Mapping of CaM, S100A1 and PIP2-Binding Epitopes in the Intracellular N- and C-Termini of TRPM4

 , ,
, ,
Abstract
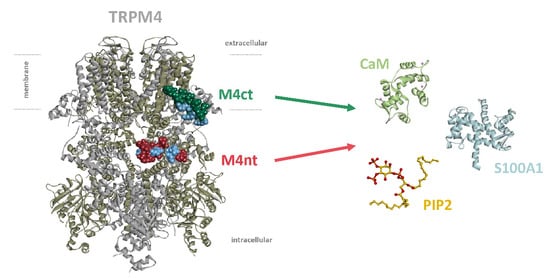
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Design of Peptides Representing Potential TRPM4-Binding Epitopes for CaM, S100A1 and PIP2
2.2. CaM and S100A1 Form Complexes with M4nt_WT and M4ct_WT
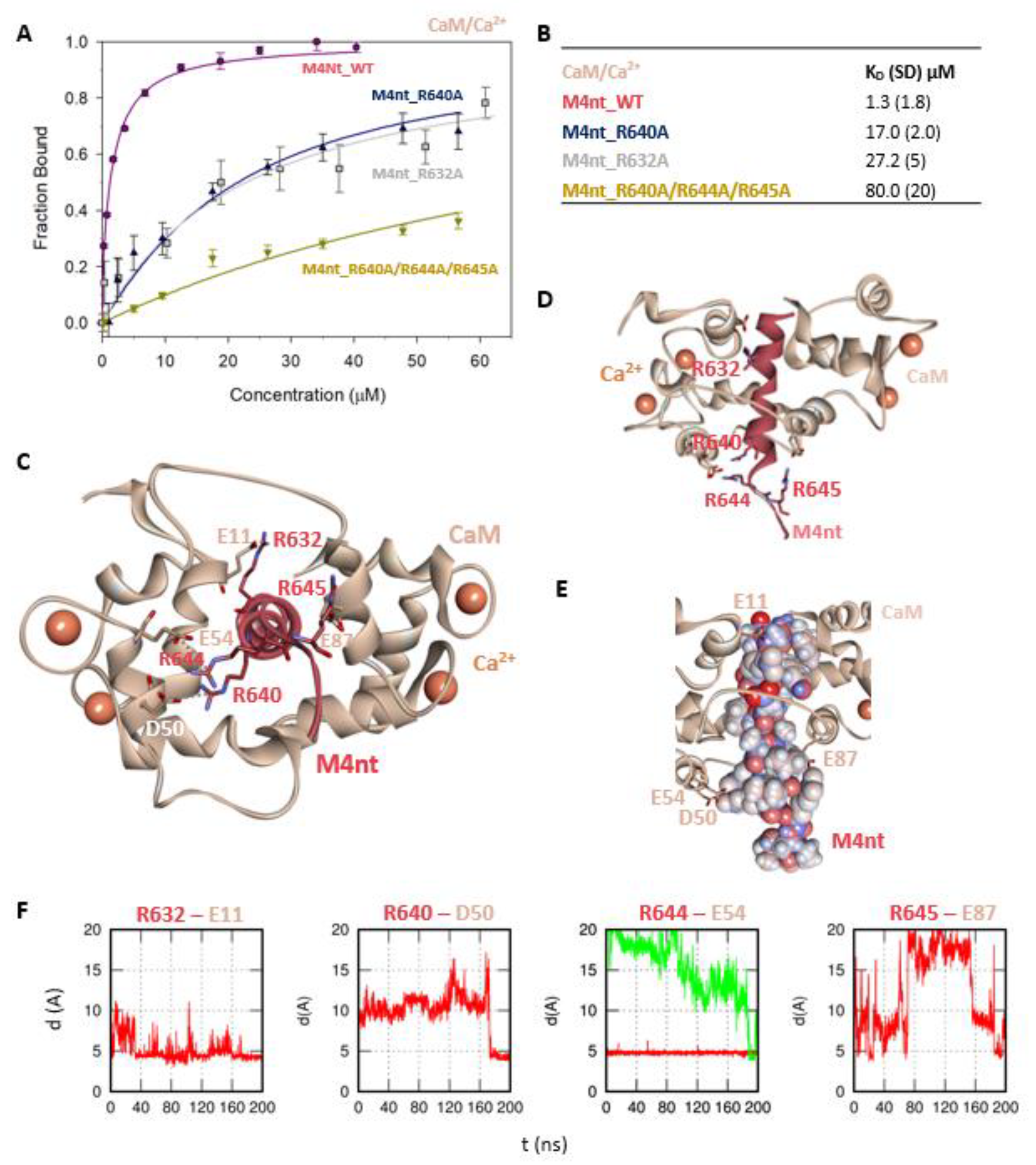
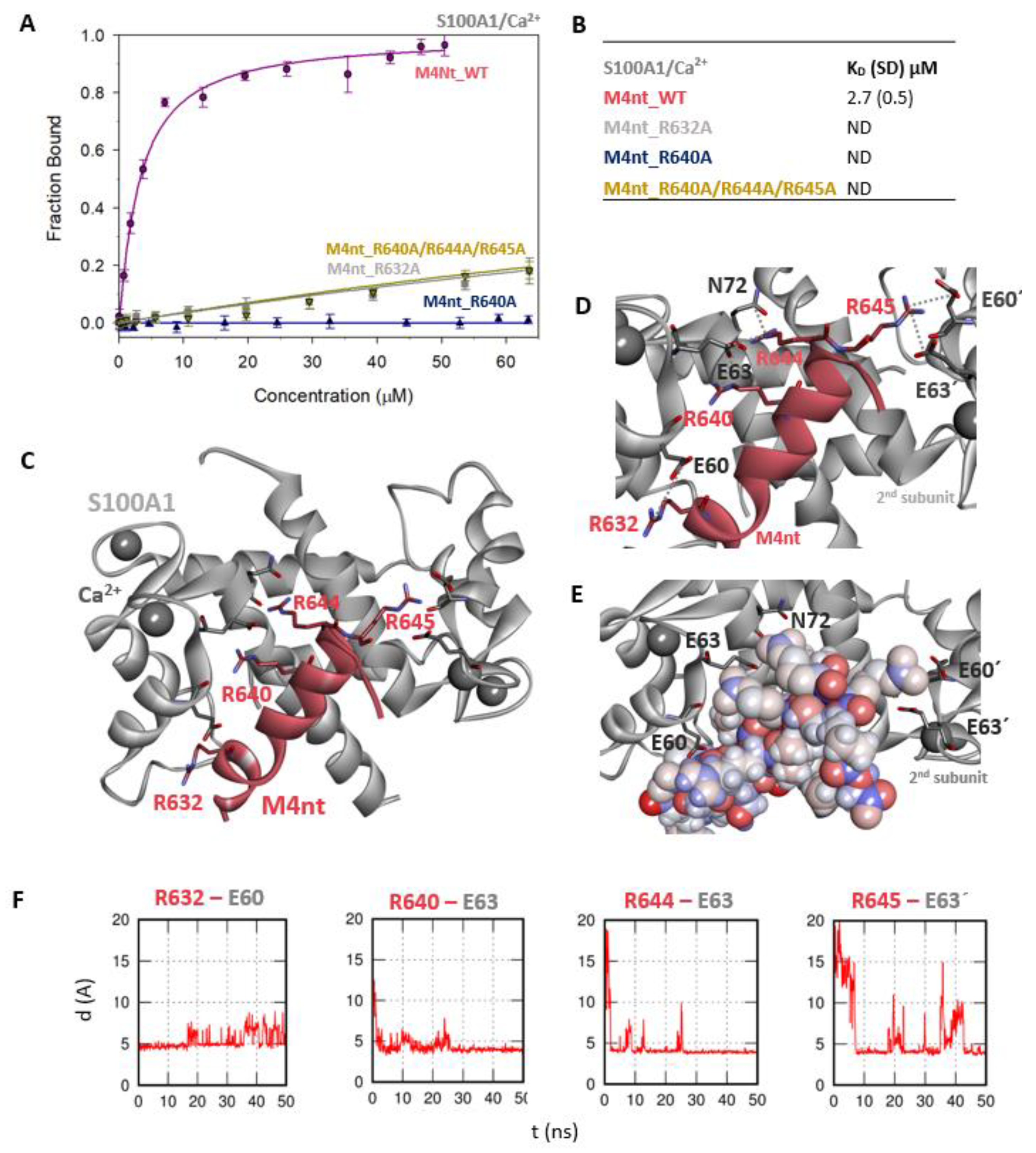
2.3. The Basic Amino Acids of M4nt_WT and M4ct_WT are Crucial for Binding to CaM and S100A1
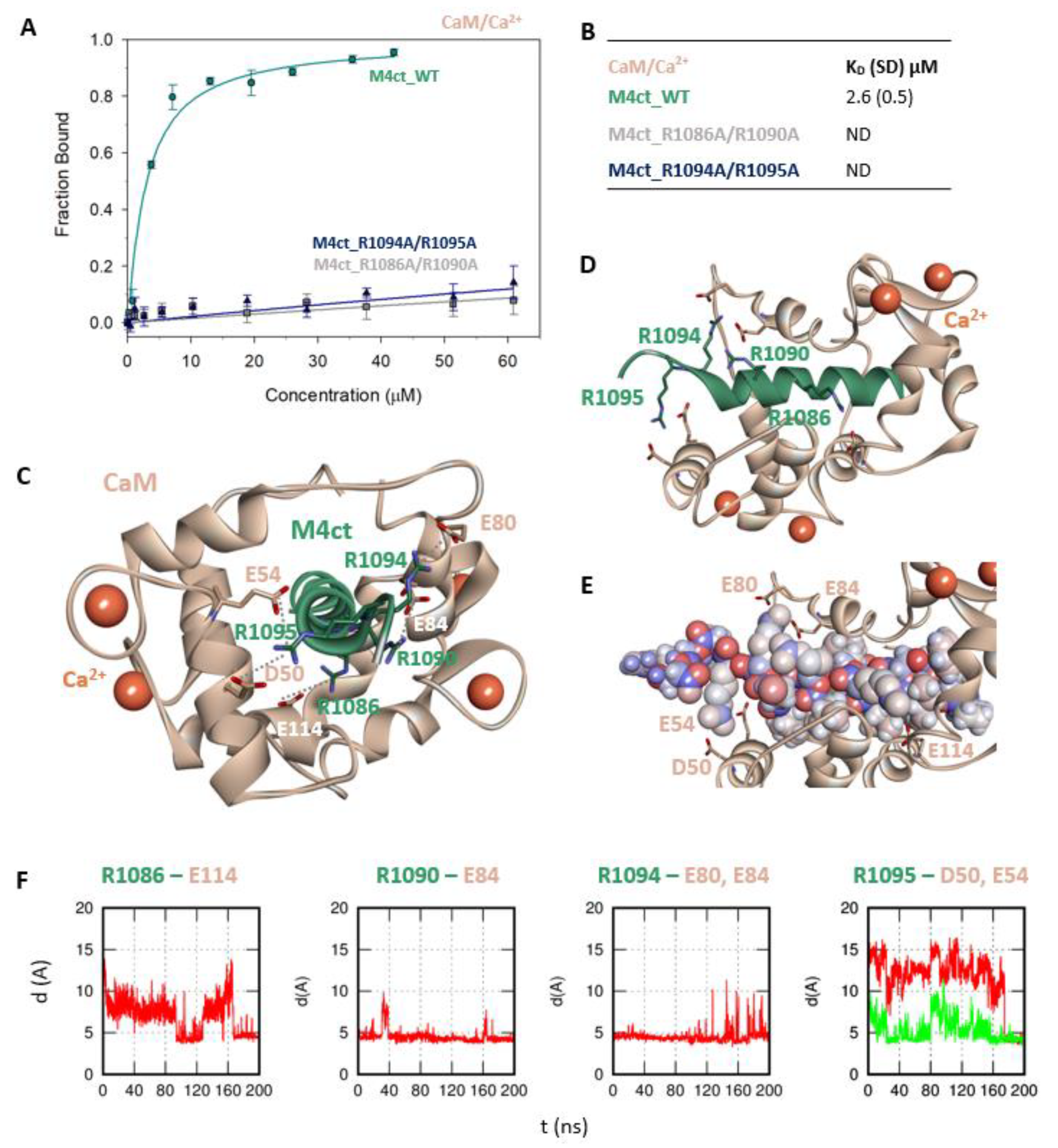
2.4. The Binding Interfaces of M4nt_WT/CaM and M4ct_WT/CaM Complexes
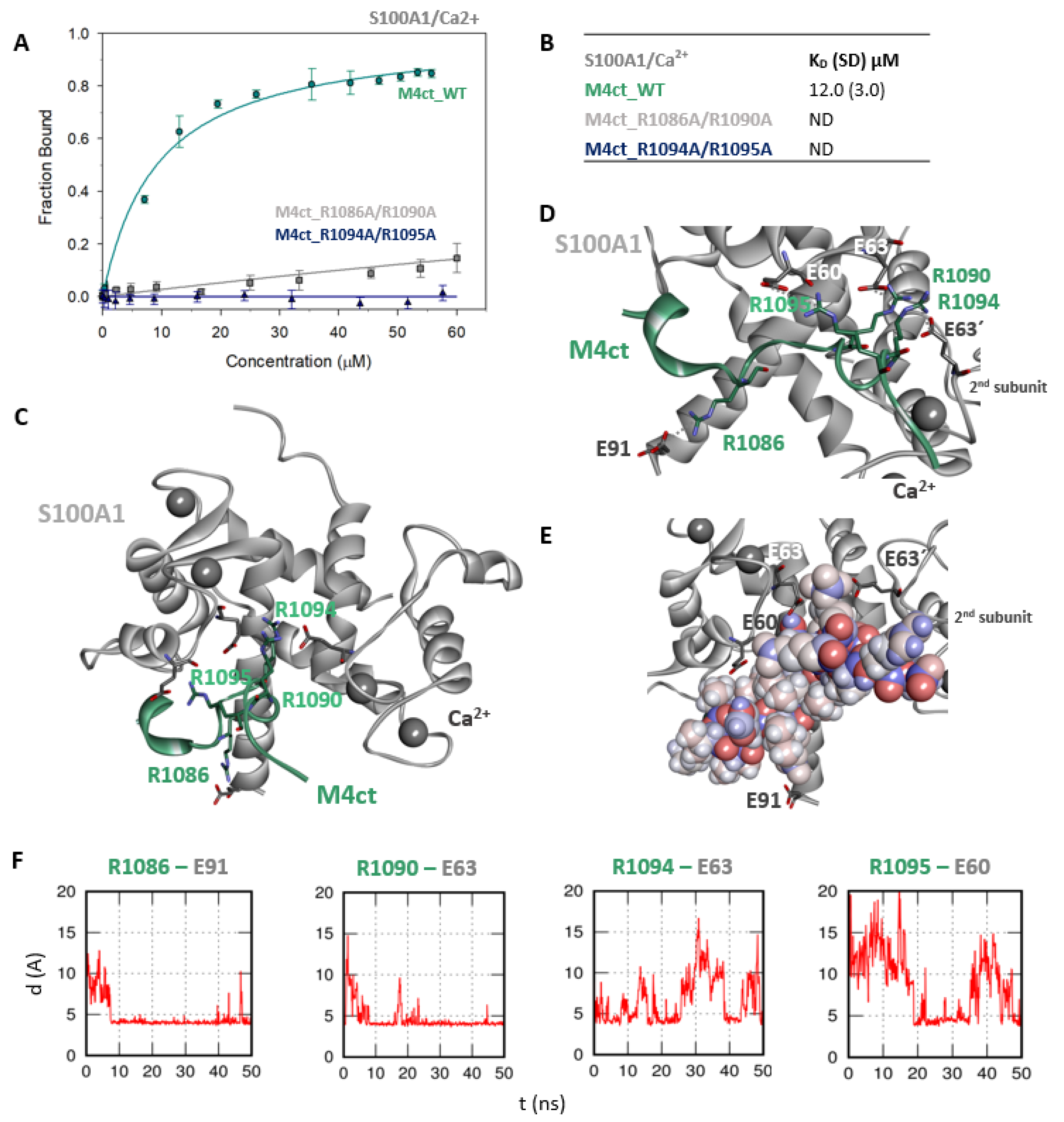
2.5. The Binding Interfaces of M4nt_WT/S100A1 and M4ct_WT/S100A1 Complexes
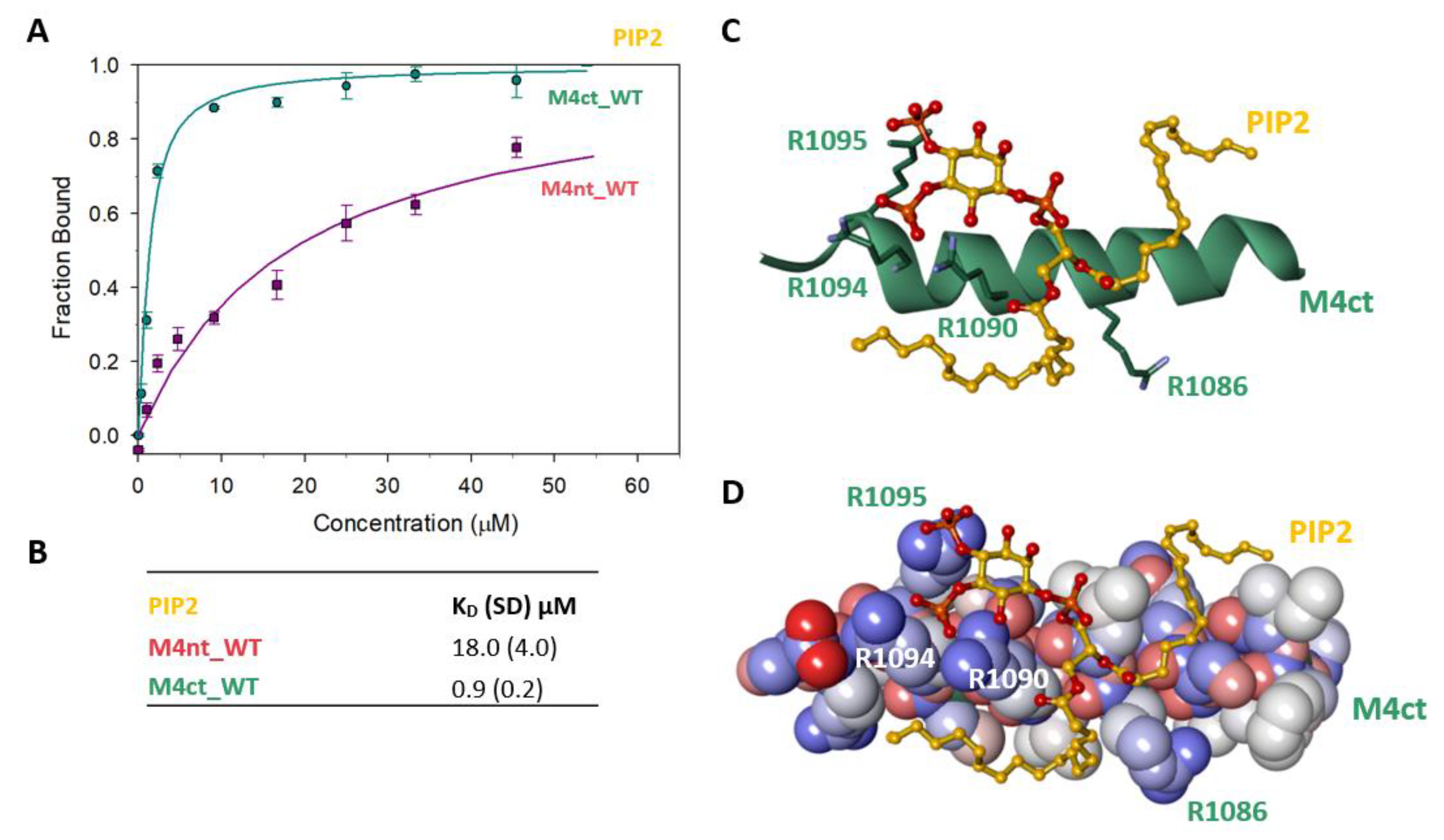
2.6. M4nt_WT and M4ct_WT Bind PIP2
2.7. The Binding Interface of the PIP2/M4ct_WT Complex
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Design of TRPM4 N- and C- Terminal Binding Epitopes
5.2. M4nt and M4ct Peptide Synthesis and Site-Directed Mutagenesis
5.3. CaM, S100A1 Purification and PIP2 Preparation
5.4. Steady-State Fluorescence Anisotropy
5.5. Time-Resolved Fluorescence Measurements
5.6. Protein–Protein Docking
5.7. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| CaM | Calmodulin |
| EM | Electron microscopy |
| FA | Fluorescence anisotropy |
| FFT | Fourier transform |
| MD | Molecular dynamics |
| PBC | Periodic boundary conditions |
| PH | Pleckstrin homology |
| PIP2 | phosphatidylinositol 4, 5-bisphosphate |
| PME | particle mesh Ewald |
| S100A1 | S100 calcium-binding protein A1 |
| TRPs | transient receptor potential (channels) |
| TRPC | TRP canonical |
| TRPV | TRP vaniloid |
| TRPM4 | TRP melastatin 4 |
References
- Nilius, B.; Prenen, J.; Tang, J.; Wang, C.; Owsianik, G.; Janssens, A.; Voets, T.; Zhu, M.X. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem. 2005, 280, 6423–6433. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E.; Runnels, L.W.; Strübing, C. The TRP ion channel family. Nat. Rev. Neurosci. 2001, 2, 387. [Google Scholar] [CrossRef] [PubMed]
