Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Lyso-Gb1 Quantification in Dried Blood Spots Is a Valid Laboratory Approach Including Excellent Precision Characteristics
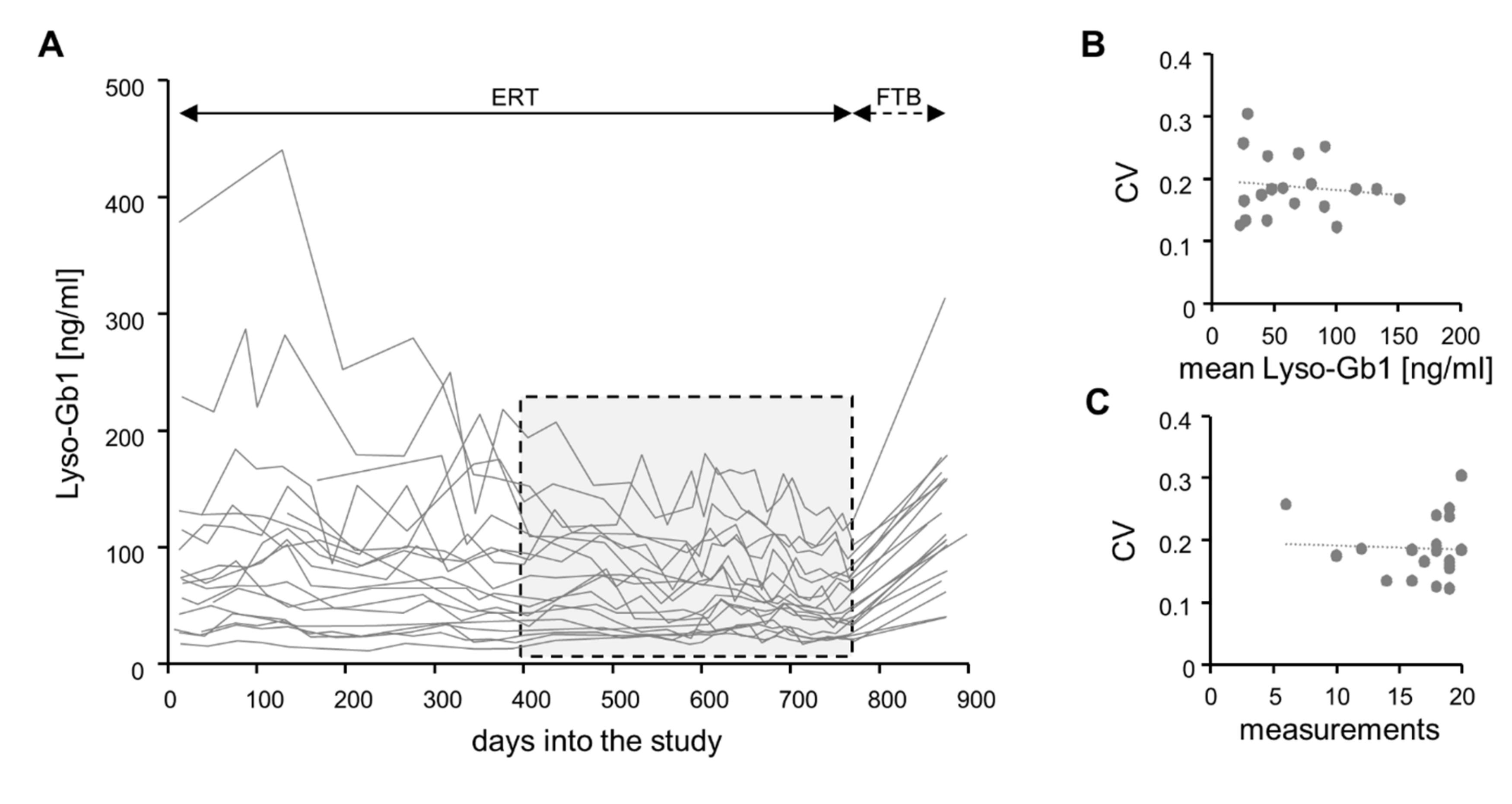
2.2. Under Continuous Treatment, Lyso-Gb1 Levels Vary over Time in a Patient-Specific Manner
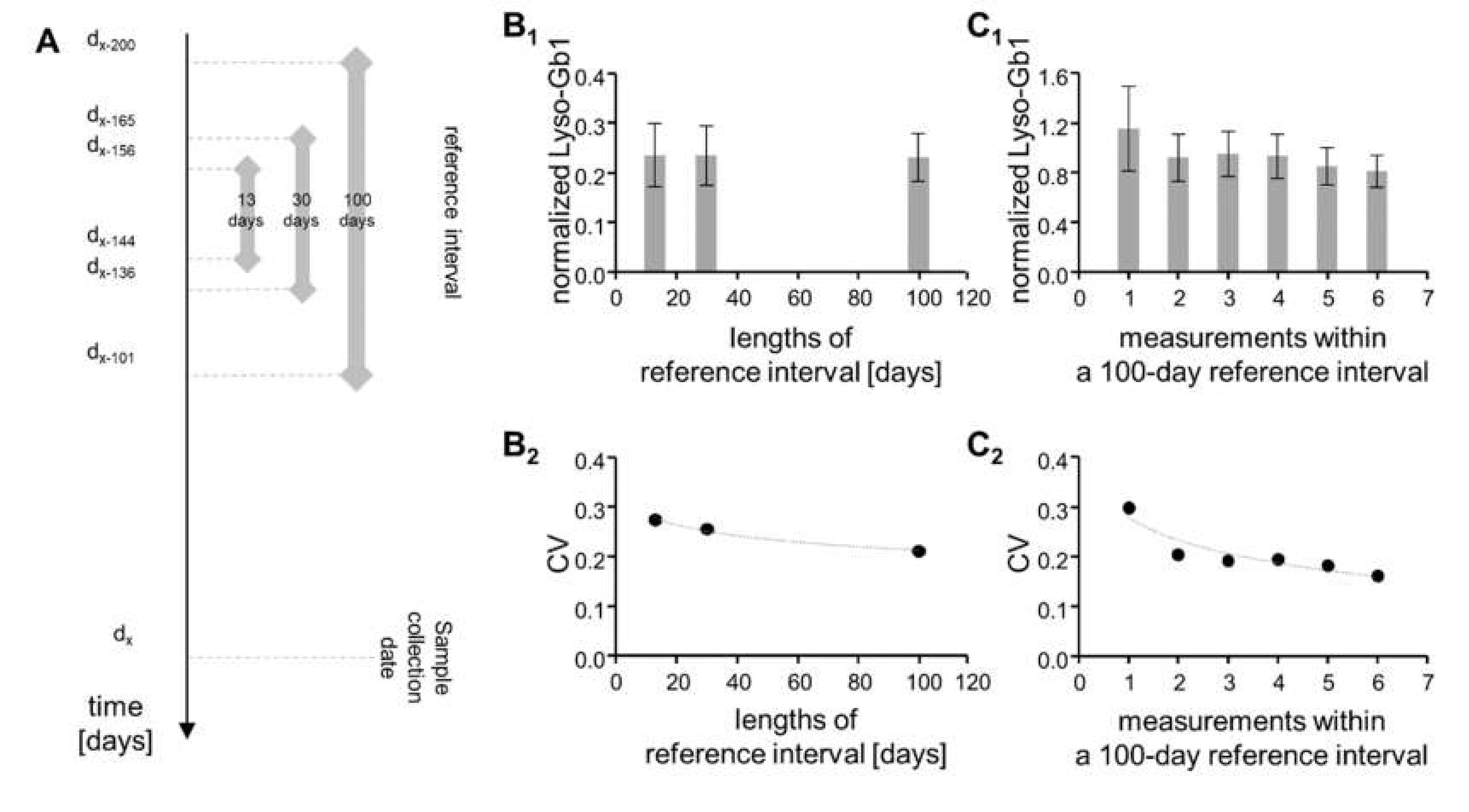
2.3. Temporal Fluctuation Is Leveled Out by Frequent Measurements over Long Reference Intervals
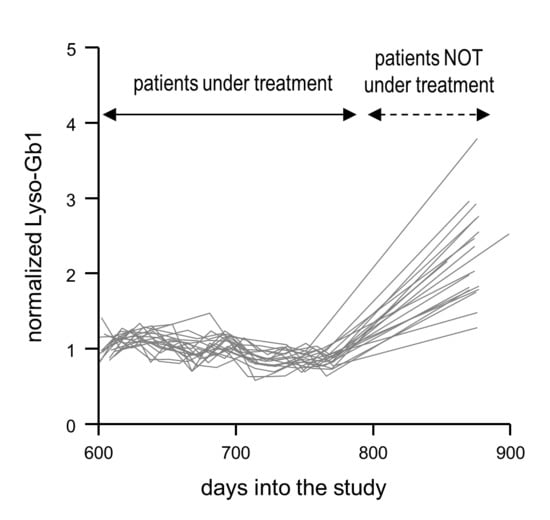
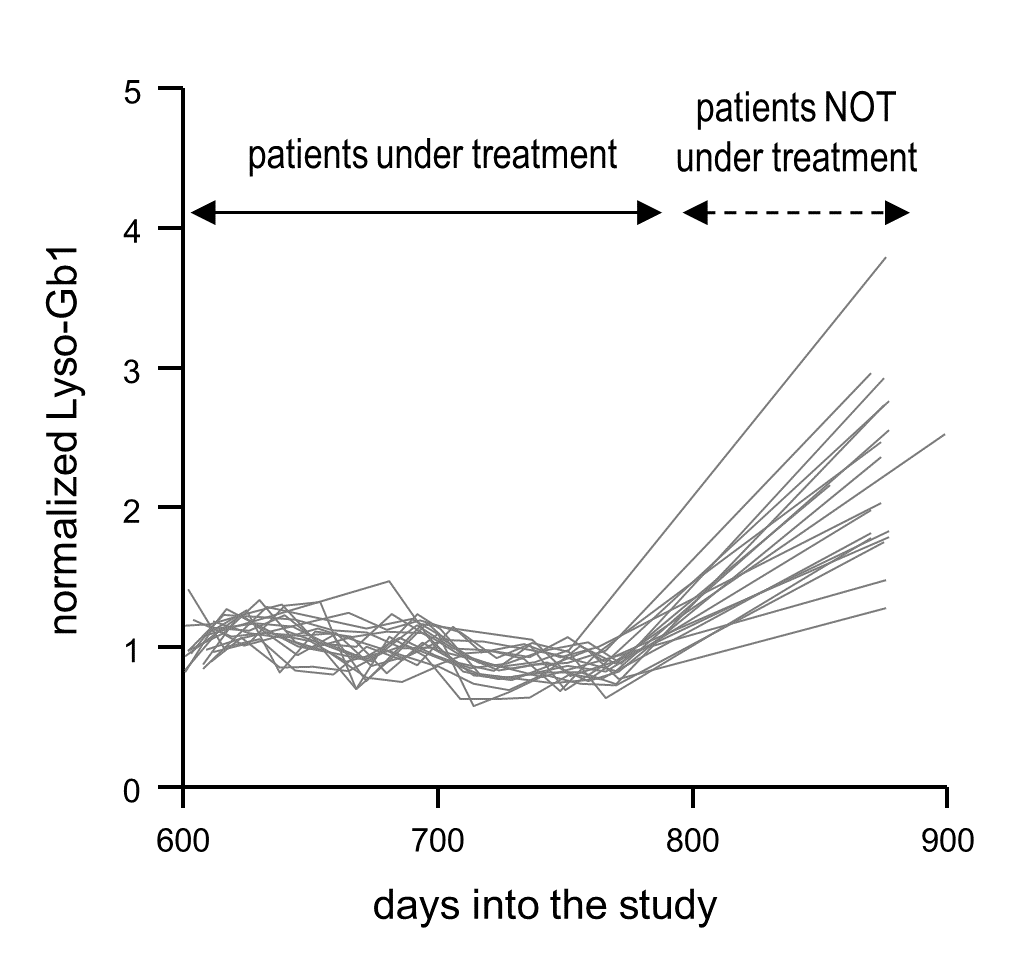
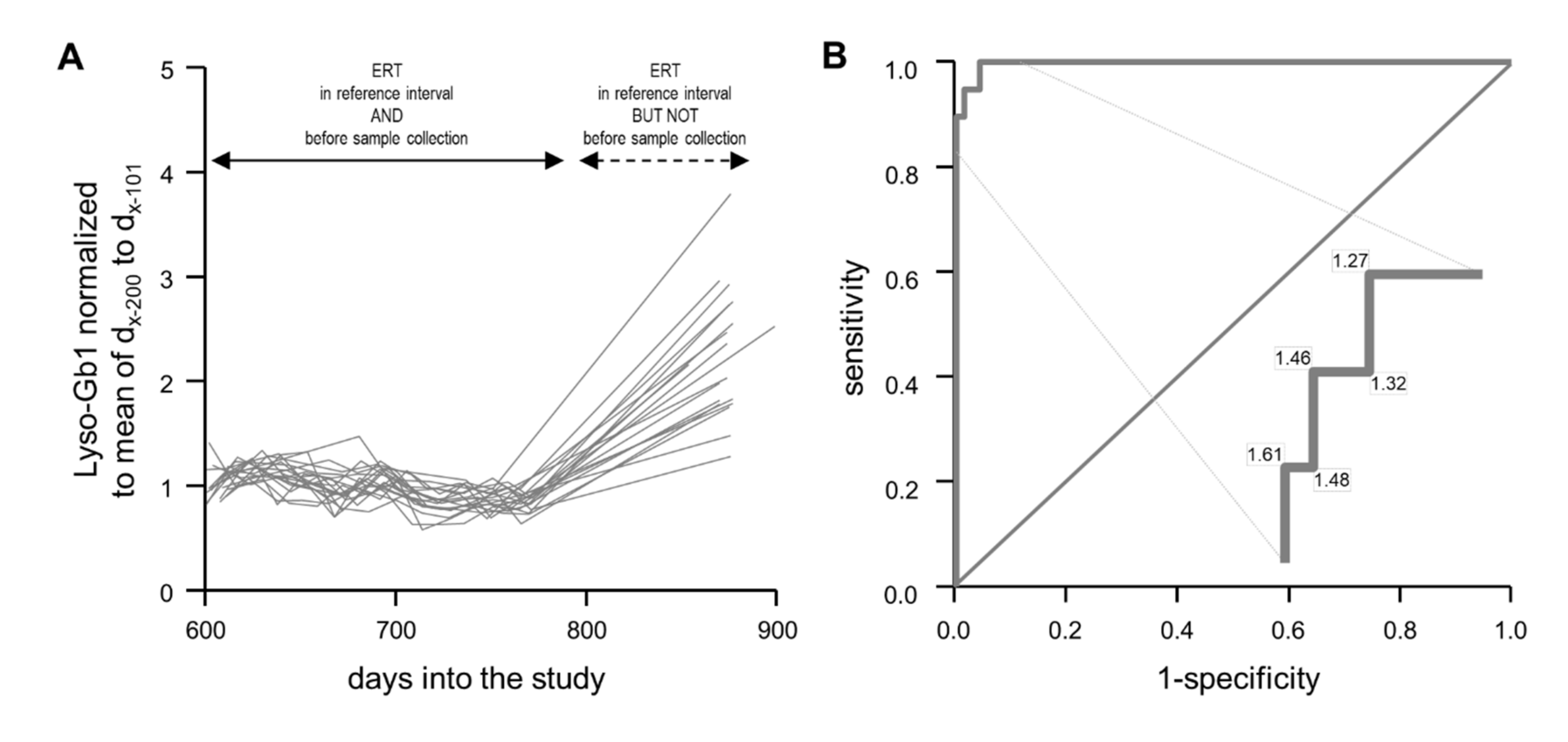
2.4. Normalized Lyso-Gb1 Values Are Highly Sensitive and Specific for Flagging Absent Treatment
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Quantification of Lyso-Gb1
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Hughes, D.A. Gaucher Disease. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; Springer: Boston, MA, USA, 1993. [Google Scholar]
- Weinreb, N.J.; Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: A report from the Gaucher Registry. Am. J. Med. 2002, 113, 112–119. [Google Scholar] [CrossRef]
- Raghavan, S.S.; Mumford, R.A.; Kanfer, J.N. Isolation and characterization of glucosylsphingosine from Gaucher’s spleen. J. Lipid Res. 1974, 15, 484–490. [Google Scholar]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Orvisky, E.; Sidransky, E.; McKinney, C.E.; Lamarca, M.E.; Samimi, R.; Krasnewich, D.; Martin, B.M.; Ginns, E.I. Glucosylsphingosine accumulation in mice and patients with type 2 Gaucher disease begins