78 kDa Glucose-Regulated Protein Attenuates Protein Aggregation and Monocyte Adhesion Induced by Angiotensin II in Vascular Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
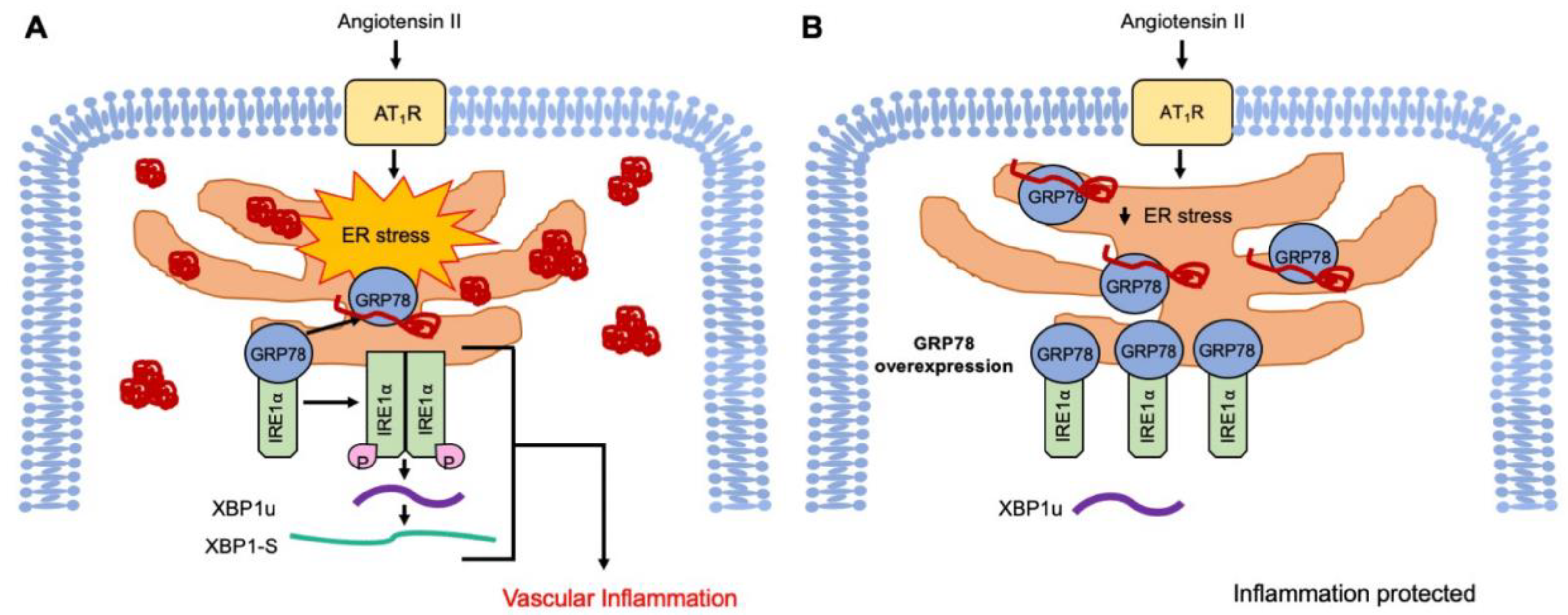
Abstract
:1. Introduction
2. Results
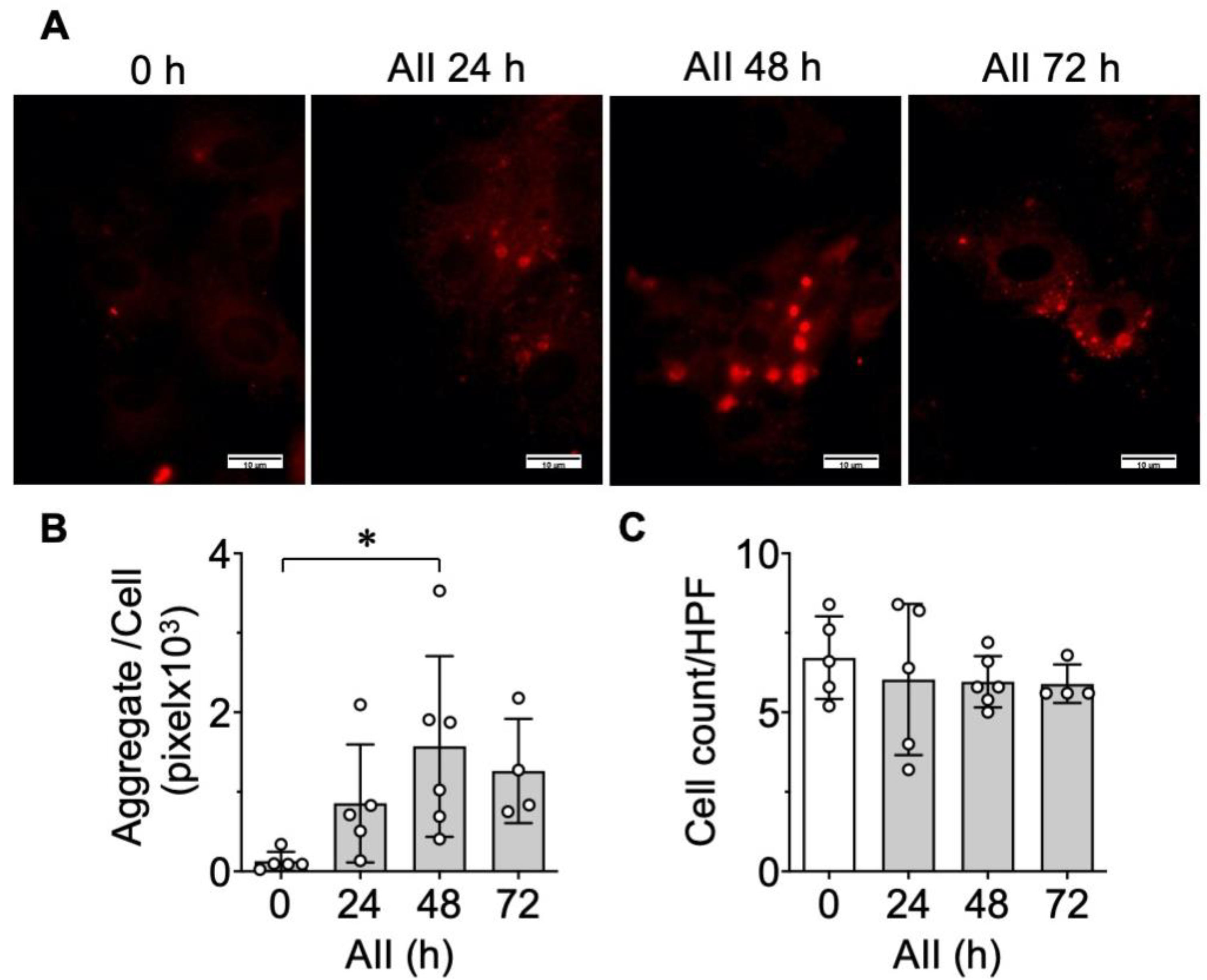
2.1. Protein Aggregate Accumulation Is Induced by AngII
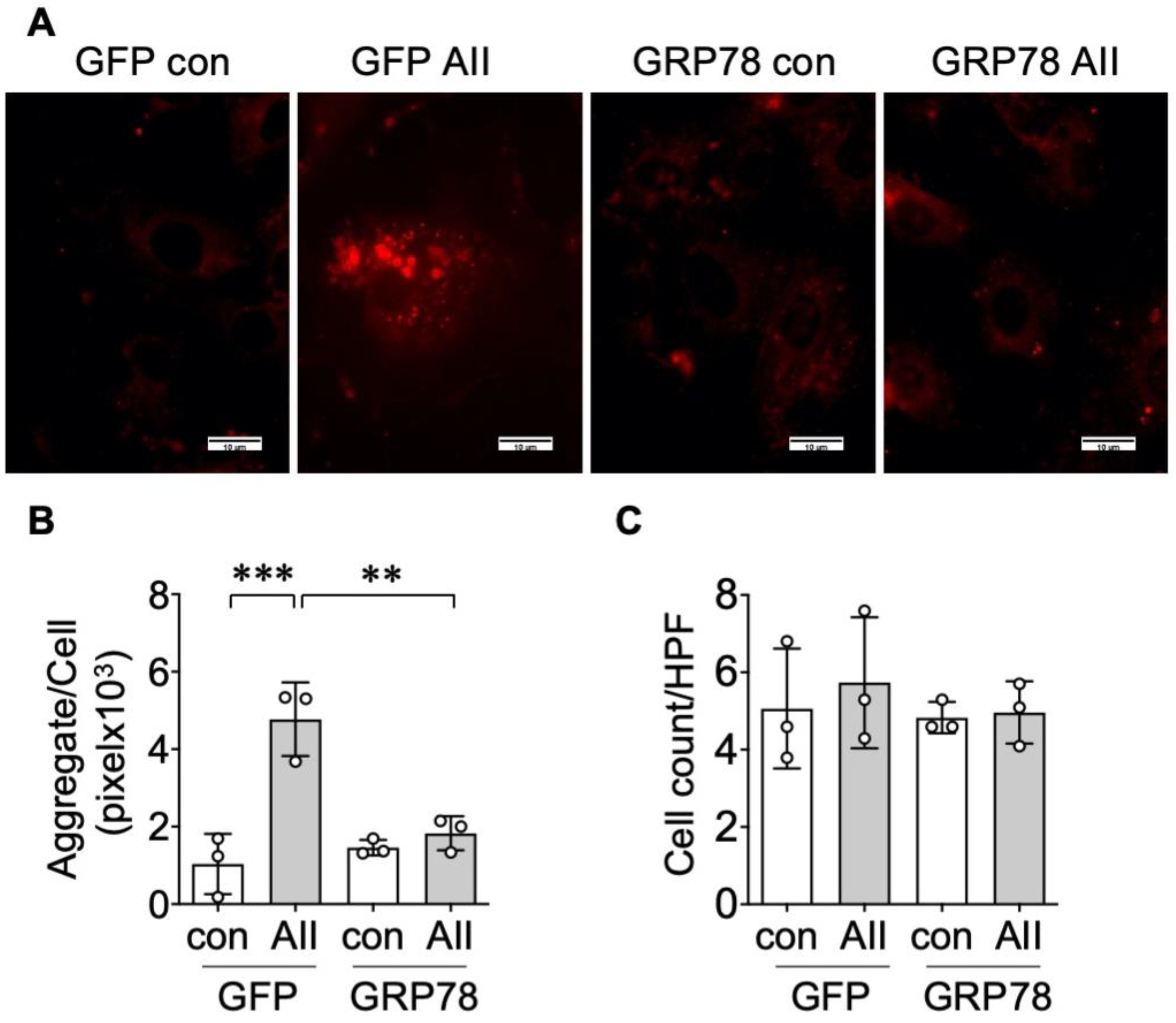
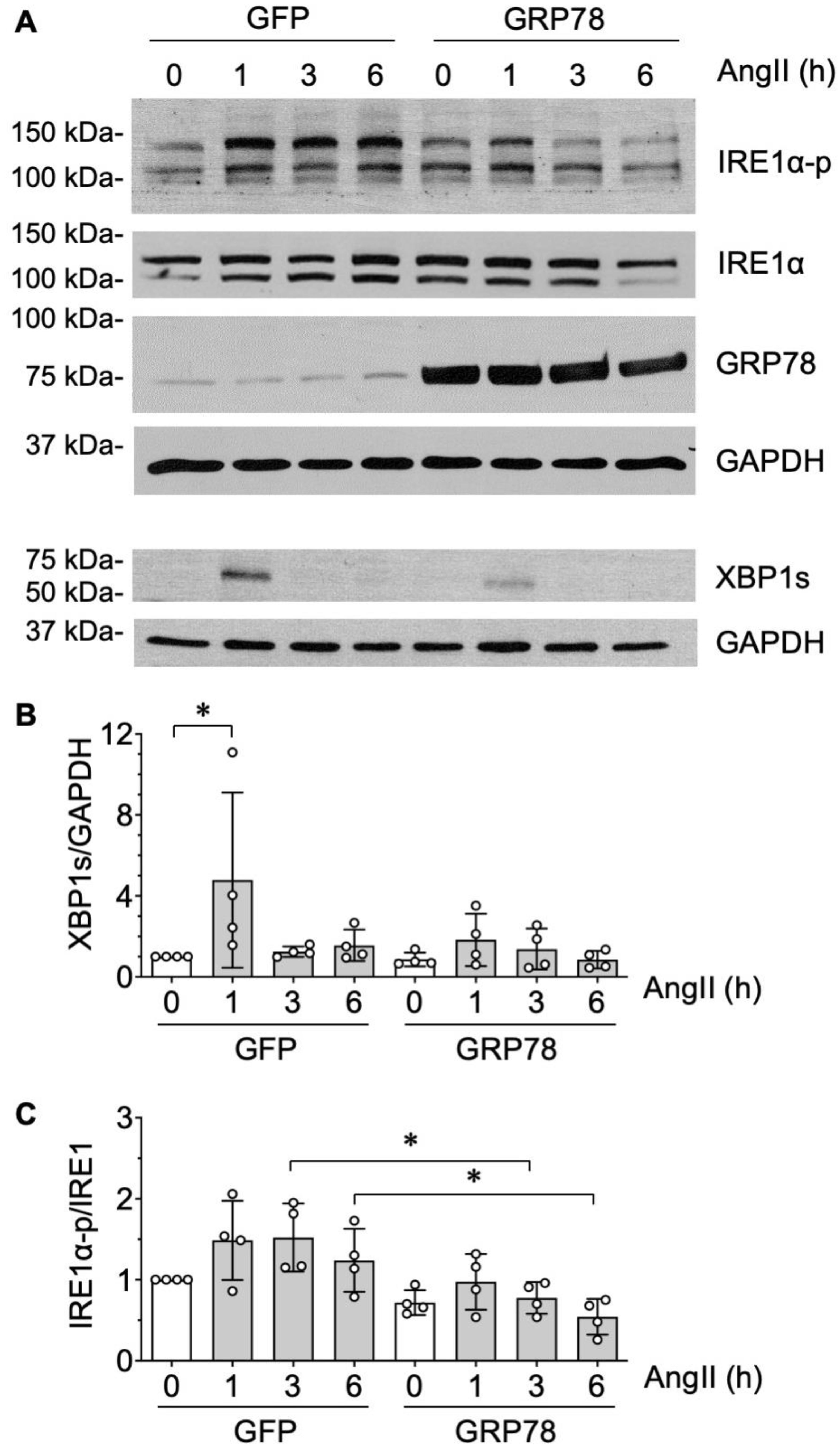
2.2. GRP78 Chaperoning Reduces AngII-Induced Protein Aggregation and UPR in VSMCs
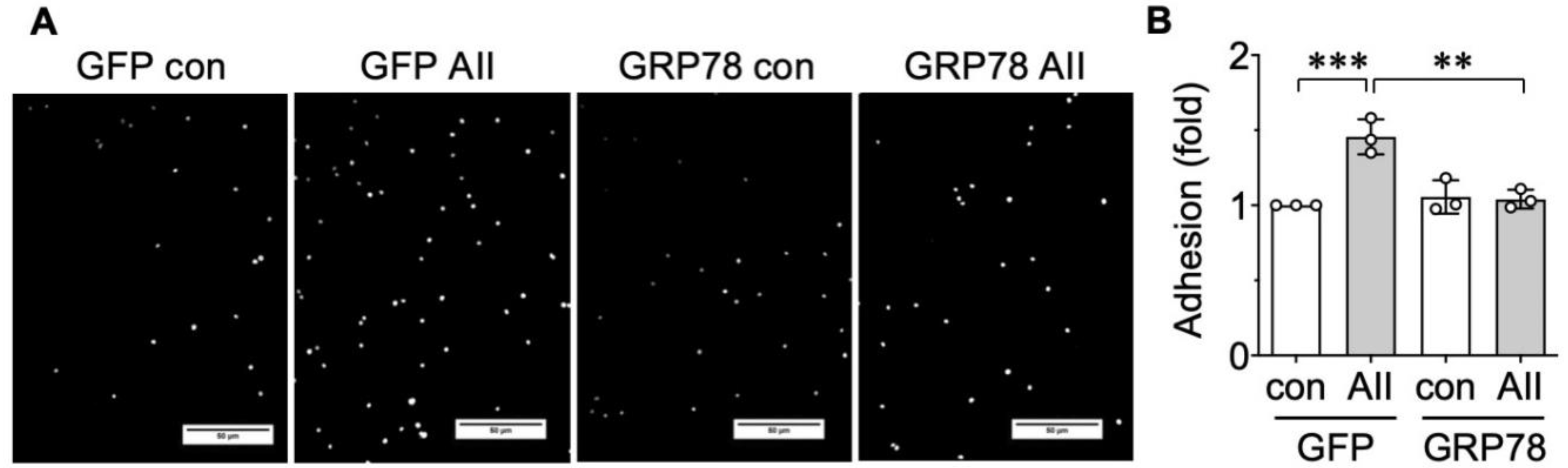
2.3. AngII-Induced Proinflammatory Phenotype Is Mitigated by GRP78
3. Discussion
4. Materials and Methods
4.1. Culture of Rat Aortic VSMCs and Adenoviral Transduction
4.2. Proteostat Immunofluorescent Staining
4.3. Immunoblotting
4.4. Monocyte Adhesion Assay
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AngII | Angiotensin II |
| ATF | Activating transcription factor |
| BSA | Bovine serum albumin |
| CVD | Cardiovascular diseases |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DMSO | Dimethyl sulfoxide |
| ECs | Endothelial cells |
| ER | Endoplasmic reticulum |
| FBS | Fetal bovine serum |
| GFP | Green fluorescent protein |
| GRP78 | 78-kDa glucose-regulated protein |
| HPF | High power field |
| IRE1α | Inositol-requiring enzyme 1 α |
| moi | Multiplicity of infection |
| PBS | Phosphate buffered saline |
| PERK | Protein kinase R-like ER kinase |
| SD | Standard deviation |
| TBS | Tris buffered saline |
| UPR | Unfolded protein response |
| VSMCs | Vascular smooth muscle cells |
| XBP1s | X-box-binding-protein 1 spliced isoform |
