Chitosan-Based Coacervate Polymers for Propolis Encapsulation: Release and Cytotoxicity Studies

 ,
,  and
and 
Abstract
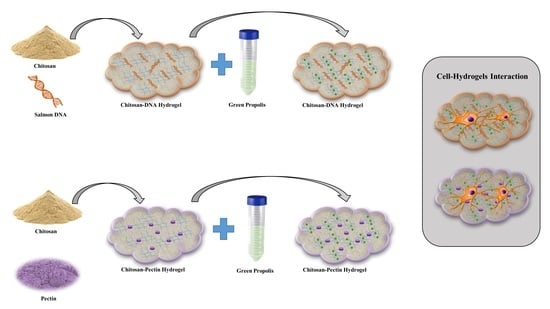
:
1. Introduction
2. Results and Discussion
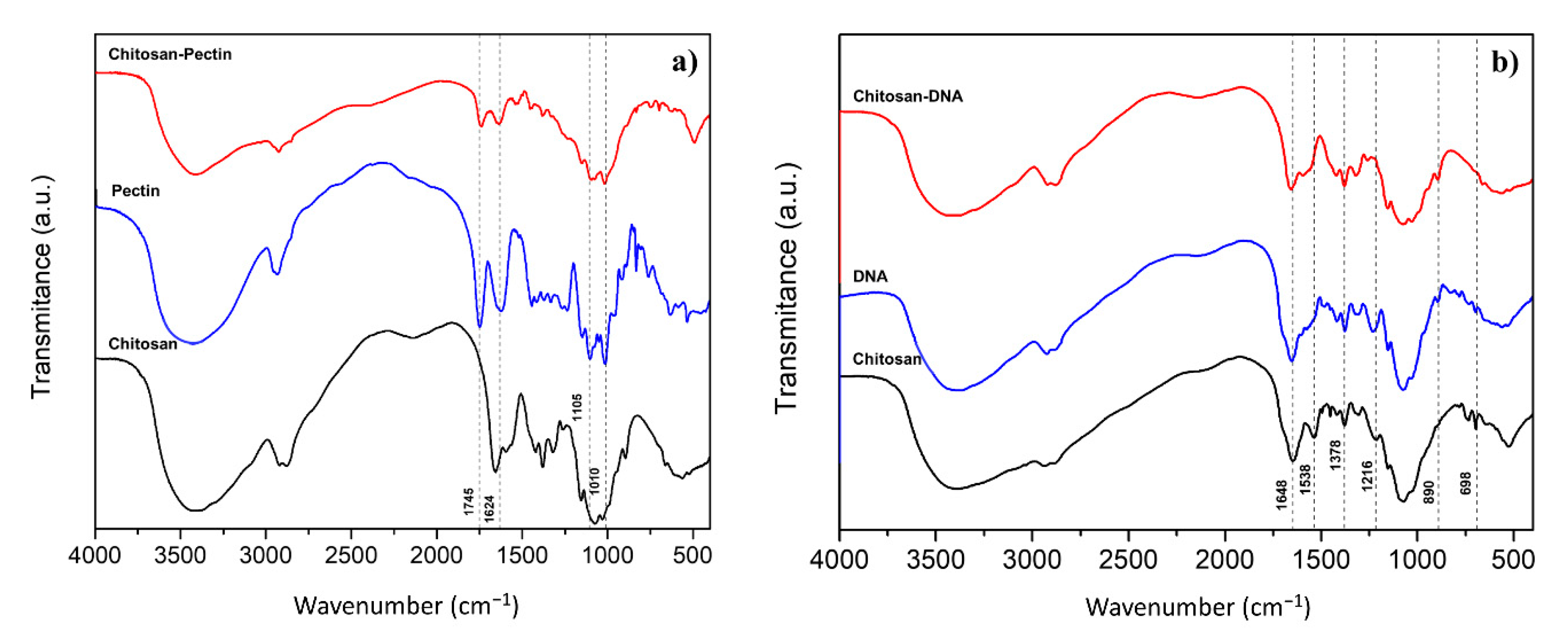
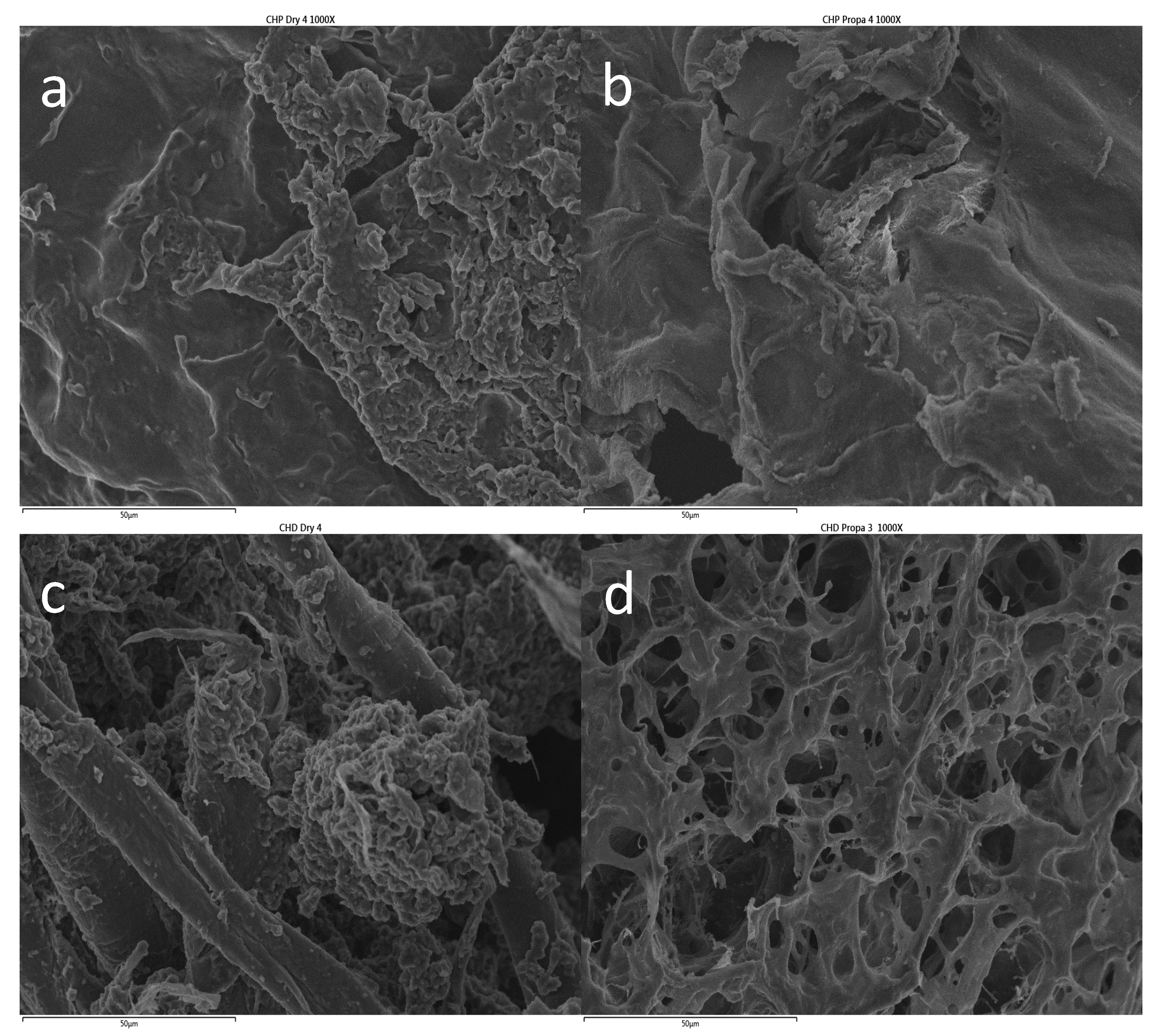
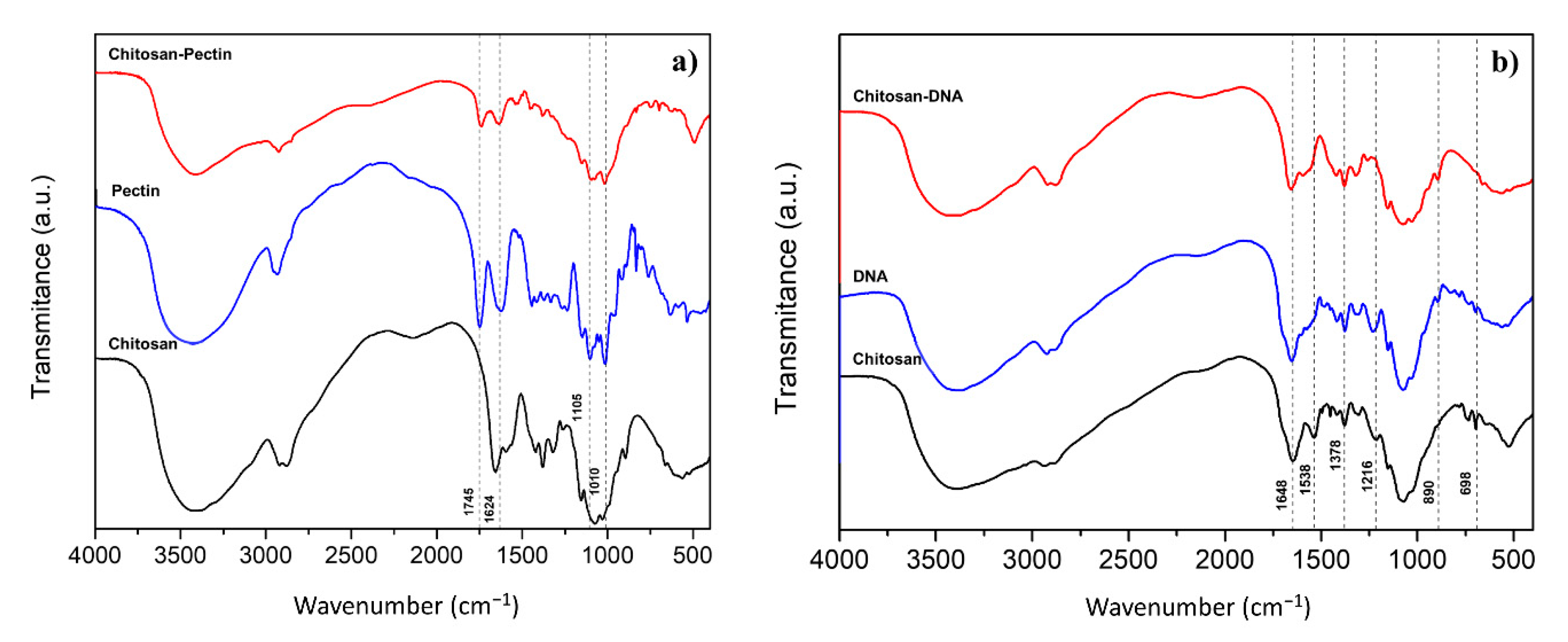
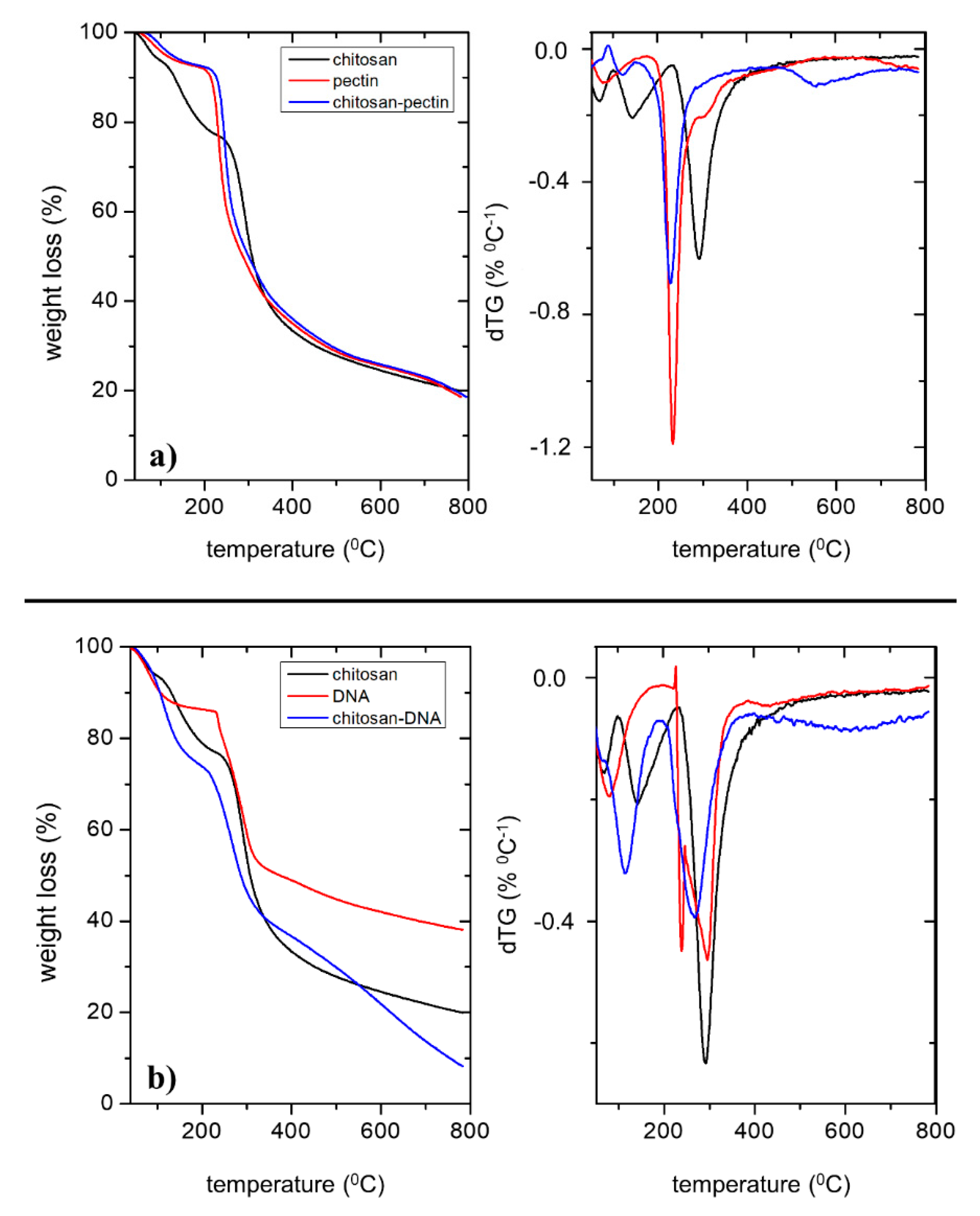
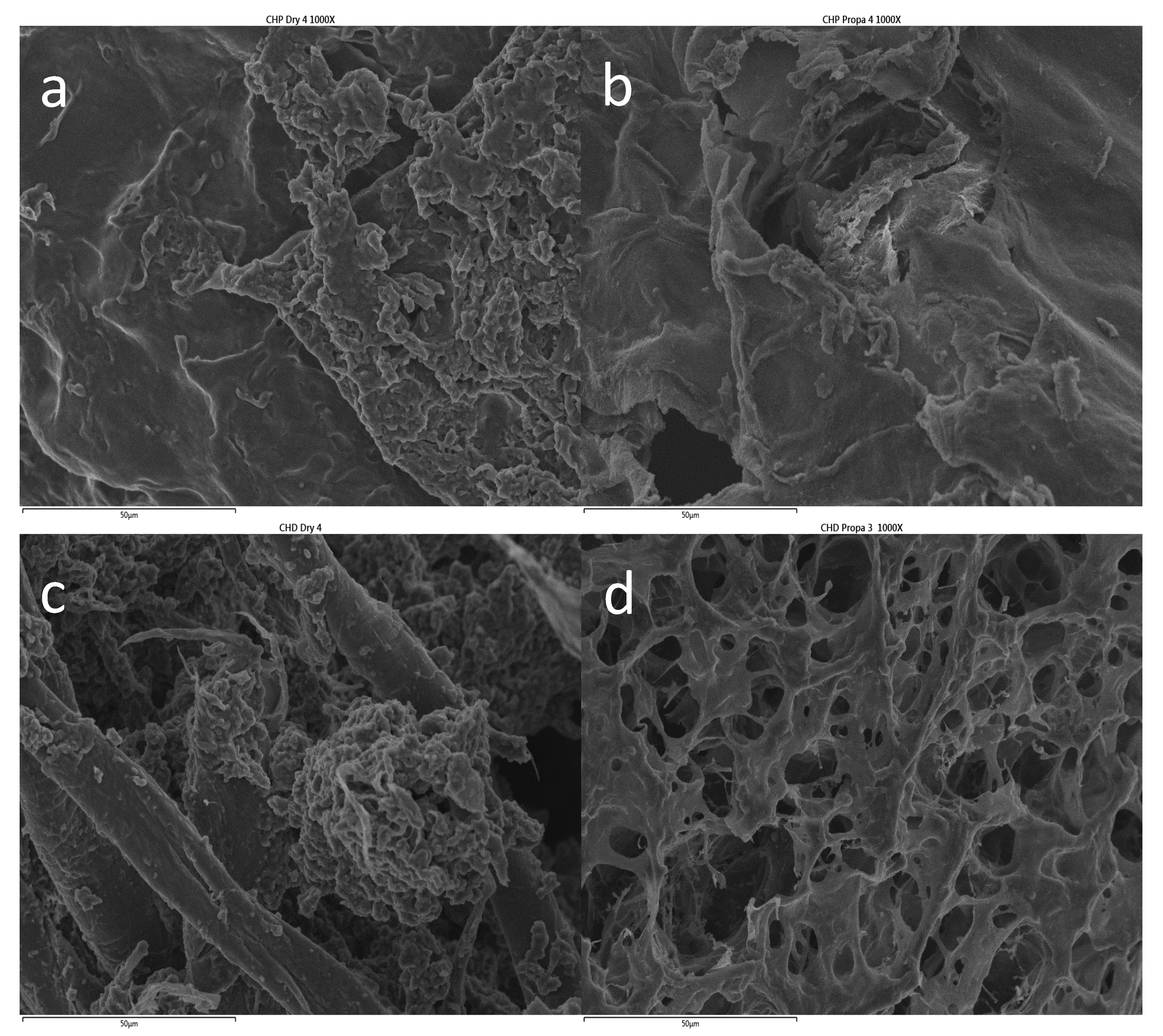
2.1. Gel Characterization
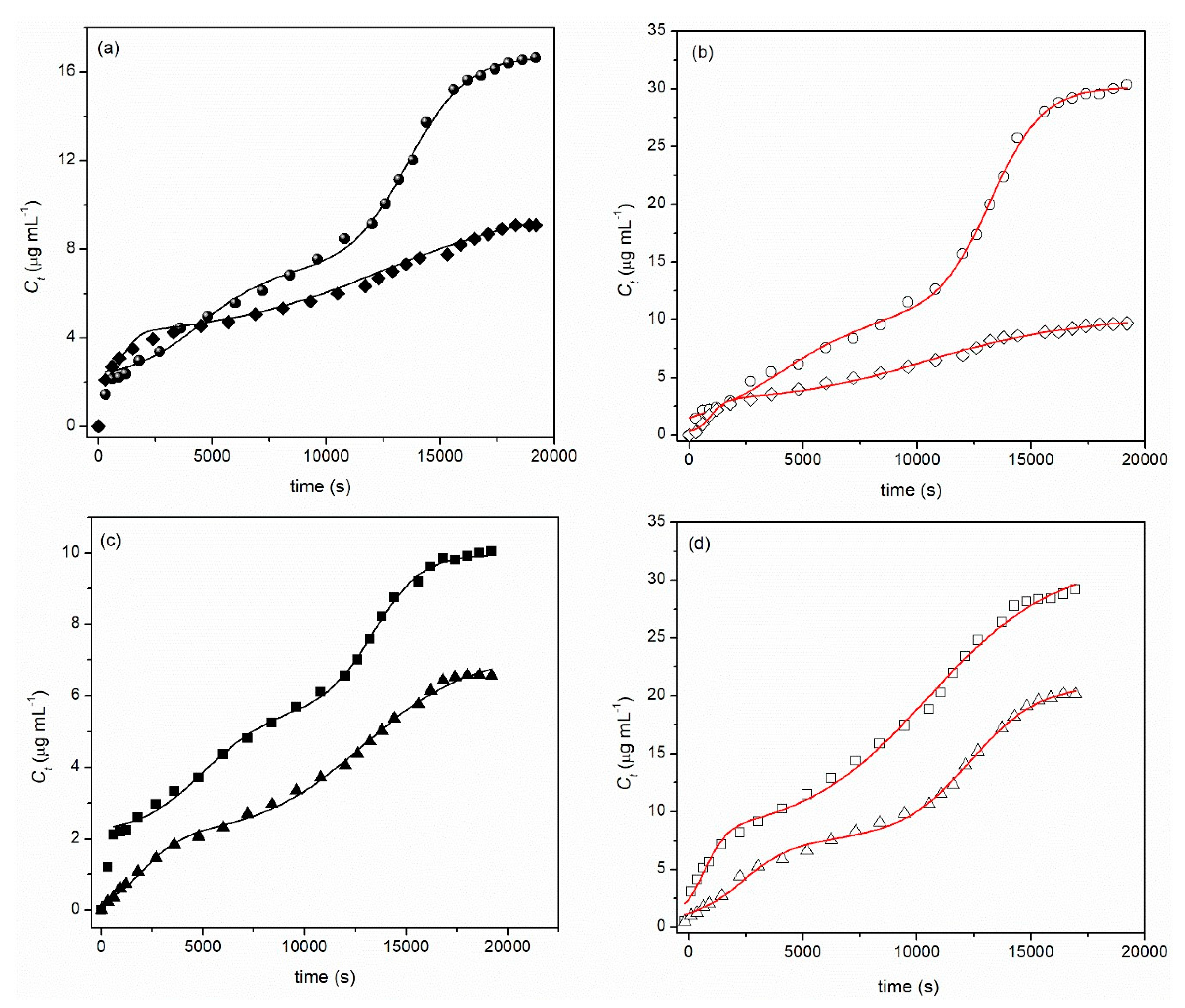
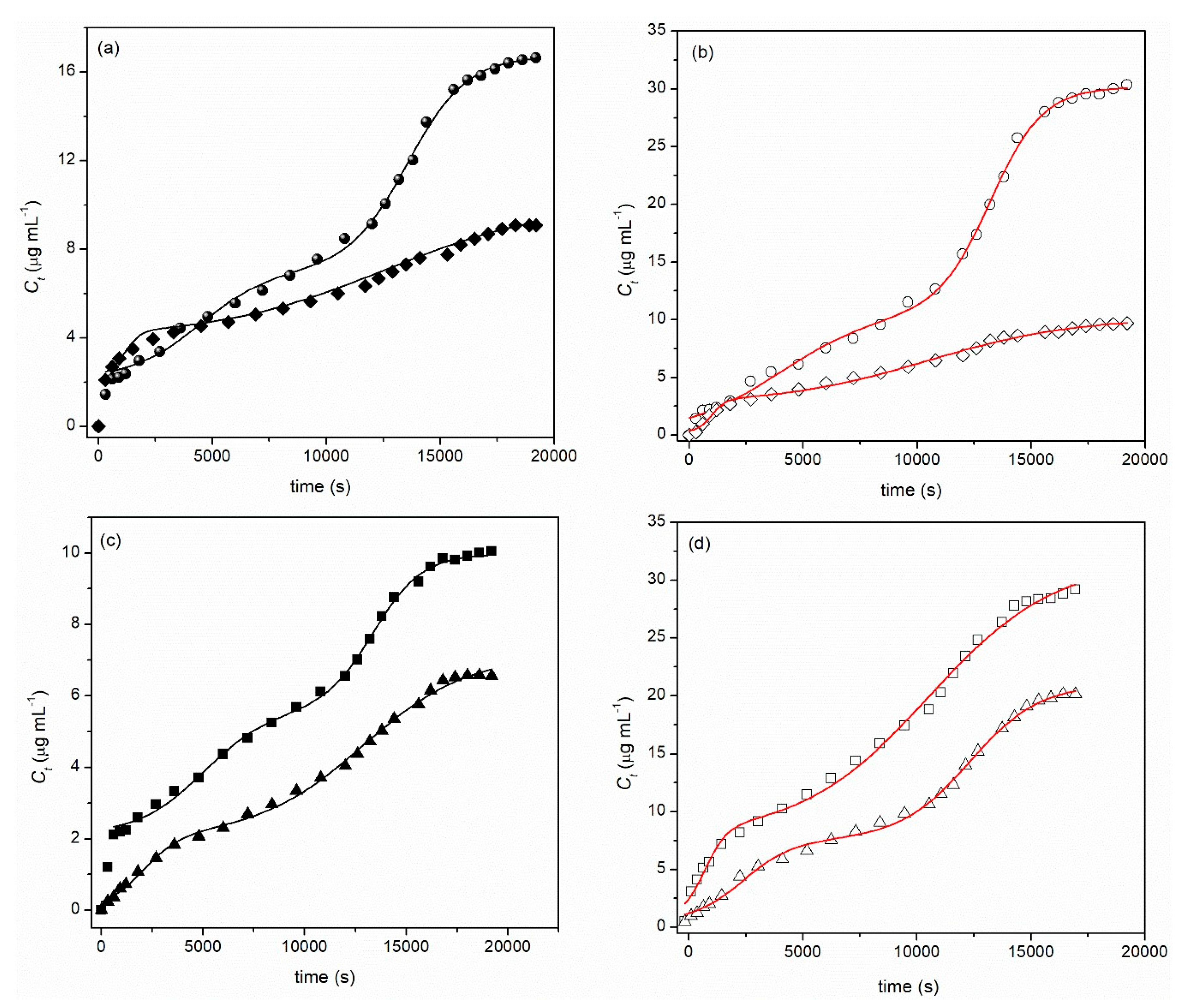
2.2. Loading and Drug Release
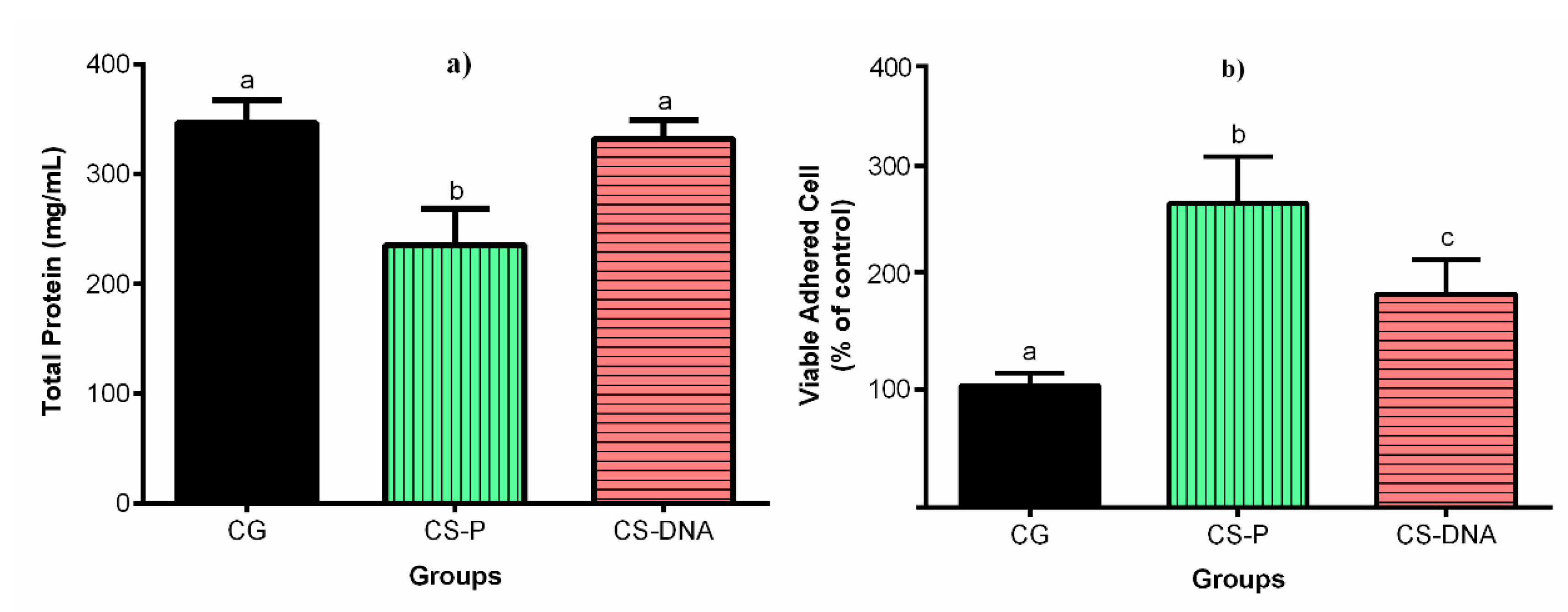
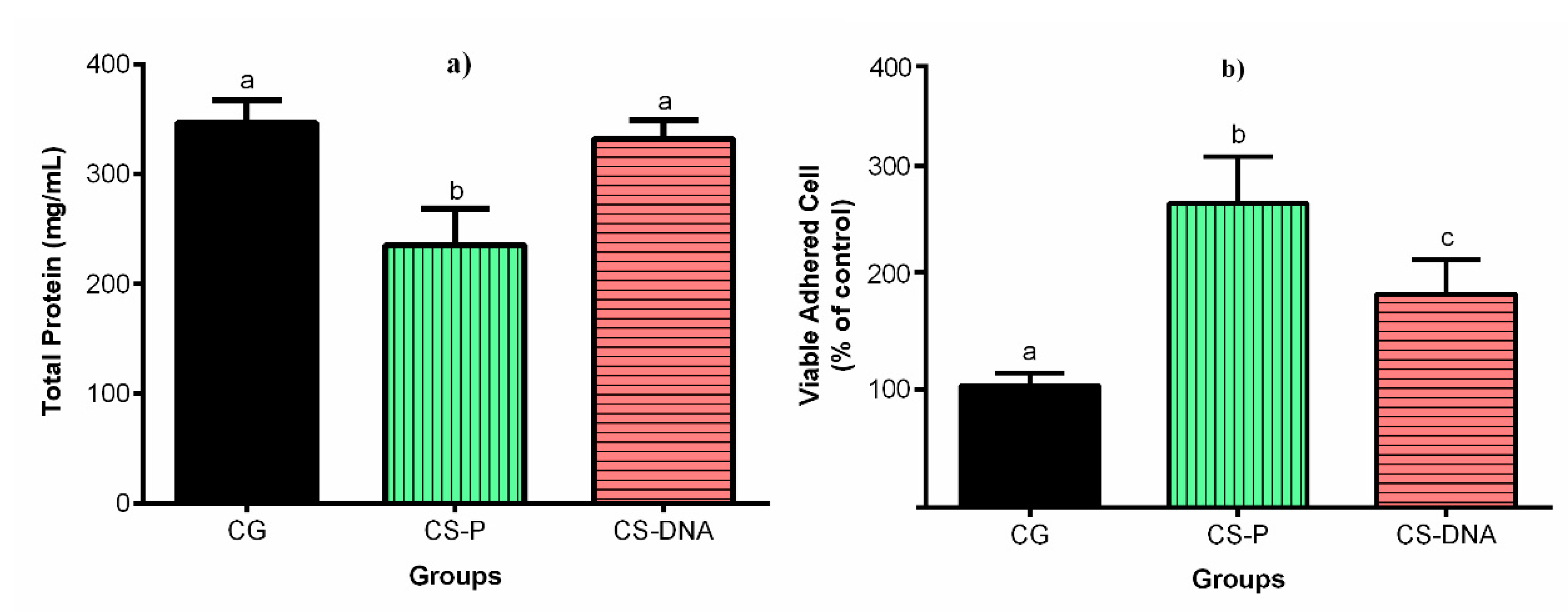
2.3. Biological Analysis
3. Materials and Methods
3.1. Materials
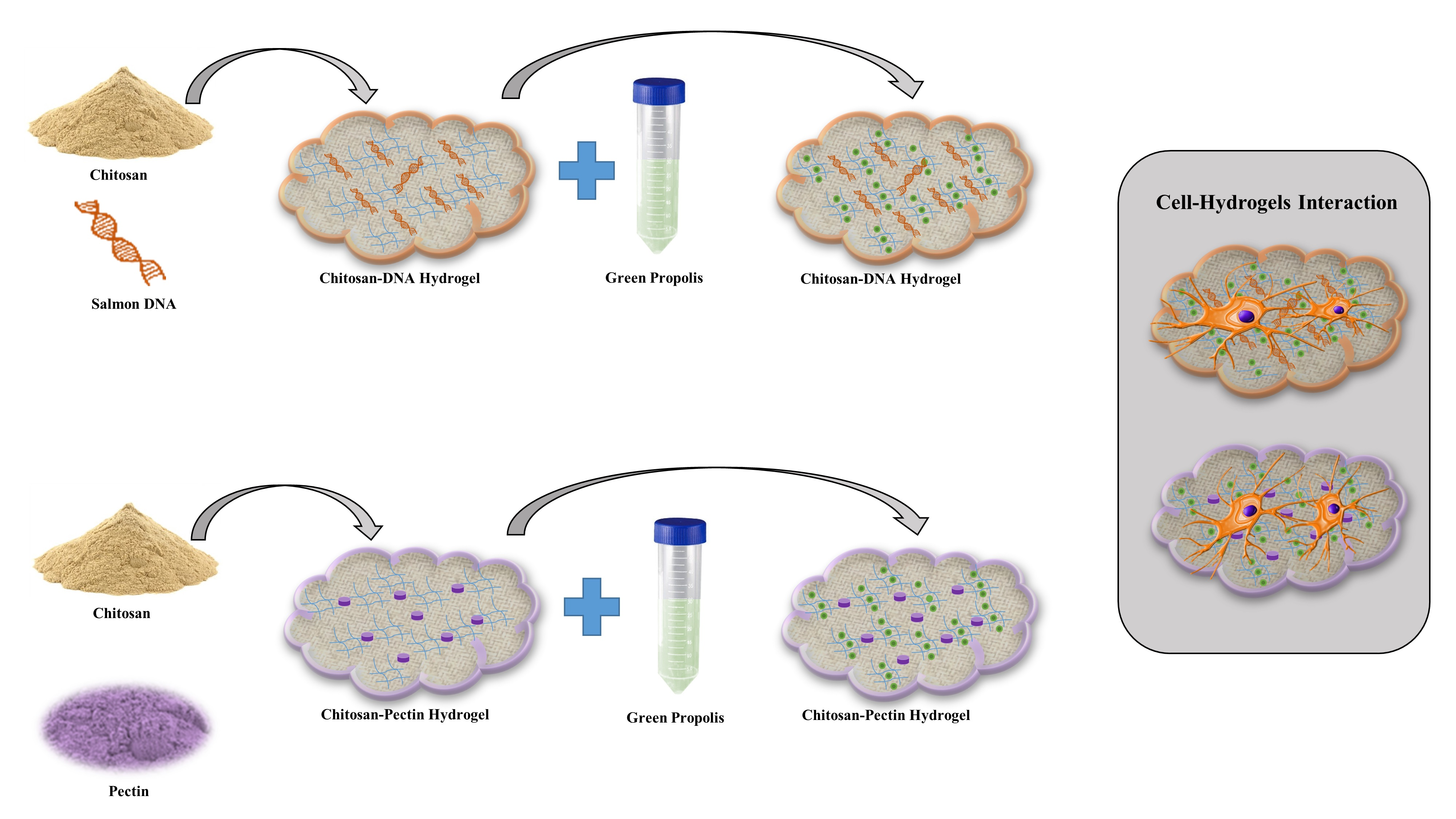
3.2. Hydrogel Preparation
3.3. Hydrogel Characterization
3.4. Loading and Release of Green Propolis
3.5. Biological Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CS | Chitosan |
| P | Pectin |
| DNA | Deoxyribonucleic acid |
| GP | Green propolis |
| TGA | Thermogravimetric analysis |
| dTG | Derivative thermogravimetry |
| PVA | Polyvinyl alcohol |
| MTT | 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide |
References
- Peppas, N.A. Hydrogels in Medicine and Pharmacy: Fundamentals; CRC Press: Boca Raton, FL, USA, 1986; Volume 1. [Google Scholar]
- Ma, S.; Yu, B.; Pei, X.; Zhou, F. Structural hydrogels. Polymer 2016, 98, 516–535. [Google Scholar] [CrossRef]
- Gong, J.P. Friction and lubrication of hydrogels—Its richness and complexity. Soft Matter 2006, 2, 544–552. [Google Scholar] [CrossRef]
- Costa, D.; Albuquerque, T.; Queiroz, J.A.; Valente, A.J.M. A co-delivery platform based on plasmid DNA peptide-surfactant complexes: Formation, characterization and release behavior. Colloids Surf B Biointerfaces 2019, 178, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Croisfelt, F.M.; Tundisi, L.L.; Ataide, J.A.; Silveira, E.; Tambourgi, E.B.; Jozala, A.F.; Souto, E.M.B.; Mazzola, P.G. Modified-release topical hydrogels: A ten-year review. J. Mater. Sci. 2019, 1, 1–21. [Google Scholar] [CrossRef]
- Agnihotri, S.; Singhal, R. Synthesis and characterization of novel poly (acrylic acid/sodium alginate/sodium humate) superabsorbent hydrogels. Part II: The effect of SH variation on Cu 2+, Pb 2+, Fe 2+ metal ions, MB, CV dye adsorption study. J. Polym. Environ. 2018, 26, 383–395. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, H.; Wang, Z.; Jin, Y. Magnetic chitosan/graphene oxide composite loaded with novel photosensitizer for enhanced photodynamic therapy. RSC Adv. 2018, 8, 10376–10388. [Google Scholar] [CrossRef] [Green Version]
