The Mitochondrial Genome Assembly of Fennel (Foeniculum vulgare) Reveals Two Different atp6 Gene Sequences in Cytoplasmic Male Sterile Accessions

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
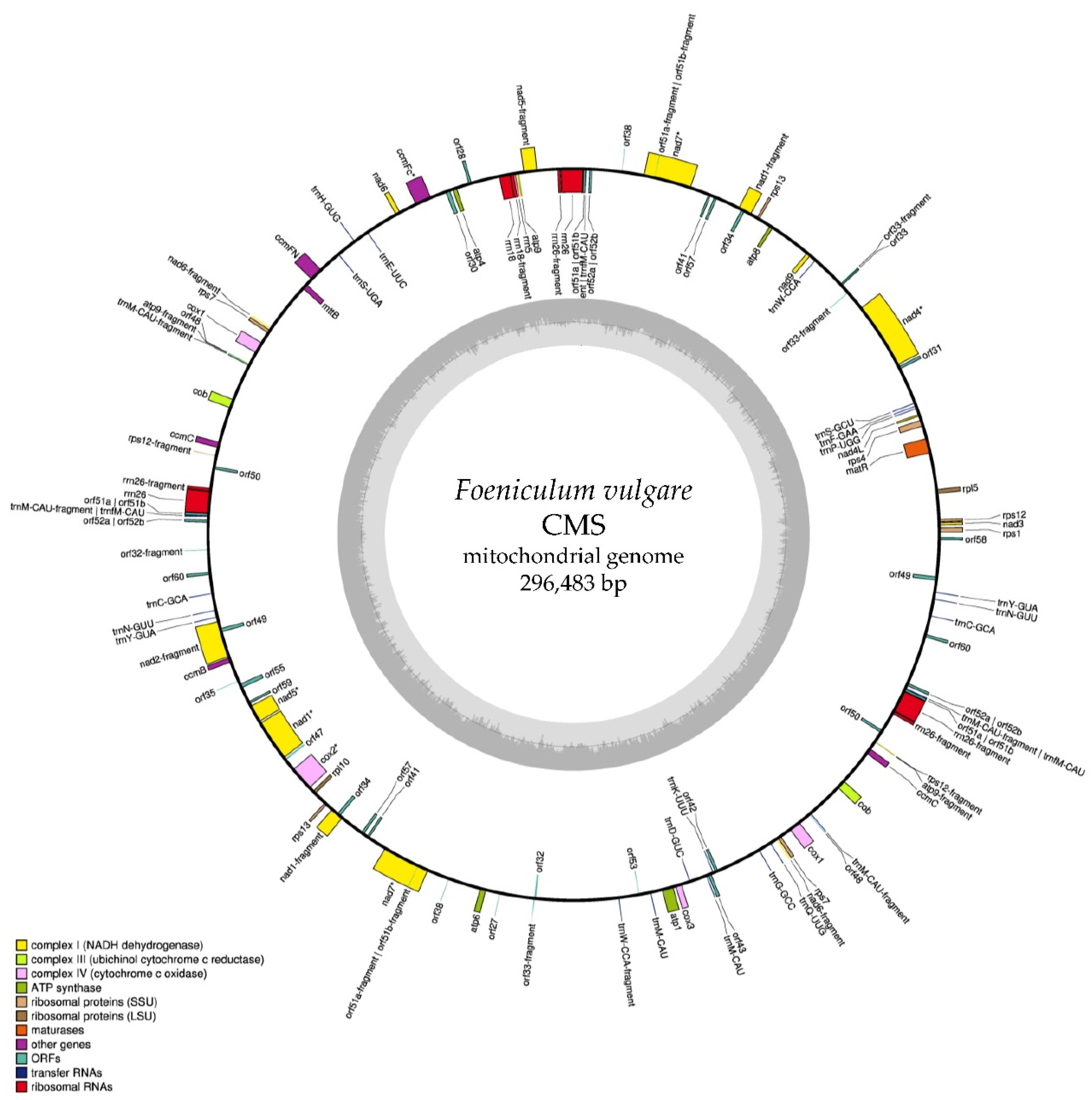
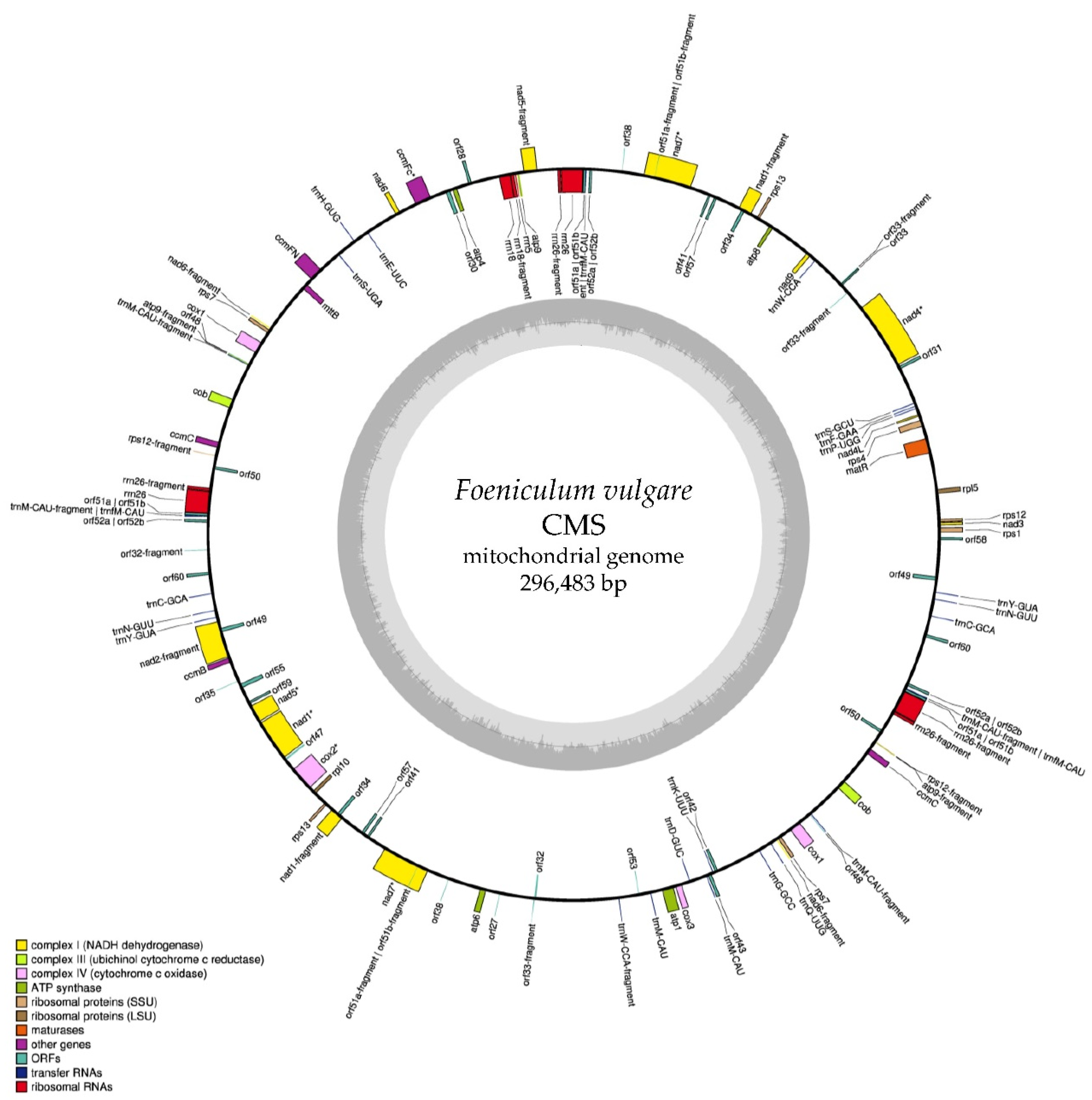
2.1. Mitochondrial Genome Sequence and Annotation
2.2. SNP and InDel Detection
2.3. Selection of Candidate Gene Controlling Cytoplasmic Male Sterility
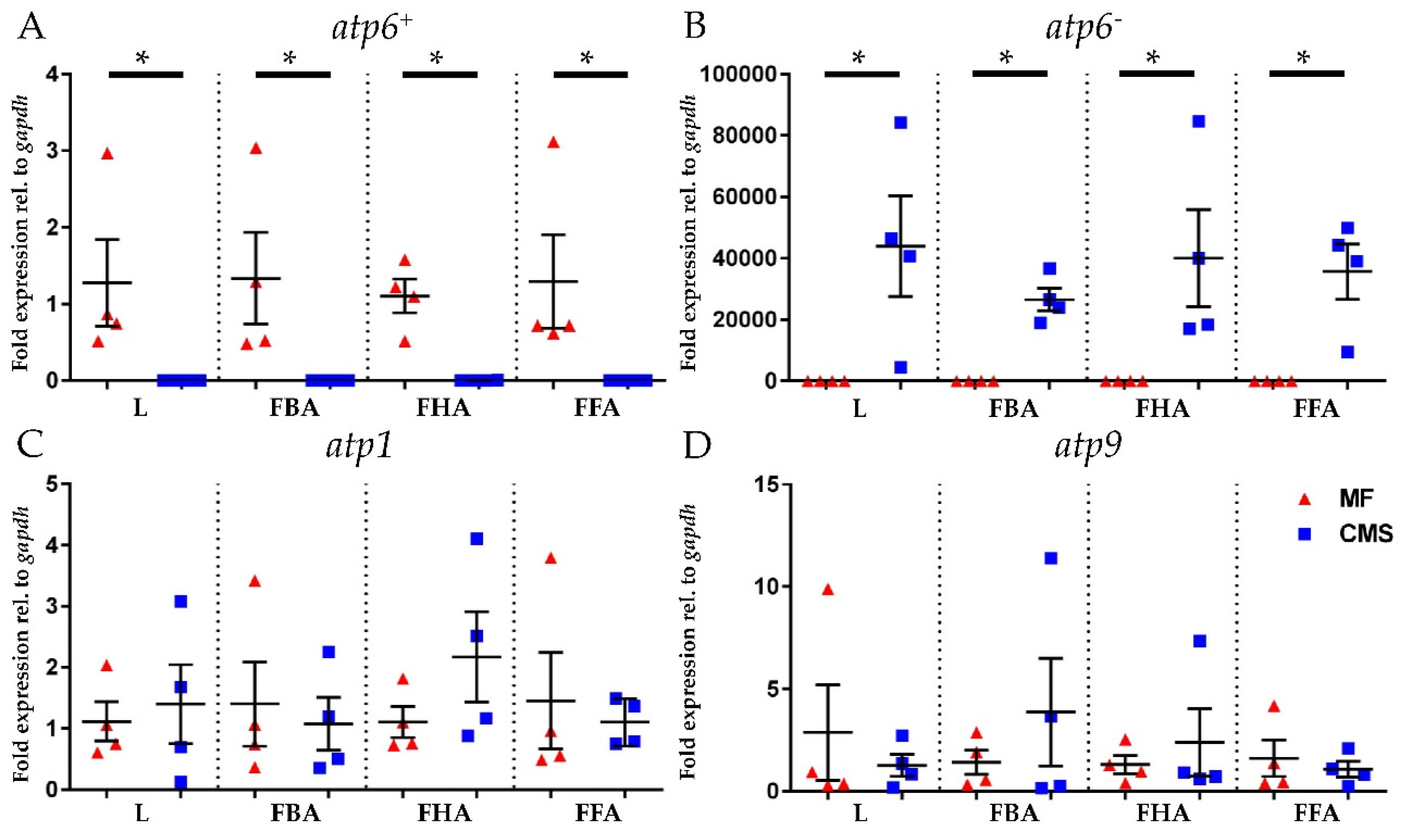
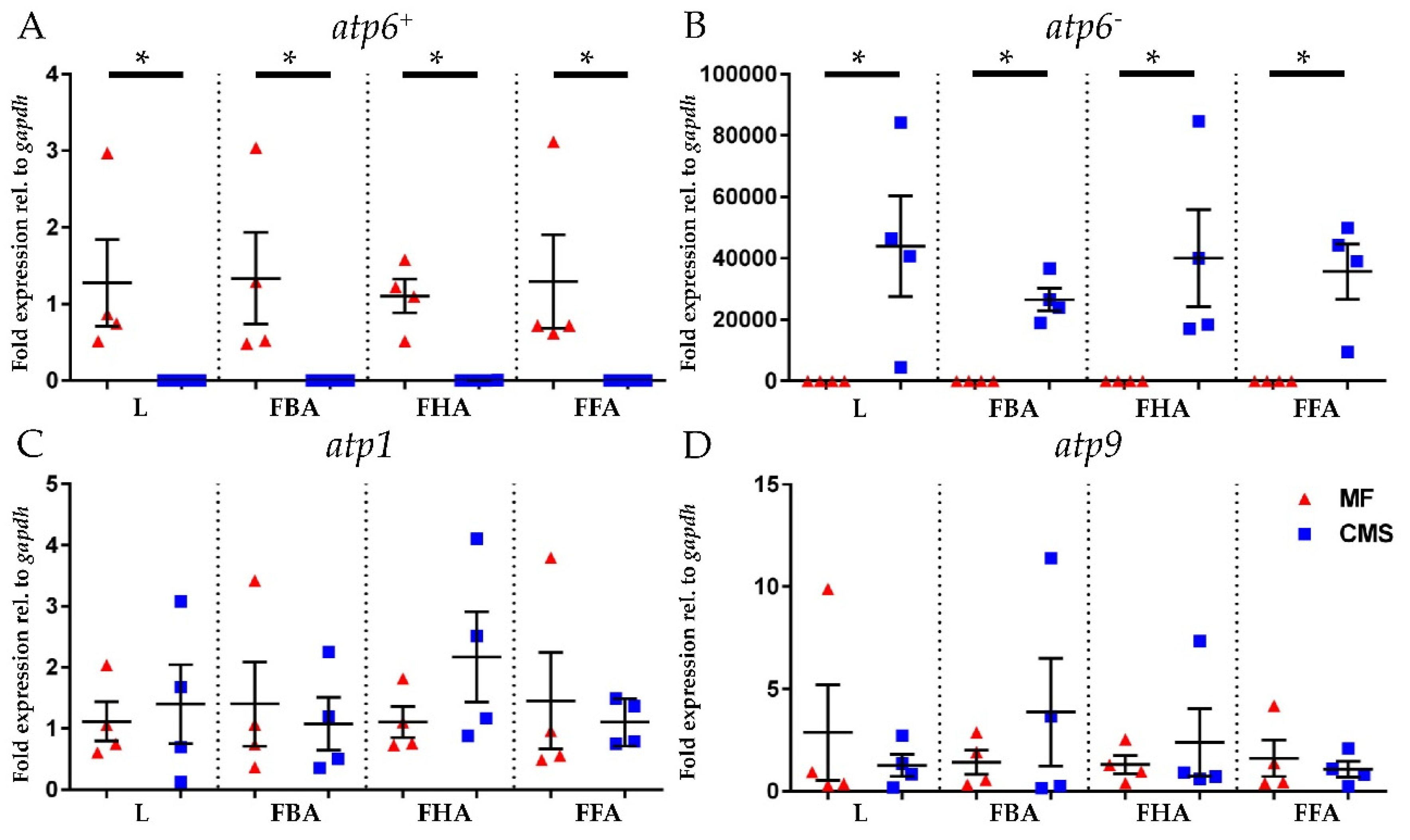
2.4. Expression Analyses of atp6+, atp6−, atp1, and atp9
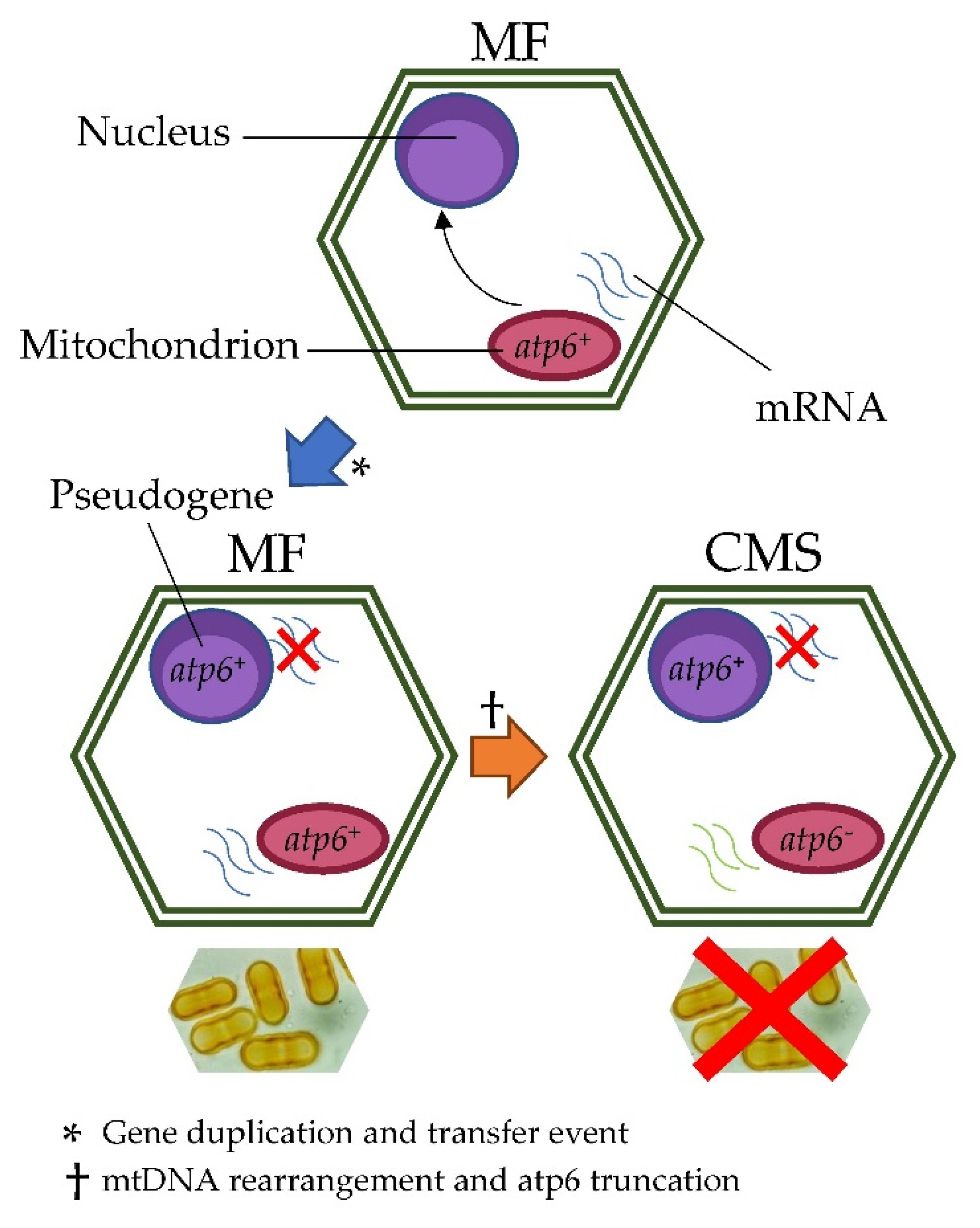
3. Discussion
4. Materials and Methods
4.1. Assembly of Mitochondrial DNA
4.2. Variant Calling and Sanger DNA Sequencing Validation
4.3. RNA Isolation and Quantitative Real-Time PCR