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension 2018, 72, 537–548. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Hong, Z.; Reeves, K.J.; Sun, Z.; Li, Z.; Brown, N.J.; Meininger, G.A. Vascular smooth muscle cell stiffness and adhesion to collagen I modified by vasoactive agonists. PLoS ONE 2015, 10, e0119533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Forrester, S.J.; O’Brien, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125 Pt A, 4–13. [Google Scholar] [CrossRef]
- Cooper, H.A.; Scalia, R.; Rizzo, V.; Eguchi, S. Angiotensin II- and alzheimer-type cardiovascular aging. Circ. Res. 2018, 123, 651–653. [Google Scholar] [CrossRef]
- Spitler, K.M.; Webb, R.C. Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms. Hypertension 2014, 63, e40–e45. [Google Scholar] [CrossRef] [Green Version]
- Camargo, L.L.; Harvey, A.P.; Rios, F.J.; Tsiropoulou, S.; Da Silva, R.N.O.; Cao, Z.; Graham, D.; McMaster, C.; Burchmore, R.J.; Hartley, R.C.; et al. Vascular Nox (NADPH Oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension 2018, 72, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, R.E.; Werner, K.E.; Yum, V.; Lu, C.; Tat, V.; Memon, M.; No, Y.; Ask, K.; Dickhout, J.G. Endoplasmic reticulum stress inhibition reduces hypertension through the preservation of resistance blood vessel structure and function. J. Hypertens. 2016, 34, 1556–1569. [Google Scholar] [CrossRef]
- Takayanagi, T.; Kawai, T.; Forrester, S.J.; Obama, T.; Tsuji, T.; Fukuda, Y.; Elliott, K.J.; Tilley, D.G.; Davisson, R.L.; Park, J.Y.; et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 2015, 65, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 2015, 16, 