early in gestation. Pediatr. Res. 2000, 48, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Cozma, C.; Yang, F.; Kramp, G.; Meyer, A.; Nesslauer, A.M.; Eichler, S.; Bottcher, T.; Witt, M.; Brauer, A.U.; et al. Glucosylsphingosine Causes Hematological and Visceral Changes in Mice-Evidence for a Pathophysiological Role in Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 2192. [Google Scholar] [CrossRef] [Green Version]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Smid, B.E.; Ferraz, M.J.; Verhoek, M.; Mirzaian, M.; Wisse, P.; Overkleeft, H.S.; Hollak, C.E.; Aerts, J.M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 2016, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Elstein, D.; Mellgard, B.; Dinh, Q.; Lan, L.; Qiu, Y.; Cozma, C.; Eichler, S.; Bottcher, T.; Zimran, A. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naive and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 2017, 122, 113–120. [Google Scholar] [CrossRef]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Yum, M.S.; Heo, S.H.; Kim, T.; Jin, H.K.; Bae, J.S.; Seo, G.H.; Oh, A.; Yoon, H.M.; Lim, H.T.; et al. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J. Med. Genet 2020, 57, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. J. Clin. Med. 2019, 8, 1662. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.D.; Rosman, L.M.; Ratcliff, J.D.; Strickland, P.T.; Graham, D.R.; Silbergeld, E.K. State of the Science in Dried Blood Spots. Clin. Chem. 2018, 64, 656–679. [Google Scholar] [CrossRef] [Green Version]