- Kyzas, G.Z.; Bikiaris, D.N. Recent modifications of chitosan for adsorption applications: A critical and systematic review. Mar. Drugs 2015, 13, 312–337. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.F.; Bueno, P.V.; Almeida, E.A.; Rodrigues, F.H.; Rubira, A.F.; Muniz, E.C. Characterization of N-trimethyl chitosan/alginate complexes and curcumin release. Int. J. Biol. Macromol. 2013, 57, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Yu, S.; Sun, B.; Gao, S.; Guo, S.; Zhao, K. Biomedical applications of chitosan and its derivative nanoparticles. Polymers 2018, 10, 462. [Google Scholar] [CrossRef] [Green Version]
- Chien, K.B.; Chung, E.J.; Shah, R.N. Investigation of soy protein hydrogels for biomedical applications: Materials characterization, drug release, and biocompatibility. J. Biomater. Appl. 2014, 28, 1085–1096. [Google Scholar] [CrossRef]
- Pakzad, Y.; Ganji, F. Thermosensitive hydrogel for periodontal application: In vitro drug release, antibacterial activity and toxicity evaluation. J. Biomater. Appl. 2016, 30, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Han, B.; Liu, W.; Peng, Y. Evaluation on biological compatibility of carboxymethyl chitosan as biomaterials for antitumor drug delivery. J. Biomater. Appl. 2017, 31, 985–994. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Jiang, L.J.; Cao, P.P.; Li, J.B.; Chen, X.G. Glycerophosphate-based chitosan thermosensitive hydrogels and their biomedical applications. Carbohydr. Polym. 2015, 117, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Sci. 2004, 57, 19–34. [Google Scholar] [CrossRef]
- Aydınoğlu, D.; Ünal, M. Evaluation of the influence of spirulina microalgae on the drug delivery characteristics of genipin cross-linked chitosan hydrogels. Int. J. Polym. Mater. 2019, 68, 1020–1033. [Google Scholar] [CrossRef]
- Reyna-Urrutia, V.A.; Mata-Haro, V.; Cauich-Rodriguez, J.V.; Herrera-Kao, W.A.; Cervantes-Uc, J.M. Effect of two crosslinking methods on the physicochemical and biological properties of the collagen-chitosan scaffolds. Eur. Polym. J. 2019, 117, 424–433. [Google Scholar] [CrossRef]