7193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassan, M.; Galan, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.M.H.; Lau, Y.S.; Miller, A.A.; Ku, J.M.; Potocnik, S.; Ye, J.M.; Woodman, O.L.; Herbert, T.P. Angiotensin II causes beta-cell dysfunction through an er stress-induced proinflammatory response. Endocrinology 2017, 158, 3162–3173. [Google Scholar] [CrossRef] [Green Version]
- Menikdiwela, K.R.; Ramalingam, L.; Allen, L.; Scoggin, S.; Kalupahana, N.S.; Moustaid-Moussa, N. Angiotensin II increases endoplasmic reticulum stress in adipose tissue and adipocytes. Sci. Rep. 2019, 9, 8481. [Google Scholar] [CrossRef] [Green Version]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity, and cardiac dysfunction: Therapeutic and molecular approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Wang, X.; Gillette, T.G.; Deng, Y.; Wang, Z.V. Unfolded protein response as a therapeutic target in cardiovascular disease. Curr. Top. Med. Chem. 2019, 19, 1902–1917. [Google Scholar] [CrossRef]
- Jiang, Y.; Lv, H.; Liao, M.; Xu, X.; Huang, S.; Tan, H.; Peng, T.; Zhang, Y.; Li, H. GRP78 counteracts cell death and protein aggregation caused by mutant huntingtin proteins. Neurosci. Lett. 2012, 516, 182–187. [Google Scholar] [CrossRef]
- Park, K.W.; Eun Kim, G.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The endoplasmic reticulum chaperone grp78/bip modulates prion propagation in vitro and in vivo. Sci. Rep. 2017, 7, 44723. [Google Scholar] [CrossRef] [Green Version]
- Giri, R.; Shen, Y.; Stins, M.; Du Yan, S.; Schmidt, A.M.; Stern, D.; Kim, K.S.; Zlokovic, B.; Kalra, V.K. Beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am. J. Physiol. Cell Physiol. 2000, 279, C1772–C1781. [Google Scholar] [CrossRef] [PubMed]
