Discrimination between G/C Binding Sites by Olivomycin A Is Determined by Kinetics of the Drug-DNA Interaction

 , ,
, ,
Abstract
:1. Introduction
2. Results
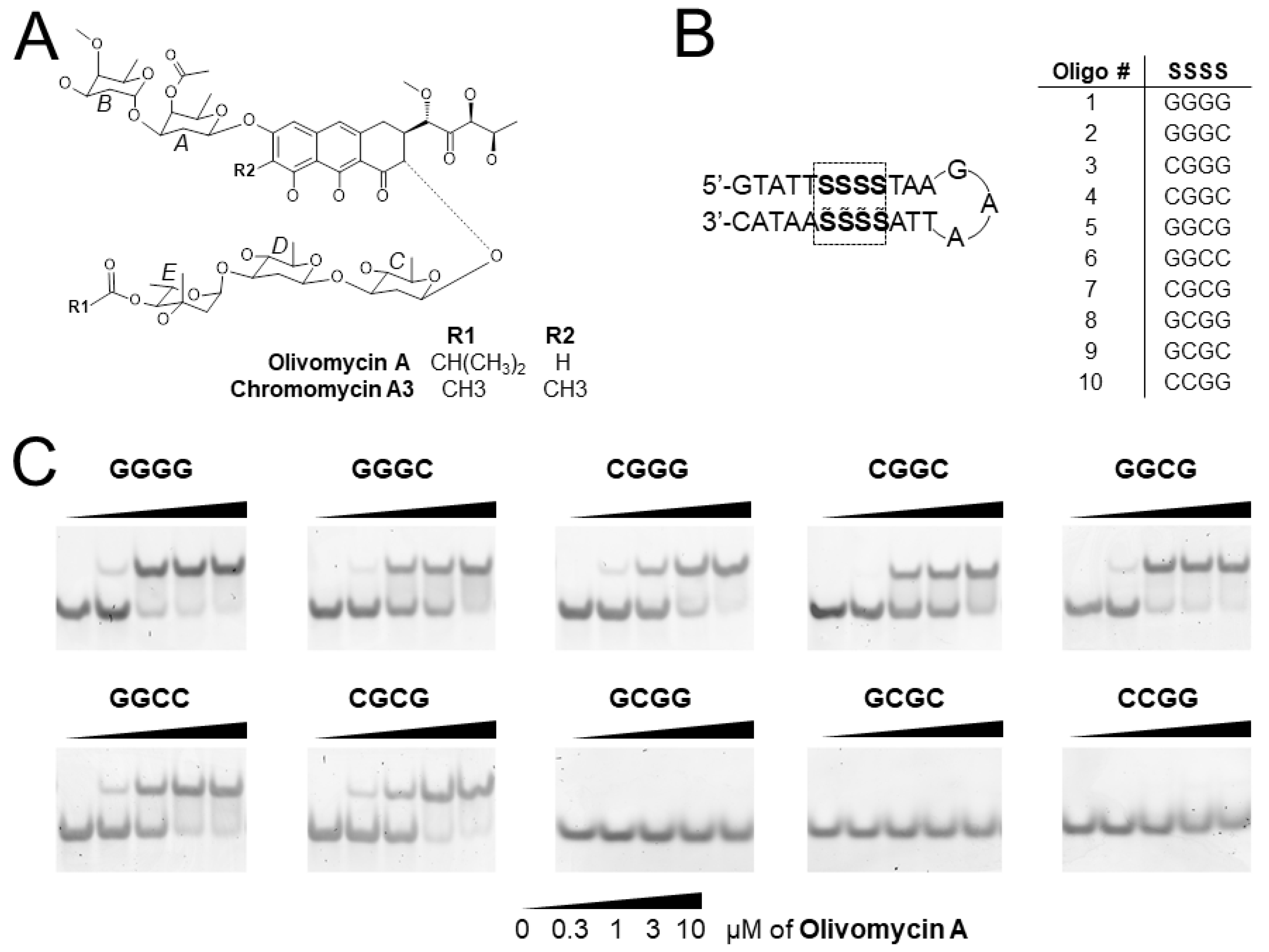
2.1. Screening of G/C Tetranucleotide Sites for OA Binding by EMSA
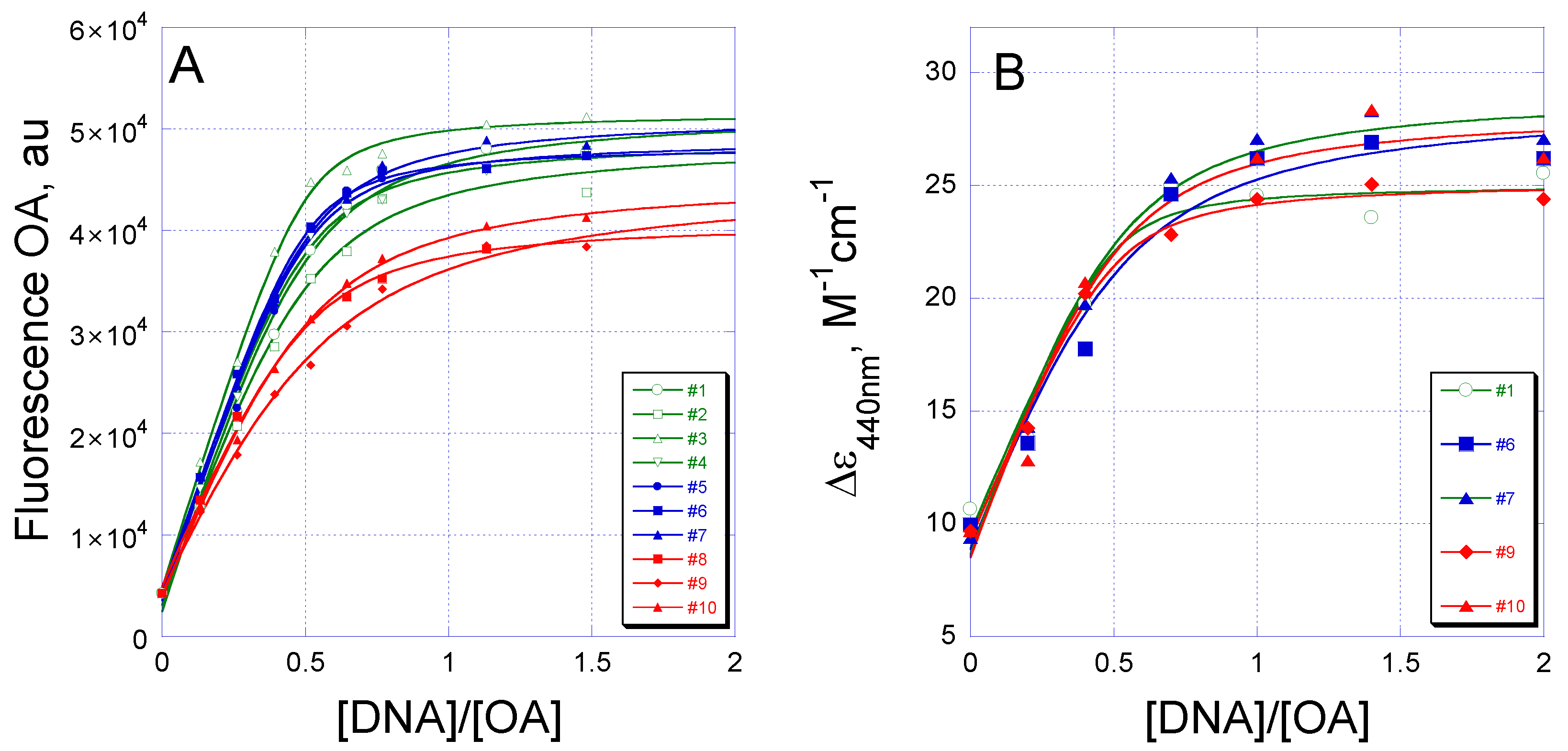
2.2. Equilibrium Binding of OA to G/C Sites
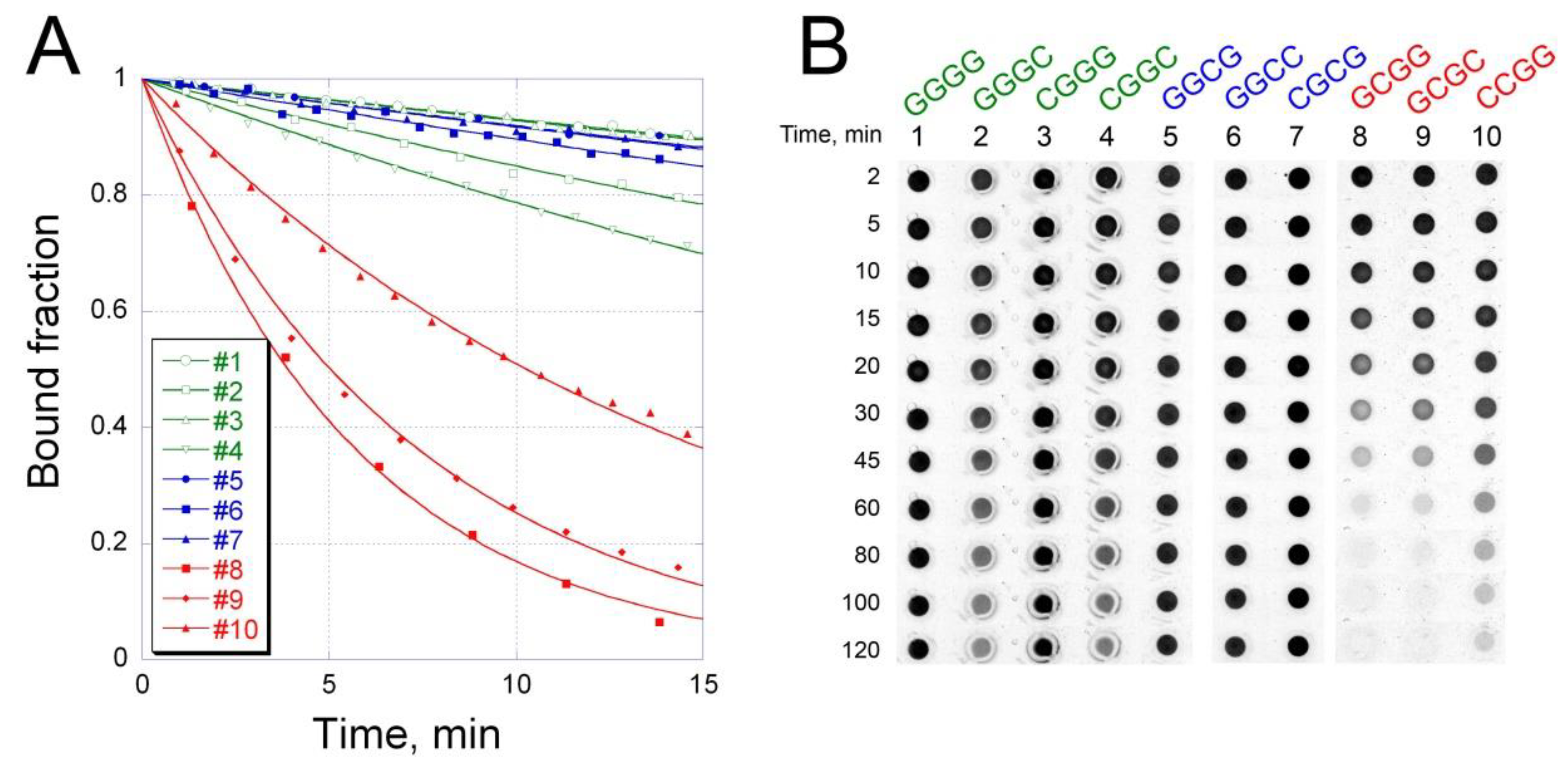
2.3. Kinetics of OA Interaction with Different G/C Sites
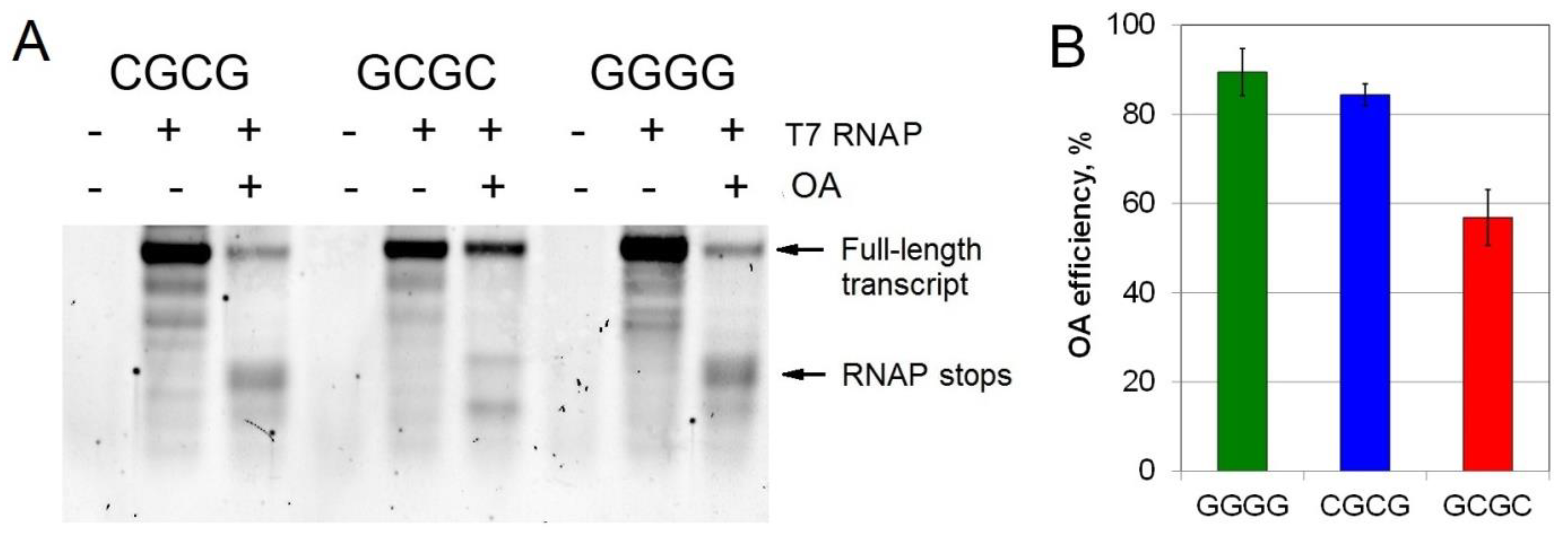
2.4. Inhibition of T7 Transcription by OA at Different G/C Sites
3. Discussion
4. Materials and Methods
4.1. Chemicals and DNA Oligonucleotides
4.2. Electrophoretic Mobility Shift Assays (EMSA)
4.3. Fluorescence
4.4. CD Spectroscopy
4.5. ITC
4.6. Kinetics of OA-DNA Complex Formation and Dissociation
4.7. Diffusion in PAAG
4.8. T7 RNA Polymerase Mediated Transcription