- Uzumcu, A.T.; Guney, O.; Okay, O. Nanocomposite DNA hydrogels with temperature sensitivity. Polymer 2016, 100, 169–178. [Google Scholar] [CrossRef]
- Murakami, Y.; Maeda, M. DNA-responsive hydrogels that can shrink or swell. Biomacromolecules 2005, 6, 2927–2929. [Google Scholar] [CrossRef]
- Ishizuka, N.; Hashimoto, Y.; Matsuo, Y.; Ijiro, K. Highly expansive DNA hydrogel films prepared with photocrosslinkable poly (vinyl alcohol). Colloids. Surf. A Physicochem. Eng. Asp. 2006, 284, 440–443. [Google Scholar] [CrossRef]
- Costa, D.; Valente, A.J.; Miguel, M.G.; Queiroz, J. Gel network photodisruption: A new strategy for the codelivery of plasmid DNA and drugs. Langmuir 2011, 27, 13780–13789. [Google Scholar] [CrossRef]
- Um, S.H.; Lee, J.B.; Park, N.; Kwon, S.Y.; Umbach, C.C.; Luo, D. Enzyme-catalysed assembly of DNA hydrogel. Nat. Mater. 2006, 5, 797. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Valente, A.J.M.; Miguel, M.G.; Queiroz, J. Plasmid DNA hydrogels for biomedical applications. Adv. Colloid. Interfac. 2014, 205, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Valente, A.J.; Queiroz, J. Stimuli-responsive polyamine-DNA blend nanogels for co-delivery in cancer therapy. Colloids Surf. B Biointerfaces 2015, 132, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.F.; Dias, R.S.; Pereira, J.C.; Pais, A.A.C.C. DNA Condensation by pH-Responsive Polycations. Biomacromolecules 2010, 11, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.F.; Morán, M.C.; Vinardell, M.P.; Pereira, J.C.; Dias, R.S.; Pais, A.A. Ternary complexes DNA–polyethylenimine–Fe (iii) with linear and branched polycations: Implications on condensation, size, charge and in vitro biocompatibility. Soft Matter. 2013, 9, 10799–10810. [Google Scholar] [CrossRef]
- Bravo-Anaya, L.M.; Rinaudo, M.; Martínez, F.A.S. Conformation and rheological properties of calf-thymus DNA in solution. Polymers 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Anaya, L.M.; Soltero, J.F.A.; Rinaudo, M. DNA/chitosan electrostatic complex. Int. J. Biol. Macromol. 2016, 88, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, L.; Bianco-Peled, H. Pectin–chitosan physical hydrogels as potential drug delivery vehicles. Int. J. Biol. Macromol. 2017, 101, 852–861. [Google Scholar] [CrossRef]