- Ehara, T.; Noma, A.; Ono, K. Calcium-activated non-selective cation channel in ventricular cells isolated from adult guinea-pig hearts. J. Physiol. 1988, 403, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Fleig, A.; Perraud, A.-L.; Scharenberg, A.M.; Penner, R.; Kinet, J.-P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; An, X.; Fu, M. Transient receptor potential melastatin 4 cation channel in pediatric heart block. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 79–84. [Google Scholar] [PubMed]
- Nilius, B.; Prenen, J.; Droogmans, G.; Voets, T.; Vennekens, R.; Freichel, M.; Wissenbach, U.; Flockerzi, V. Voltage dependence of the Ca2+-activated cation channel TRPM4. J. Biol. Chem. 2003, 278, 30813–30820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathar, I.; Jacobs, G.; Kecskes, M.; Menigoz, A.; Philippaert, K.; Vennekens, R. Trpm4. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Springer: Berlin, Germany, 2014; pp. 461–487. [Google Scholar]
- Duan, J.; Li, Z.; Li, J.; Santa-Cruz, A.; Sanchez-Martinez, S.; Zhang, J.; Clapham, D.E. Structure of full-length human TRPM4. Proc. Natl. Acad. Sci. USA 2018, 115, 2377–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Winkler, P.A.; Huang, Y.; Sun, W.; Du, J.; Lü, W. Electron cryo-microscopy structure of a human TRPM4 channel. Nature 2017, 552, 200. [Google Scholar] [CrossRef]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.-c.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Mahieu, F.; Prenen, J.; Janssens, A.; Owsianik, G.; Vennekens, R.; Voets, T. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4, 5-biphosphate. EMBO J. 2006, 25, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Okawa, H.; Wang, Y.; Liman, E.R. Phosphatidylinositol 4, 5-bisphosphate rescues TRPM4 channels from desensitization. J. Biol. Chem. 2005, 280, 39185–39192. [Google Scholar] [CrossRef] [Green Version]
