. Joachim, H. Burkhard Dick, Andreas Faissner, and Vinodh Kakkassery. 2020. "Expression Changes and Impact of the Extracellular Matrix on Etoposide Resistant Human Retinoblastoma Cell Lines" International Journal of Molecular Sciences 21, no. 12: 4322. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124322