- Elstein, D.; Abrahamov, A.; Hadas-Halpern, I.; Zimran, A. Withdrawal of enzyme replacement therapy in Gaucher’s disease. Br. J. Haematol. 2000, 110, 488–492. [Google Scholar] [CrossRef]
- vom Dahl, S.; Poll, L.W.; Haussinger, D. Clinical monitoring after cessation of enzyme replacement therapy in M. Gaucher. Br. J. Haematol. 2001, 113, 1084–1087. [Google Scholar] [CrossRef]
- Hollak, C.E.; vom Dahl, S.; Aerts, J.M.; Belmatoug, N.; Bembi, B.; Cohen, Y.; Collin-Histed, T.; Deegan, P.; van Dussen, L.; Giraldo, P.; et al. Force majeure: Therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells Mol. Dis. 2010, 44, 41–47. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Elstein, D. Nonprecipitous changes upon withdrawal from imiglucerase for Gaucher disease because of a shortage in supply. Blood Cells Mol. Dis. 2011, 46, 111–114. [Google Scholar] [CrossRef]
- Giraldo, P.; Irun, P.; Alfonso, P.; Dalmau, J.; Fernandez-Galan, M.A.; Figueredo, A.; Hernandez-Rivas, J.M.; Julia, A.; Luno, E.; Marin-Jimenez, F.; et al. Evaluation of Spanish Gaucher disease patients after a 6-month imiglucerase shortage. Blood Cells Mol. Dis. 2011, 46, 115–118. [Google Scholar] [CrossRef]
- Stirnemann, J.; Rose, C.; Serratrice, C.; Dalbies, F.; Lidove, O.; Masseau, A.; Pers, Y.M.; Baron, C.; Belmatoug, N. Impact of imiglucerase supply constraint on the therapeutic management and course of disease in French patients with Gaucher disease type 1. Orphanet J. Rare Dis. 2015, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biegstraaten, M.; Cox, T.M.; Belmatoug, N.; Berger, M.G.; Collin-Histed, T.; Vom Dahl, S.; Di Rocco, M.; Fraga, C.; Giona, F.; Giraldo, P.; et al. Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czartoryska, B.; Tylki-Szymanska, A.; Lugowska, A. Changes in serum chitotriosidase activity with cessation of replacement enzyme (cerebrosidase) administration in Gaucher disease. Clin. Biochem. 2000, 33, 147–149. [Google Scholar] [CrossRef]
- Schwartz, I.V.; Karam, S.; Ashton-Prolla, P.; Michelin, K.; Coelho, J.; Pires, R.F.; Pereira, M.L.; Giugliani, R. Effects of imilglucerase withdrawal on an adult with Gaucher disease. Br. J. Haematol. 2001, 113, 1089. [Google Scholar] [CrossRef]
- Machaczka, M.; Kampe Bjorkvall, C.; Wieremiejczyk, J.; Paucar Arce, M.; Myhr-Eriksson, K.; Klimkowska, M.; Hagglund, H.; Svenningsson, P. Impact of imiglucerase supply shortage on clinical and laboratory parameters in Norrbottnian patients with Gaucher disease type 3. Arch. Immunol. Ther. Exp. 2015, 63, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Perez-Calvo, J.; Giraldo, P.; Pastores, G.M.; Fernandez-Galan, M.; Martin-Nunez, G.; Pocovi, M. Extended interval between enzyme therapy infusions for adult patients with Gaucher’s disease type 1. J. Postgrad Med. 2003, 49, 127–131. [Google Scholar]
- Raskovalova, T.; Deegan, P.B.; Yang, R.; Pavlova, E.; Stirnemann, J.; Labarere, J.; Zimran, A.; Mistry, P.K.; Berger, M. Plasma chitotriosidase activity versus CCL18 level for assessing type I Gaucher disease severity: Protocol for a systematic review with meta-analysis of individual participant data. Syst. Rev. 2017, 6, 87. [Google Scholar] [CrossRef]