- Vennekens, R.; Nilius, B. Insights into TRPM4 function, regulation and physiological role. In Transient Receptor Potential (TRP) Channels; Springer: Berlin, Germany, 2007; pp. 269–285. [Google Scholar]
- Singh, A.K.; McGoldrick, L.L.; Twomey, E.C.; Sobolevsky, A.I. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci. Adv. 2018, 4, eaau6088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, T.; Kovalevskaya, N.V.; Verkaart, S.; Schilderink, N.; Felici, M.; van der Hagen, E.A.; Bindels, R.J.; Vuister, G.W.; Hoenderop, J.G. The molecular mechanisms of calmodulin action on TRPV5 and the modulation by parathyroid hormone. Mol. Cell. Biol. 2011, 31, 2845–2853. [Google Scholar] [CrossRef] [Green Version]
- Villalobo, A.; González-Muñoz, M.; Berchtold, M.W. Proteins with calmodulin-like domains: Structures and functional roles. Cell. Mol. Life Sci. 2019, 76, 2299–2328. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, L.; Taylor, D.A.; Chandross, R.J.; VanBerkum, M.F.; Means, A.R.; Quiocho, F.A.; Sack, J.S. The structure of a calmodulin mutant with a deletion in the central helix: Implications for molecular recognition and protein binding. Structure 1997, 5, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [Google Scholar] [CrossRef]
- Zhu, M.X. Multiple roles of calmodulin and other Ca2+-binding proteins in the functional regulation of TRP channels. Pflügers Archiv. 2005, 451, 105–115. [Google Scholar] [CrossRef]
- Hasan, R.; Zhang, X. Ca2+ regulation of TRP ion channels. Int. J. Mol. Sci. 2018, 19, 1256. [Google Scholar] [CrossRef] [Green Version]
- Rohacs, T.; Nilius, B. Regulation of transient receptor potential (TRP) channels by phosphoinositides. Pflügers Archiv. Eur. J. Physiol. 2007, 455, 157–168. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M.; O’Brien, R.; Sigler, P.B.; Schlessinger, J. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. USA 1995, 92, 10472–10476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Tanimoto, A.; Iwasa, S.; Otsuguro, K.-i. TRPM4 and TRPM5 Channels Share Crucial Amino Acid Residues for Ca2+ Sensitivity but Not Significance of PI (4, 5) P2. Int. J. Mol. Sci. 2019, 20, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousova, K.; Jirku, M.; Bumba, L.; Bednarova, L.; Sulc, M.; Franek, M.; Vyklicky, L.; Vondrasek, J.; Teisinger, J. PIP2 and PIP3 interact with N-terminus region of TRPM4 channel. Biophys. Chem. 2015, 205, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Ufret-Vincenty, C.A.; Klein, R.M.; Hua, L.; Angueyra, J.; Gordon, S.E. Localization of the PIP2 sensor of TRPV1 ion channels. J. Biol. Chem. 2011, 286, 9688–9698. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Hughes, T.E.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 4198. [Google Scholar] [CrossRef] [Green Version]
- Fine, M.; Schmiege, P.; Li, X. Structural basis for PtdInsP 2-mediated human TRPML1 regulation. Nat. Commun. 2018, 9, 4192. [Google Scholar] [CrossRef]
- Stokum, J.A.; Kwon, M.S.; Woo, S.K.; Tsymbalyuk, O.; Vennekens, R.; Gerzanich, V.; Simard, J.M. SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia 2018, 66, 108–125. [Google Scholar] [CrossRef]
- Woo, S.K.; Kwon, M.S.; Ivanov, A.; Gerzanich, V.; Simard, J.M. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J. Biol. Chem. 2013, 288, 3655–3667. [Google Scholar] [CrossRef] [Green Version]
- Pratt, E.B.; Tewson, P.; Bruederle, C.E.; Skach, W.R.; Shyng, S.-L. N-terminal transmembrane domain of SUR1 controls gating of Kir6. 2 by modulating channel sensitivity to PIP2. J. Gen. Physiol. 2011, 137, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Galizia, L.; Pizzoni, A.; Fernandez, J.; Rivarola, V.; Capurro, C.; Ford, P. Functional interaction between AQP2 and TRPV4 in renal cells. J. Cell. Biochem. 2012, 113, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Kim, J.; Truong, K.; Sherman, M.; Yuan, T.; Ikura, M. Calmodulin target database. J. Struct. Funct. Genom. 2000, 1, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.V.; Törnroth-Horsefield, S. Aquaporin protein-protein interactions. Int. J. Mol. Sci. 2017, 18, 2255. [Google Scholar] [CrossRef] [PubMed]