- Cesar Filho, M.; Bueno, P.V.; Matsushita, A.F.; Rubira, A.F.; Muniz, E.C.; Durães, L.; Murtinho, D.M.; Valente, A.J. Synthesis, characterization and sorption studies of aromatic compounds by hydrogels of chitosan blended with β-cyclodextrin-and PVA-functionalized pectin. RSC Adv. 2018, 8, 14609–14622. [Google Scholar] [CrossRef] [Green Version]
- Mohnen, D. Pectin structure and biosynthesis. Curr. Opin. Plant Biol. 2008, 11, 266–277. [Google Scholar] [CrossRef]
- Thakur, B.R.; Singh, R.K.; Handa, A.K.; Rao, M. Chemistry and uses of pectin—a review. Crit. Rev. Food Sci. Nutr. 1997, 37, 47–73. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef] [PubMed]
- De Barros, M.P.; Sousa, J.P.B.; Bastos, J.K.; de Andrade, S.F. Effect of Brazilian green propolis on experimental gastric ulcers in rats. J. Ethnopharmacol. 2007, 110, 567–571. [Google Scholar] [CrossRef]
- Santos, V.; Pimenta, F.; Aguiar, M.; Do Carmo, M.; Naves, M.; Mesquita, R. Oral candidiasis treatment with Brazilian ethanol propolis extract. Phytother. Res. 2005, 19, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Shimazawa, M.; Mishima, S.; Hara, H. Water extract of propolis and its main constituents, caffeoylquinic acid derivatives, exert neuroprotective effects via antioxidant actions. Life Sci. 2007, 80, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Bankova, V.S.; de Castro, S.L.; Marcucci, M.C. Propolis: Recent advances in chemistry and plant origin. Apidologie 2000, 31, 3–15. [Google Scholar] [CrossRef]
- Fernandes Queiroz, M.; Melo, K.R.; Sabry, D.A.; Sassaki, G.L.; Rocha, H.A. Does the use of chitosan contribute to oxalate kidney stone formation? Mar. Drugs 2014, 13, 141–158. [Google Scholar] [CrossRef]
- Souza, N.L.G.D.; Salles, T.F.; Brandão, H.M.; Edwards, H.G.M.; de Oliveira, L.F.C. Synthesis, Vibrational Spectroscopic and Thermal Properties of Oxocarbon Cross Linked Chitosan. J. Braz. Chem. Soc. 2015, 26, 1247–1256. [Google Scholar] [CrossRef]
- Krishnaveni, B.; Ragunathan, R. Extraction and characterization of chitin and chitosan from F. solani CBNR BKRR, Synthesis of their bionanocomposites and study of their productive application. Int. J. Pharm. Sci. Rev. Res. 2015, 7, 197. [Google Scholar]
- Gnanasambandam, R.; Proctor, A. Determination of pectin degree of esterification by diffuse reflectance Fourier transform infrared spectroscopy. Food Chem. 2000, 68, 327–332. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C. Spectrometric identification of organic compounds. J. Chem. Educ. 1962, 39, 546. [Google Scholar] [CrossRef]