- Guo, Y.; He, W.; Boer, A.M.; Wevers, R.A.; de Bruijn, A.M.; Groener, J.E.; Hollak, C.E.; Aerts, J.M.; Galjaard, H.; van Diggelen, O.P. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J. Inherit. Metab. Dis. 1995, 18, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Schutyser, E.; Richmond, A.; Van Damme, J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J. Leukoc. Biol. 2005, 78, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Boot, R.G.; Renkema, G.H.; Verhoek, M.; Strijland, A.; Bliek, J.; de Meulemeester, T.M.; Mannens, M.M.; Aerts, J.M. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998, 273, 25680–25685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, G.A. Gaucher disease: Gene frequencies and genotype/phenotype correlations. Genet Test 1997, 1, 5–12. [Google Scholar] [CrossRef]
- Santamaria, R.; Michelakakis, H.; Moraitou, M.; Dimitriou, E.; Dominissini, S.; Grossi, S.; Sanchez-Olle, G.; Chabas, A.; Pittis, M.G.; Filocamo, M.; et al. Haplotype analysis suggests a single Balkan origin for the Gaucher disease [D409H;H255Q] double mutant allele. Hum. Mutat. 2008, 29, E58–E67. [Google Scholar] [CrossRef] [PubMed]
- ISO 15189:2012. Medical Laboratories — Requirements for Quality and Competence. Available online: https://www.iso.org/standard/56115.html (accessed on 27 June 2020).