- Bily, J.; Grycova, L.; Holendova, B.; Jirku, M.; Janouskova, H.; Bousova, K.; Teisinger, J. Characterization of the S100A1 protein binding site on TRPC6 C-terminus. PLoS ONE 2013, 8, e62677. [Google Scholar] [CrossRef] [Green Version]
- Grycova, L.; Holendova, B.; Bumba, L.; Bily, J.; Jirku, M.; Lansky, Z.; Teisinger, J. Integrative binding sites within intracellular termini of TRPV1 receptor. PLoS ONE 2012, 7, e48437. [Google Scholar] [CrossRef] [Green Version]
- Holakovska, B.; Grycova, L.; Bily, J.; Teisinger, J. Characterization of calmodulin binding domains in TRPV2 and TRPV5 C-tails. Amino Acids 2011, 40, 741–748. [Google Scholar] [CrossRef]
- Prosser, B.L.; Hernández-Ochoa, E.O.; Schneider, M.F. S100A1 and calmodulin regulation of ryanodine receptor in striated muscle. Cell Calcium 2011, 50, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Bousova, K.; Herman, P.; Vecer, J.; Bednarova, L.; Monincova, L.; Majer, P.; Vyklicky, L.; Vondrasek, J.; Teisinger, J. Shared CaM-and S100A1-binding epitopes in the distal TRPM 4 N terminus. FEBS J. 2018, 285, 599–613. [Google Scholar] [CrossRef]
- Lau, S.-Y.; Procko, E.; Gaudet, R. Distinct properties of Ca2+–calmodulin binding to N-and C-terminal regulatory regions of the TRPV1 channel. J. Gen. Physiol. 2012, 140, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science 1992, 257, 1251–1255. [Google Scholar] [CrossRef]
- Maximciuc, A.A.; Putkey, J.A.; Shamoo, Y.; MacKenzie, K.R. Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode. Structure 2006, 14, 1547–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, N.T.; Prosser, B.L.; Varney, K.M.; Zimmer, D.B.; Schneider, M.F.; Weber, D.J. S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J. Biol. Chem. 2008, 283, 26676–26683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, B.L.; Wright, N.T.; Hernandez-Ochoa, E.O.; Varney, K.M.; Liu, Y.; Olojo, R.O.; Zimmer, D.B.; Weber, D.J.; Schneider, M.F. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J. Biol. Chem. 2008, 283, 5046–5057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grycova, L.; Holendova, B.; Lansky, Z.; Bumba, L.; Jirku, M.; Bousova, K.; Teisinger, J. Ca2+ Binding Protein S100A1 Competes with Calmodulin and PIP2 for Binding Site on the C-Terminus of the TPRV1 Receptor. ACS Chem. Neurosci. 2014, 6, 386–392. [Google Scholar] [CrossRef]
- Holakovska, B.; Grycova, L.; Jirku, M.; Sulc, M.; Bumba, L.; Teisinger, J. Calmodulin and S100A1 protein interact with N terminus of TRPM3 channel. J. Biol. Chem. 2012, 287, 16645–16655. [Google Scholar] [CrossRef] [Green Version]
- Jirku, M.; Lansky, Z.; Bednarova, L.; Sulc, M.; Monincova, L.; Majer, P.; Vyklicky, L.; Vondrasek, J.; Teisinger, J.; Bousova, K. The characterization of a novel S100A1 binding site in the N-terminus of TRPM1. Int. J. Biochem. Cell Biol. 2016, 78, 186–193. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Valdez, F.; Azumaya, C.M.; Romero, L.O.; Nakagawa, T.; Cordero-Morales, J.F. Structure–function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating. J. Biol. Chem. 2018, 293, 16102–16114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, K.W.; Cohen, M.R.; Jiang, J.; Samanta, A.; Lodowski, D.T.; Zhou, Z.H.; Moiseenkova-Bell, V.Y. Structure of the full-length TRPV2 channel by cryo-EM. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zubcevic, L.; Le, S.; Yang, H.; Lee, S.-Y. Conformational plasticity in the selectivity filter of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 2018, 25, 405–415. [Google Scholar] [CrossRef]