- Papancea, A.; Valente, A.J.; Patachia, S.; Miguel, M.G.; Lindman, B. PVA− DNA cryogel membranes: Characterization, swelling, and transport studies. Langmuir 2008, 24, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, M.; Gräslund, A. An FTIR and CD study of the structural effects of G-tract length and sequence context on DNA conformation in solution. J. Mol. Biol. 2001, 314, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Ede, S.R.; Ramadoss, A.; Anantharaj, S.; Nithiyanantham, U.; Kundu, S. Enhanced catalytic and supercapacitor activities of DNA encapsulated β-MnO 2 nanomaterials. Phys. Chem. Chem. Phys. 2014, 16, 21846–21859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froehlich, E.; Mandeville, J.; Weinert, C.; Kreplak, L.; Tajmir-Riahi, H. Bundling and aggregation of DNA by cationic dendrimers. Biomacromolecules 2010, 12, 511–517. [Google Scholar] [CrossRef]
- Rachini, A.; Le Troedec, M.; Peyratout, C.; Smith, A. Comparison of the thermal degradation of natural, alkali-treated and silane-treated hemp fibers under air and an inert atmosphere. J. Appl. Polym. Sci. 2009, 112, 226–234. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Y.; Ding, G.; Geng, Q.; Zhu, J.; Guo, M.; Duan, Y.; Wang, B.; Cao, Y. Synthesis, characterization, and application of microbe-triggered controlled-release kasugamycin–pectin conjugate. J. Agric. Food Chem. 2015, 63, 4263–4268. [Google Scholar] [CrossRef]
- Corazzari, I.; Nisticò, R.; Turci, F.; Faga, M.G.; Franzoso, F.; Tabasso, S.; Magnacca, G. Advanced physico-chemical characterization of chitosan by means of TGA coupled on-line with FTIR and GCMS: Thermal degradation and water adsorption capacity. Polym. Degrad. Stab. 2015, 112, 1–9. [Google Scholar] [CrossRef]
- Safaee, M.M.; Gravely, M.; Lamothe, A.; McSweeney, M.; Roxbury, D. Enhancing the Thermal Stability of Carbon Nanomaterials with DNA. Sci. Rep. 2019, 9, 11926. [Google Scholar] [CrossRef]
- Stealey, S.; Guo, X.; Ren, L.; Bryant, E.; Kaltchev, M.; Chen, J.; Kumpaty, S.; Hua, X.; Zhang, W. Stability improvement and characterization of bioprinted pectin-based scaffold. J. Appl. Biomater. Func. 2019, 17, 2280800018807108. [Google Scholar] [CrossRef] [Green Version]