- Tanna, S.; Lawson, G. Self-sampling and quantitative analysis of DBS: Can it shift the balance in over-burdened healthcare systems? Bioanalysis 2015, 7, 1963–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielczarek, P.; Silberring, J.; Smoluch, M. Miniaturization in Mass Spectrometry. Mass Spectrom. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Sample | Control Sample Spiked with Low Levels of Lyso-Gb1 | Control Sample Spiked with High Levels of Lyso-Gb1 | ||
|---|---|---|---|---|
| Fresh sample | Mean a [ng/µL] | 5.3 | 29.7 | 217.2 |
| SD [ng/µL] | 0.3 | 1.8 | 10.6 | |
| CV [%] | 4.8 | 5.9 | 4.9 | |
| After 3 months of storage | Mean a [ng/µL] | 5.6 | 27.0 | 218.5 |
| SD [ng/µL] | 0.2 | 1.4 | 10.2 | |
| CV [%] | 3.1 | 5.3 | 4.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int. J. Mol. Sci. 2020, 21, 4577. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134577
Cozma C, Cullufi P, Kramp G, Hovakimyan M, Velmishi V, Gjikopulli A, Tomori S, Fischer S, Oppermann S, Grittner U, et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. International Journal of Molecular Sciences. 2020; 21(13):4577. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134577
Chicago/Turabian StyleCozma, Claudia, Paskal Cullufi, Guido Kramp, Marina Hovakimyan, Virtut Velmishi, Agim Gjikopulli, Sonila Tomori, Steffen Fischer, Sebastian Oppermann, Ulrike Grittner, and et al. 2020. "Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots" International Journal of Molecular Sciences 21, no. 13: 4577. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134577