- Driggin, E.; Helmke, S.; De Los Santos, J.; Teruya, S.; Guadalupe, S.; Goldsmith, J.; Maurer, M.S. Markers of nutritional status and inflammation in transthyretin cardiac amyloidosis: Association with outcomes and the clinical phenotype. Amyloid 2020, 27, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.; Zaccagnini, G.; Fuschi, P.; Voellenkle, C.; Carrara, M.; Sadeghi, I.; Bearzi, C.; Maimone, B.; Castelvecchio, S.; Stellos, K.; et al. Increased BACE1-AS long noncoding RNA and beta-amyloid levels in heart failure. Cardiovasc. Res. 2017, 113, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Mercanti, F.; Wang, X.; Mackintosh, S.G.; Tackett, A.J.; Prayaga, S.V.; Romeo, F.; Shmookler Reis, R.J.; Mehta, J.L. Age-and hypertension-associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension 2016, 67, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [Green Version]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.; May, H.I.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.; et al. Endoplasmic reticulum chaperone grp78 protects heart from ischemia/reperfusion injury through akt activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tatebe, H.; Taguchi, K.; Endo, Y.; Tokuda, T.; Mizuno, T.; Nakagawa, M.; Tanaka, M. p62/SQSTM1-dependent autophagy of Lewy body-like alpha-synuclein inclusions. PLoS ONE 2012, 7, e52868. [Google Scholar] [CrossRef]
- Gonzalez-Teuber, V.; Albert-Gasco, H.; Auyeung, V.C.; Papa, F.R.; Mallucci, G.R.; Hetz, C. Small molecules to improve er proteostasis in disease. Trends Pharmacol. Sci. 2019, 40, 684–695. [Google Scholar] [CrossRef]