- Cazorla-Luna, R.; Notario-Pérez, F.; Martín-Illana, A.; Ruiz-Caro, R.; Tamayo, A.; Rubio, J.; Veiga, M.D. Chitosan-based mucoadhesive vaginal tablets for controlled release of the Anti-HIV drug tenofovir. Pharmaceutics 2019, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Varaprasad, K.; Vimala, K.; Ravindra, S.; Reddy, N.N.; Reddy, G.S.M.; Raju, K.M. Biodegradable chitosan hydrogels for in vitro drug release studies of 5-flurouracil an anticancer drug. J. Polym. Environ. 2012, 20, 573–582. [Google Scholar] [CrossRef]
- Macha, I.J.; Ben-Nissan, B.; Vilchevskaya, E.N.; Abali, B.E.; Mueller, W.H.; Rickert, W. Drug delivery from polymer-based nanopharmaceuticals—An experimental study complemented by a simulation of selected diffusion processes. Front. Bioeng. Biotechnol. 2019, 7, 37. [Google Scholar] [CrossRef]
- Issa, M.G.; Pessole, L.; Takahashi, A.I.; Andréo Filho, N.; Ferraz, H.G. Physicochemical and dissolution profile characterization of pellets containing different binders obtained by the extrusion-spheronization process. Braz. J. Pharm. Sci. 2012, 48, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Liu, L.; Hu, S.; Wang, Y.; Yang, S.; Qu, J. Optimizing the synthesis of core/shell structure Au@ Cu 2 S nanocrystals as contrast-enhanced for bioimaging detection. Sci. Rep. 2018, 8, 1–9. [Google Scholar]
- Baker, J.P.; Stephens, D.R.; Blanch, H.W.; Prausnitz, J.M. Swelling equilibria for acrylamide-based polyampholyte hydrogels. Macromolecules 1992, 25, 1955–1958. [Google Scholar] [CrossRef] [Green Version]
- Valente, A.J.; Cruz, S.M.; Murtinho, D.M.; Miguel, M.G.; Muniz, E.C. DNA–poly (vinyl alcohol) gel matrices: Release properties are strongly dependent on electrolytes and cationic surfactants. Colloids Surf. B. Biointerfaces 2013, 101, 111–117. [Google Scholar] [CrossRef]
- Siepmann, J.; Podual, K.; Sriwongjanya, M.; Peppas, N.; Bodmeier, R. A new model describing the swelling and drug release kinetics from hydroxypropyl methylcellulose tablets. J. Pharm. Sci. 1999, 88, 65–72. [Google Scholar] [CrossRef]
- Faisant, N.; Siepmann, J.; Benoit, J. PLGA-based microparticles: Elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur. J. Pharm. Sci. 2002, 15, 355–366. [Google Scholar] [CrossRef]
- Polishchuk, A.Y.; Zaikov, G.E. Multicomponent transport in polymer systems for controlled release; CRC Press: Boca Raton, FL, USA, 1997. [Google Scholar]