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Liu, Y.-G. Male Sterility and Fertility Restoration in Crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, F.; Galla, G.; Vitulo, N.; Barcaccia, G. First draft genome sequencing of fennel (Foeniculum vulgare Mill.): Identification of simple sequence repeats and their application in marker-assisted breeding. Mol. Breed. 2018, 38, 122. [Google Scholar] [CrossRef]
- Tester, M.; Langridge, P. Breeding Technologies to Increase Crop Production in a Changing World. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Prasannan, A. Hybrid Seeds Market by Crop, Duration, Seed Treatment, Farm Type and Acreage: Global Opportunity Analysis and Industry Forecast 2017-2023; Allied Market Research: Portland, OR, USA, 2017. [Google Scholar]
- Wang, P.; Lu, Q.; Ai, Y.; Wang, Y.; Li, T.; Wu, L.; Liu, J.; Cheng, Q.; Sun, L.; Shen, H. Candidate gene selection for cytoplasmic male sterility in pepper (Capsicum annuum L.) through whole mitochondrial genome sequencing. Int. J. Mol. Sci. 2019, 20, 578. [Google Scholar] [CrossRef] [Green Version]
- Budar, F.; Pelletier, G. Male sterility in plants: Occurrence, determinism, significance and use. Comptes Rendus l’Academie des Sci. Ser. III 2001, 324, 543–550. [Google Scholar] [CrossRef]
- Feng, X.; Kaur, A.P.; MacKenzie, S.A.; Dweikat, I.M. Substoichiometric shifting in the fertility reversion of cytoplasmic male sterile pearl millet. Theor. Appl. Genet. 2009, 118, 1361–1370. [Google Scholar] [CrossRef]
- Priyadarshan, P.M. Male sterility. In Plant breeding: Classical to Modern; Priyadarshan, P.M., Ed.; Springer: Singapore, 2019; pp. 105–129. ISBN 9789811370953. [Google Scholar]
- Kim, B.; Yang, T.J.; Kim, S. Identification of a gene responsible for cytoplasmic male-sterility in onions (Allium cepa L.) using comparative analysis of mitochondrial genome sequences of two recently diverged cytoplasms. Theor. Appl. Genet. 2019, 132, 313–322. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, Y.P.; Lee, J.; Choi, B.S.; Kim, S.; Yang, T.J. Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in radish (Raphanus sativus L.) containing DCGMS cytoplasm. Theor. Appl. Genet. 2013, 126, 1763–1774. [Google Scholar] [CrossRef]
- Nakajima, Y.; Yamamoto, T.; Muranaka, T.; Oeda, K. A novel orfB-related gene of carrot mitochondrial genomes that is associated with homeotic cytoplasmic male sterility (CMS). Plant Mol. Biol. 2001, 46, 99–107. [Google Scholar] [CrossRef]
- Jing, B.; Heng, S.; Tong, D.; Wan, Z.; Fu, T.; Tu, J.; Ma, C.; Yi, B.; Wen, J.; Shen, J. A male sterility-associated cytotoxic protein ORF288 in Brassica juncea causes aborted pollen development. J. Exp. Bot. 2012, 63, 1285–1295. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.P.; Kubo, T.; Mikami, T. The 5′-leader sequence of sugar beet mitochondrial atp6 encodes a novel polypeptide that is characteristic of Owen cytoplasmic male sterility. Mol. Genet. Genomics 2005, 273, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zou, Y.; Li, X.; Zhang, Q.; Chen, L.; Wu, H.; Su, D.; Chen, Y.; Guo, J.; Luo, D.; et al. Cytoplasmic Male Sterility of Rice with Boro II Cytoplasm Is Caused by a Cytotoxic Peptide and is restored by Two Related PPR Motif Genes via Distinct Modes of mRNA Silencing. Plant Cell 2006, 18, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.V.; Pring, D.R.; Shaw, L.C.; Salazar, R.A.; Muza, F.R.; Yan, B.; Schertz, K.F. Transcript processing internal to a mitochondrial open reading frame is correlated with fertility restoration in male-sterile sorghum. Plant J. 1996, 10, 123–133. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine National Center for Biotechnology Information. Nucleotide Database. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/nucleotide/ (accessed on 30 June 2020).