- Jwa, M.; Chang, P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK-and IRE1alpha-mediated unfolded protein response. Nat. Cell. Biol. 2012, 14, 1223–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chavez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Conza, G.; Ho, P.C. Er stress responses: An emerging modulator for innate immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, H.A.; Cicalese, S.; Preston, K.J.; Kawai, T.; Okuno, K.; Choi, E.T.; Kasahara, S.; Uchida, H.A.; Otaka, N.; Scalia, R.; et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Spitler, K.M.; Matsumoto, T.; Webb, R.C. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H344–H353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, C.G.; Wenceslau, C.F.; Webb, R.C.; Joe, B. Novel Contributors and mechanisms of cellular senescence in hypertension-associated premature vascular aging. Am. J. Hypertens. 2019, 32, 709–719. [Google Scholar] [CrossRef]
- Yeager, M.E.; Belchenko, D.D.; Nguyen, C.M.; Colvin, K.L.; Ivy, D.D.; Stenmark, K.R. Endothelin-1, the unfolded protein response, and persistent inflammation: Role of pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 14–22. [Google Scholar] [CrossRef]
- Kawanami, D.; Matoba, K.; Okada, R.; Tsukamoto, M.; Kinoshita, J.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Fasudil inhibits ER stress-induced VCAM-1 expression by modulating unfolded protein response in endothelial cells. Biochem. Biophys. Res. Commun. 2013, 435, 171–175. [Google Scholar] [CrossRef]