- Dinescu, S.; Ionita, M.; Ignat, S.-R.; Costache, M.; Hermenean, A. Graphene oxide enhances chitosan-Based 3D scaffold properties for bone tissue engineering. Int. J. Mol. Sci. 2019, 20, 5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Qiao, Z.; Yu, F.; Hu, H.; Huang, Y.; Xiang, Q. Transforming growth factor-beta3/chitosan sponge (TGF-beta3/CS) facilitates osteogenic differentiation of human periodontal ligament stem cells. Int. J. Mol. Sci. 2019, 20, 20. [Google Scholar]
- Singh, Y.P.; Dasgupta, S.; Bhaskar, R. Preparation, characterization and bioactivities of nano anhydrous calcium phosphate added gelatin–chitosan scaffolds for bone tissue engineering. J. Biomater. Sci. Polym. Ed. 2019, 30, 1756–1778. [Google Scholar] [CrossRef] [PubMed]
- Normalizacyjny, P.P.K. Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity (ISO 10993-5:2009); Polski Komitet Normalizacyjny: Warsaw, Poland, 2009. [Google Scholar]
- Hwang, C.M.; Sant, S.; Masaeli, M.; Kachouie, N.N.; Zamanian, B.; Lee, S.-H.; Khademhosseini, A. Fabrication of three-dimensional porous cell-laden hydrogel for tissue engineering. Biofabrication 2010, 2, 035003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, M.L.; Phillips, R.W.; Daoud El Tannir, M. Tarnish of certain dental alloys. J. Dent. Res. 1958, 37, 837–847. [Google Scholar] [CrossRef]
- Fusayama, T.; Katayori, T.; Nomoto, S. Corrosion of gold and amalgam placed in contact with each other. J. Dent. Res. 1963, 42, 1183–1197. [Google Scholar] [CrossRef]
- Nomura, D.; Saito, M.; Takahashi, Y.; Takahashi, Y.; Takakura, Y.; Nishikawa, M. Development of Orally-deliverable DNA Hydrogel by Microemulsification and Chitosan Coating. Int. J. Pharm. 2018, 547, 556–562. [Google Scholar] [CrossRef]
- Grande, R.; Carvalho, A.J. Compatible ternary blends of chitosan/poly (vinyl alcohol)/poly (lactic acid) produced by oil-in-water emulsion processing. Biomacromolecules 2011, 12, 907–914. [Google Scholar] [CrossRef]
- do Prado, R.F.; de Oliveira, F.S.; Nascimento, R.D.; de Vasconcellos, L.M.R.; Carvalho, Y.R.; Cairo, C.A.A. Osteoblast response to porous titanium and biomimetic surface: In vitro analysis. Mat. Sci. Eng. C-Mater. 2015, 52, 194–203. [Google Scholar] [CrossRef]
- De Oliveira, P.T.; Nanci, A. Nanotexturing of titanium-based surfaces upregulates expression of bone sialoprotein and osteopontin by cultured osteogenic cells. Biomaterials 2004, 25, 403–413. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CS-P | CS-DNA | |||