- Liu, C.G.; Hou, N.; Liu, L.K.; Liu, J.C.; Kang, X.S.; Zhang, A.M. A YA-type cytoplasmic male-sterile source in common wheat. Plant Breed. 2006, 125, 437–440. [Google Scholar] [CrossRef]
- Huang, J.Z.; Zhi-Guo, E.; Zhang, H.L.; Shu, Q.Y. Workable male sterility systems for hybrid rice: Genetics, biochemistry, molecular biology, and utilization. Rice 2014, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gabay-Laughnan, S.; Laughnan, J.R. Male sterility and restorer genes in maize. In The Maize Handbook; Freeling, M., Walbot, V., Eds.; Springer-Verlag: New York, NY, USA, 1994; pp. 418–423. [Google Scholar]
- Zhao, N.; Xu, X.; Wamboldt, Y.; Mackenzie, S.A.; Yang, X.; Hu, Z.; Yang, J.; Zhang, M. MutS HOMOLOG1 silencing mediates ORF220 substoichiometric shifting and causes male sterility in Brassica juncea. J. Exp. Bot. 2016, 67, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Heng, S.; Liu, S.; Xia, C.; Tang, H.Y.; Xie, F.; Fu, T.; Wan, Z. Morphological and genetic characterization of a new cytoplasmic male sterility system (oxa CMS) in stem mustard (Brassica juncea). Theor. Appl. Genet. 2018, 131, 59–66. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Hao, W.; Li, J.; Qi, M.; Zhang, L. Mitochondrial Genome Sequencing Reveals orf463a May Induce Male Sterility in NWB Cytoplasm. Genes 2020, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Hans Köhler, R.; Horn, R.; Lössl, A.; Zetsche, K. Cytoplasmic male sterility in sunflower is correlated with the co-transcription of a new open reading frame with the atpA gene. Mol. Gen. Genet. 1991, 227, 369–376. [Google Scholar] [CrossRef]
- Quagliotti, L.; Accati, E.; Milanesio, M. Preliminary report on a functional type of male-sterility in fennel. In Proceedings of the 5th Eucarpia Congress, Milan, Italy, 30 September–2 October 1968; pp. 359–363. [Google Scholar]
- Ramanujam, S.; Tewari, V.P. Heterosis breeding in fennel. Indian J. Genet. Plant Breed. 1970, 30, 732–737. [Google Scholar]
- Bruins, M. Modernising fennel. Available online: https://european-seed.com/2015/10/modernising-fennel/. (accessed on 30 June 2020).
- Food and Agriculture Organization (FAO), FAO Food and Agriculture Organization of the United Nations: Value of Agricultural Production. Available online: http://www.fao.org/faostat/ (accessed on 30 June 2020).
- Peery, R.; Kuehl, J.V.; Boore, J.L.; Raubeson, L. Comparisons of three Apiaceae chloroplast genomes—coriander, dill and fennel. In Proceedings of the Botany 2006 Meeting, Chico, CA, USA, 28 July–2 August 2006. [Google Scholar]
- Palumbo, F.; Vannozzi, A.; Vitulo, N.; Lucchin, M.; Barcaccia, G. The leaf transcriptome of fennel (Foeniculum vulgare Mill.) enables characterization of the t-anethole pathway and the discovery of microsatellites and single-nucleotide variants. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Z.; Zhao, N.; Wang, Y.; Nie, H.; Hua, J. The comparison of four mitochondrial genomes reveals cytoplasmic male sterility candidate genes in cotton. BMC Genomics 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Allen, J.O.; Fauron, C.M.; Minx, P.; Roark, L.; Oddiraju, S.; Guan, N.L.; Meyer, L.; Sun, H.; Kim, K.; Wang, C.; et al. Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics 2007, 177, 1173–1192. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI), Organelle Genome Resources. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genome/organelle/ (accessed on 30 June 2020).