- Miyao, M.; Cicalese, S.; Kawai, T.; Cooper, H.A.; Boyer, M.J.; Elliott, K.J.; Forrester, S.J.; Kuroda, R.; Rizzo, V.; Hashimoto, T.; et al. Involvement of senescence and mitochondrial fission in endothelial cell pro-inflammatory phenotype induced by angiotensin ii. Int. J. Mol. Sci. 2020, 21, 3112. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, X.; Shen, B.; Ma, J.; Zhao, W. Is vascular amyloidosis intertwined with arterial aging, hypertension and atherosclerosis? Front. Genet. 2017, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robblee, M.M.; Kim, C.C.; Porter Abate, J.; Valdearcos, M.; Sandlund, K.L.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated fatty acids engage an ire1alpha-dependent pathway to activate the nlrp3 inflammasome in myeloid cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, S.; Hirata, Y.; Imai, T.; Kanno, K.; Marumo, F. Phenotypic change of endothelin receptor subtype in cultured rat vascular smooth muscle cells. Endocrinology 1994, 134, 222–228. [Google Scholar] [CrossRef]
- Elliott, K.J.; Eguchi, S. In vitro analysis of hypertensive signal transduction: Kinase activation, kinase manipulation, and physiologic outputs. Methods Mol. Biol. 2017, 1527, 201–211. [Google Scholar] [CrossRef]
- Takayanagi, T.; Bourne, A.M.; Kimura, K.; Takaguri, A.; Elliott, K.J.; Eguchi, K.; Eguchi, S. Constitutive stimulation of vascular smooth muscle cells by angiotensin II derived from an adenovirus encoding a furin-cleavable fusion protein. Am. J. Hypertens. 2012, 25, 280–283. [Google Scholar] [CrossRef] [Green Version]
- Mokhashi, N.; Choi, R.Y.; Cicalese, S.; Eguchi, K.; Boyer, M.J.; Cooper, H.A.; Kimura, Y.; Akiyama, T.; Scalia, R.; Rizzo, V.; et al. Transduction efficiency of adenovirus vectors in endothelial cells and vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2020, 75, 603–607. [Google Scholar] [CrossRef]