- Zhu, M.X.; Tang, J. TRPC channel interactions with calmodulin and IP3 receptors. Novartis Found. Symp. 2004, 258, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.M.; Yoshioka, C.; Rex, E.A.; Fay, J.F.; Xie, Q.; Whorton, M.R.; Chen, J.Z.; Shyng, S.-L. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife 2017, 6, e24149. [Google Scholar] [CrossRef] [PubMed]
- Subbotina, E.; Williams, N.; Sampson, B.A.; Tang, Y.; Coetzee, W.A. Functional characterization of TRPM4 variants identified in sudden unexpected natural death. Forensic Sci. Int. 2018, 293, 37–46. [Google Scholar] [CrossRef]
- Lindsay, C.; Sitsapesan, M.; Chan, W.M.; Venturi, E.; Welch, W.; Musgaard, M.; Sitsapesan, R. Promiscuous attraction of ligands within the ATP binding site of RyR2 promotes diverse gating behaviour. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Kohda, D. “Multiple partial recognitions in dynamic equilibrium” in the binding sites of proteins form the molecular basis of promiscuous recognition of structurally diverse ligands. Biophys. Rev. 2018, 10, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Brix, J.; Dietmeier, K.; Pfanner, N. Differential recognition of preproteins by the purified cytosolic domains of the mitochondrial import receptors Tom20, Tom22, and Tom70. J. Biol. Chem. 1997, 272, 20730–20735. [Google Scholar] [CrossRef] [Green Version]
- Hainzl, T.; Huang, S.; Meriläinen, G.; Brännström, K.; Sauer-Eriksson, A.E. Structural basis of signal-sequence recognition by the signal recognition particle. Nat. Struct. Mol. Biol. 2011, 18, 389. [Google Scholar] [CrossRef]
- Hansen, S.B.; Tao, X.; MacKinnon, R. Structural basis of PIP 2 activation of the classical inward rectifier K+ channel Kir2. 2. Nature 2011, 477, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, B.-C.; Hille, B. Regulation of ion channels by phosphatidylinositol 4, 5-bisphosphate. Curr. Opin. Neurobiol. 2005, 15, 370–378. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Ray, K.; Chowdhury, M.; Szmacinski, H.; Fu, Y.; Zhang, J.; Nowaczyk, K. Plasmon-controlled fluorescence: A new paradigm in fluorescence spectroscopy. Analyst 2008, 133, 1308–1346. [Google Scholar] [CrossRef] [Green Version]