- Wu, Z.; Hu, K.; Yan, M.; Song, L.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; Yi, B.; Tu, J. Mitochondrial genome and transcriptome analysis of five alloplasmic male-sterile lines in Brassica juncea. BMC Genomics 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Ngahang Kamte, S.L.; Ranjbarian, F.; Cianfaglione, K.; Sut, S.; Dall’Acqua, S.; Bruno, M.; Afshar, F.H.; Iannarelli, R.; Benelli, G.; Cappellacci, L.; et al. Identification of highly effective antitrypanosomal compounds in essential oils from the Apiaceae family. Ecotoxicol. Environ. Saf. 2018, 156, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Sayed-Ahmad, B.; Talou, T.; Saad, Z.; Hijazi, A.; Merah, O. The Apiaceae: Ethnomedicinal family as source for industrial uses. Ind. Crops Prod. 2017, 109, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Joh, H.J.; Kim, K.; Lee, S.C.; Kim, N.H.; Park, J.Y.; Park, H.S.; Park, M.S.; Kim, S.; Kwak, M.; et al. Dynamic chloroplast genome rearrangement and DNA barcoding for three apiaceae species known as the medicinal herb “bang-poong”. Int. J. Mol. Sci. 2019, 20, 2196. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.Y.; Chen, Z.X.; Wang, Q.Z. The complete chloroplast genome of Torilis scabra (Apiaceae). Mitochondrial DNA Part B Resour. 2019, 4, 2914–2915. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.F.; Wang, F.; Zhang, X.Y.; Xiong, A.S. Different lengths, copies and expression levels of the mitochondrial atp6 gene in male sterile and fertile lines of carrot (Daucus carota L.). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2018, 29, 446–454. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Eric Schranz, M.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the “master circle” model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P.; Case, A.L.; Floro, E.R.; Willis, J.H. Evidence against equimolarity of large repeat arrangements and a predominant master circle structure of the mitochondrial genome from a monkeyflower (Mimulus guttatus) lineage with cryptic CMS. Genome Biol. Evol. 2012, 4, 670–686. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Li, W.; Gao, C.W.; Zhang, D.; Gao, L.Z. Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci. Data 2019, 6, 209. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Morley, S.A.; Nielsen, B.L. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 1023–1032. [Google Scholar]
- Park, S.; Ruhlman, T.A.; Sabir, J.S.M.; Mutwakil, M.H.Z.; Baeshen, M.N.; Sabir, M.J.; Baeshen, N.A.; Jansen, R.K. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics 2014, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Marienfeld, J.; Unseld, M.; Brandt, P.; Brennicke, A. Genomic recombination of the mitochondrial atp6 gene in Arabidopsis thaliana at the protein processing site creates two different presequences. DNA Res. 1996, 3, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Brandvain, Y.; Wade, M.J. The functional transfer of genes from the mitochondria to the nucleus: The effects of selection, mutation, population size and rate of self-fertilization. Genetics 2009, 182, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Funes, S.; Davidson, E.; Gonzalo Claros, M.; Van Lis, R.; Pérez-Martínez, X.; Vázquez-Acevedo, M.; King, M.P.; González-Halphen, D. The typically mitochondrial DNA-encoded ATP6 subunit of the F1F0-ATPase is encoded by a nuclear gene in Chlamydomonas reinhardtii. J. Biol. Chem. 2002, 277, 6051–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, A.B.; Citarella, M.R.; Kocot, K.M.; Bobkova, Y.V.; Halanych, K.M.; Moroz, L.L. Rapid evolution of the compact and unusual mitochondrial genome in the ctenophore, Pleurobrachia bachei. Mol. Phylogenet. Evol. 2012, 63, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itabashi, E.; Kazama, T.; Toriyama, K. Characterization of cytoplasmic male sterility of rice with Lead Rice cytoplasm in comparison with that with Chinsurah Boro II cytoplasm. Plant Cell Rep. 2009, 28, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Dewey, R.E.; Timothy, D.H.; Levings, C.S. Chimeric mitochondrial genes expressed in the C male-sterile cytoplasm of maize. Curr. Genet. 1991, 20, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liao, X.; Zhou, B.; Zhao, H.; Zhou, Y.; Zhou, R. Mutation in the coding sequence of atp6 are associated with male sterile cytoplasm in kenaf (Hibiscus cannabinus L.). Euphytica 2016, 207, 169–175. [Google Scholar] [CrossRef]