- Oshinbolu, S.; Shah, R.; Finka, G.; Molloy, M.; Uden, M.; Bracewell, D.G. Evaluation of fluorescent dyes to measure protein aggregation within mammalian cell culture supernatants. J. Chem. Technol. Biotechnol. 2018, 93, 909–917. [Google Scholar] [CrossRef]
- Eguchi, S.; Numaguchi, K.; Iwasaki, H.; Matsumoto, T.; Yamakawa, T.; Utsunomiya, H.; Motley, E.D.; Kawakatsu, H.; Owada, K.M.; Hirata, Y.; et al. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J. Biol. Chem. 1998, 273, 8890–8896. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicalese, S.; Okuno, K.; Elliott, K.J.; Kawai, T.; Scalia, R.; Rizzo, V.; Eguchi, S. 78 kDa Glucose-Regulated Protein Attenuates Protein Aggregation and Monocyte Adhesion Induced by Angiotensin II in Vascular Cells. Int. J. Mol. Sci. 2020, 21, 4980. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144980
Cicalese S, Okuno K, Elliott KJ, Kawai T, Scalia R, Rizzo V, Eguchi S. 78 kDa Glucose-Regulated Protein Attenuates Protein Aggregation and Monocyte Adhesion Induced by Angiotensin II in Vascular Cells. International Journal of Molecular Sciences. 2020; 21(14):4980. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144980
Chicago/Turabian StyleCicalese, Stephanie, Keisuke Okuno, Katherine J. Elliott, Tatsuo Kawai, Rosario Scalia, Victor Rizzo, and Satoru Eguchi. 2020. "78 kDa Glucose-Regulated Protein Attenuates Protein Aggregation and Monocyte Adhesion Induced by Angiotensin II in Vascular Cells" International Journal of Molecular Sciences 21, no. 14: 4980. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144980