- Harper, C.C.; Berg, J.M.; Gould, S.J. PEX5 binds the PTS1 independently of Hsp70 and the peroxin PEX12. J. Biol. Chem. 2003, 278, 7897–7901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, J.J.; Schepartz, A. Kinetic studies of fos- jun- DNA complex formation: DNA binding prior to dimerization. Biochemistry 2001, 40, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Cheatham, T.I.; Miller, J.; Fox, T.; Darden, T.; Kollman, P. Molecular dynamics simulations on solvated biomolecular systems: The particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Feenstra, K.A.; Hess, B.; Berendsen, H.J. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A., Jr. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment; Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bousova, K.; Barvik, I.; Herman, P.; Hofbauerová, K.; Monincova, L.; Majer, P.; Zouharova, M.; Vetyskova, V.; Postulkova, K.; Vondrasek, J. Mapping of CaM, S100A1 and PIP2-Binding Epitopes in the Intracellular N- and C-Termini of TRPM4. Int. J. Mol. Sci. 2020, 21, 4323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124323
Bousova K, Barvik I, Herman P, Hofbauerová K, Monincova L, Majer P, Zouharova M, Vetyskova V, Postulkova K, Vondrasek J. Mapping of CaM, S100A1 and PIP2-Binding Epitopes in the Intracellular N- and C-Termini of TRPM4. International Journal of Molecular Sciences. 2020; 21(12):4323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124323
Chicago/Turabian StyleBousova, Kristyna, Ivan Barvik, Petr Herman, Kateřina Hofbauerová, Lenka Monincova, Pavel Majer, Monika Zouharova, Veronika Vetyskova, Klara Postulkova, and Jiri Vondrasek. 2020. "Mapping of CaM, S100A1 and PIP2-Binding Epitopes in the Intracellular N- and C-Termini of TRPM4" International Journal of Molecular Sciences 21, no. 12: 4323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124323