|---|---|---|---|---|
| 25 °C | 37 °C | 25 °C | 37 °C | |
| Water | ||||
| EE (%) | 19 | 21 | 7 | 8 |
| Cumulative release (%) | 13.2 | 12.1 | 25.7 | 30 |
| Simulated Saliva | ||||
| EE (%) | 30 | 38 | 8 | 10 |
| Cumulative release (%) | 13.9 | 19.9 | 31.6 | 36.5 |
| CS-P | CS-DNA | |||
|---|---|---|---|---|
| 25 °C | 37 °C | 25 °C | 37 °C | |
| Water | ||||
| Ceq/(μg mL−1) | 10.1 (±0.4) | 10.2 (±0.3) | 7.2 (±0.2) | 9.6 (±0.2) |
| logt1/s | 1051 (±73) | 1005 (±74) | 2051 (±203) | 2851 (±214) |
| logt2/s | 12,571 (±390) | 10,460 (±360) | 12,878 (±242) | 14,005 (±158) |
| k1/s−1 | 1.2 (±0.3) × 10−3 | 1.5 (±0.5) × 10−3 | 4.7 (±0.2) × 10−4 | 3.5 (±0.4) × 10−4 |
| k2/s−1 | 1.2 (±0.2) × 10−4 | 1.3 (±0.2) × 10−4 | 1.7 (±0.2) × 10−4 | 2.5 (±0.2) × 10−4 |
| P | 0.24 (±0.04) | 0.26 (±0.04) | 0.29 (±0.04) | 0.34 (±0.02) |
| R2 | 0.9963 | 0.9952 | 0.9970 | 0.9972 |
| Simulated Saliva | ||||
| Ceq/(μg mL−1) | 16.7 (±0.2) | 30.2 (±0.3) | 10.1 (±0.6) | 14.6 (±0.6) |
| logt1/s | 4468 (±363) | 3948 (±529) | 4906 (±300) | 969 (±127) |
| logt2/s | 13,567 (±131) | 13,225 (±118) | 13,379 (±147) | 11,972 (±321) |
| k1/s−1 | 2.9 (±0.4) × 10−4 | 2.0 (±0.3) × 10−4 | 2.7 (±0.3) × 10−4 | 8 (±2) × 10−4 |
| k2/s−1 | 3.3 (±0.3) × 10−4 | 3.8 (±0.4) × 10−4 | 3.3 (±0.4) × 10−4 | 1.4 (±0.2) × 10−4 |
| p | 0.34 (±0.03) | 0.36 (±0.03) | 0.43 (±0.04) | 0.24 (±0.04) |
| R2 | 0.9974 | 0.9978 | 0.9981 | 0.9951 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, T.; Mello, D.; Vasconcellos, L.; Valente, A.J.M.; Borges, A. Chitosan-Based Coacervate Polymers for Propolis Encapsulation: Release and Cytotoxicity Studies. Int. J. Mol. Sci. 2020, 21, 4561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124561
Sato T, Mello D, Vasconcellos L, Valente AJM, Borges A. Chitosan-Based Coacervate Polymers for Propolis Encapsulation: Release and Cytotoxicity Studies. International Journal of Molecular Sciences. 2020; 21(12):4561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124561
Chicago/Turabian StyleSato, Tabata, Daphne Mello, Luana Vasconcellos, Artur J. M. Valente, and Alexandre Borges. 2020. "Chitosan-Based Coacervate Polymers for Propolis Encapsulation: Release and Cytotoxicity Studies" International Journal of Molecular Sciences 21, no. 12: 4561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124561