- Makarenko, M.S.; Usatov, A.V.; Tatarinova, T.V.; Azarin, K.V.; Logacheva, M.D.; Gavrilova, V.A.; Kornienko, I.V.; Horn, R. Organization features of the mitochondrial genome of sunflower (Helianthus annuus L.) with ANN2-type male-sterile cytoplasm. Plants 2019, 8, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducos, E.; Touzet, P.; Boutry, M. The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001, 26, 171–180. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Yang, Z.; Yi, B.; Wen, J.; Shen, J.; Tu, J.; Ma, C.; Fu, T. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genomics 2014, 15, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Sabar, M.; Gagliardi, D.; Balk, J.; Leaver, C.J. ORFB is a subunit of F1F0-ATP synthase: Insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 2003, 4, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. bioRxiv 2019, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CMS mtDNA Position | Type | Ref | Alt | ID | CMS mtDNA Position | Type | Ref | Alt | ID |
|---|---|---|---|---|---|---|---|---|---|
| 4407^4408 | Ins | - | T | 210132 | SNV | A | G | atp6 | |
| 4413^4414 | Ins | - | T | 210134..210137 | MNV | ACCC | GGAA | atp6 | |
| 9932 | Del | A | - | 210140..210141 | MNV | CC | TT | atp6 | |
| 9937 | Del | A | - | 210143 | SNV | A | G | atp6 | |
| 17054 | SNV | T | A | 210145 | SNV | C | T | atp6 | |
| 17779 | SNV | A | C | 210148..210150 | MNV | CGA | TAG | atp6 | |
| 19036 | Del | T | - | 210152..210153 | MNV | TA | GG | atp6 | |
| 23779..23780 | Del | GG | - | nad4 (intron) | 210155 | SNV | T | G | atp6 |
| 26757^26758 | Ins | - | G | nad4 (intron) | 210157..210158 | MNV | CT | AG | atp6 |
| 29433 | Del | G | - | nad4 (intron) | 210160..210163 | MNV | ATAT | CCCC | atp6 |
| 38972^38973 | Ins | - | A | 210168..210171 | MNV | GCTT | ATCC | atp6 | |
| 85033 | Del | A | - | 210176 | SNV | T | C | atp6 | |
| 97184 | Del | A | - | 210179 | SNV | T | A | atp6 | |
| 97194 | Del | C | - | 210193..210194 | MNV | AA | CC | atp6 | |
| 103048 | Del | T | - | 210218 | SNV | G | T | atp6 | |
| 106177^106178 | Ins | - | T | 210225 | SNV | G | C | atp6 | |
| 109983 | Del | G | - | 210276 | SNV | G | A | atp6 | |
| 110419 | SNV | T | G | 210291 | SNV | T | C | atp6 | |
| 175249 | SNV | T | C | rps3 | 210293 | SNV | G | A | atp6 |
| 175458 | SNV | C | G | rps3 | 210296 | SNV | T | G | atp6 |
| 176686 | SNV | G | A | rps3 | 210374 | SNV | C | T | atp6 |
| 181657 | Del | T | - | cox2 (intron) | 212667 | Del | T | - | |
| 210097..210102 | Del | CCGCTC | - | atp6 | 213187 | SNV | A | G | |
| 210103..210105 | MNV | CCA | TTT | atp6 | 213216 | SNV | A | C | |
| 210111 | SNV | C | T | atp6 | 238616^238617 | Ins | - | C | |
| 210113 | SNV | G | A | atp6 | 238643^238644 | Ins | - | G | |
| 210117 | SNV | A | G | atp6 | 240379^240380 | Ins | - | TTTATTAT | orf43 |
| 210117^210118 | Ins | - | AG | atp6 | 247260 | Del | C | - | |
| 210122 | SNV | A | G | atp6 | 248437 | Del | A | - | |
| 210124..210126 | MNV | TAA | GTG | atp6 | 293347 | Del | T | - | nad2 (intron) |
| 210129^210130 | Ins | - | CTT | atp6 | 295317 | Del | T | - | nad2 (intron) |
| 210130 | SNV | A | T | atp6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, F.; Vitulo, N.; Vannozzi, A.; Magon, G.; Barcaccia, G. The Mitochondrial Genome Assembly of Fennel (Foeniculum vulgare) Reveals Two Different atp6 Gene Sequences in Cytoplasmic Male Sterile Accessions. Int. J. Mol. Sci. 2020, 21, 4664. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134664
Palumbo F, Vitulo N, Vannozzi A, Magon G, Barcaccia G. The Mitochondrial Genome Assembly of Fennel (Foeniculum vulgare) Reveals Two Different atp6 Gene Sequences in Cytoplasmic Male Sterile Accessions. International Journal of Molecular Sciences. 2020; 21(13):4664. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134664
Chicago/Turabian StylePalumbo, Fabio, Nicola Vitulo, Alessandro Vannozzi, Gabriele Magon, and Gianni Barcaccia. 2020. "The Mitochondrial Genome Assembly of Fennel (Foeniculum vulgare) Reveals Two Different atp6 Gene Sequences in Cytoplasmic Male Sterile Accessions" International Journal of Molecular Sciences 21, no. 13: 4664. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134664