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lombo, F.; Menendez, N.; Salas, J.A.; Mendez, C. The aureolic acid family of antitumor compounds: Structure, mode of action, biosynthesis, and novel derivatives. Appl. Microbiol. Biotechnol. 2006, 73, 1–14. [Google Scholar] [CrossRef]
- Chakraborty, H.; Devi, P.G.; Sarkar, M.; Dasgupta, D. Multiple functions of generic drugs: Future perspectives of aureolic acid group of anti-cancer antibiotics and non-steroidal anti-inflammatory drugs. Mini Rev. Med. Chem. 2008, 8, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chen, Q.; Wang, Y.; Zhang, N.; Meng, P.; Liu, T.; Bu, Y. Sp1 Mediates the Constitutive Expression and Repression of the PDSS2 Gene in Lung Cancer Cells. Genes 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, P.; Eckenrode, J.M.; Mandal, A.; Jha, A.K.; Salem, S.M.; Leggas, M.; Rohr, J. Development of Mithramycin Analogues with Increased Selectivity toward ETS Transcription Factor Expressing Cancers. J. Med. Chem. 2018, 61, 8001–8016. [Google Scholar] [CrossRef] [PubMed]
- Gomez, K.; Sandoval, A.; Barragan-Iglesias, P.; Granados-Soto, V.; Delgado-Lezama, R.; Felix, R.; Gonzalez-Ramirez, R. Transcription Factor Sp1 Regulates the Expression of Calcium Channel alpha2delta-1 Subunit in Neuropathic Pain. Neuroscience 2019, 412, 207–215. [Google Scholar] [CrossRef]
- Ming, L.J. Structure and function of “metalloantibiotics”. Med. Res. Rev. 2003, 23, 697–762. [Google Scholar] [CrossRef]
- Simonova, V.S.; Samusenko, A.V.; Filippova, N.A.; Tevyashova, A.N.; Lyniv, L.S.; Kulik, G.I.; Chekhun, V.F.; Shtil, A.A. Olivomycin induces tumor cell apoptosis and suppresses p53-induced transcription. Bull. Exp. Biol. Med. 2005, 139, 455–459. [Google Scholar] [CrossRef]
- Sergeev, A.V.; Tevyashova, A.N.; Vorobyov, A.P.; Gromova, E.S. The Effect of Antitumor Antibiotic Olivomycin A and Its New Semi-synthetic Derivative Olivamide on the Activity of Murine DNA Methyltransferase Dnmt3a. Biochem. Biokhimiia 2019, 84, 62–70. [Google Scholar] [CrossRef]
- Aich, P.; Sen, R.; Dasgupta, D. Role of magnesium ion in the interaction between chromomycin A3 and DNA: Binding of chromomycin A3-Mg2+ complexes with DNA. Biochemistry 1992, 31, 2988–2997. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Bhattacharyya, D.; Dasgupta, D. Structural basis of DNA recognition by anticancer antibiotics, chromomycin A(3), and mithramycin: Roles of minor groove width and ligand flexibility. Biopolymers 2000, 56, 85–95. [Google Scholar] [CrossRef]
- Fox, K.R.; Howarth, N.R. Investigations into the sequence-selective binding of mithramycin and related ligands to DNA. Nucleic Acids Res. 1985, 13, 8695–8714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, M.H.; Robinson, H.; Gao, Y.G.; Wang, A.H. Crystal structure of the [Mg2+-(chromomycin A3)2]-d(TTGGCCAA)2 complex reveals GGCC binding specificity of the drug dimer chelated by a metal ion. Nucleic Acids Res. 2004, 32, 2214–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.L.; Mirau, P.; Patel, D.J. Structure refinement of the chromomycin dimer-DNA oligomer complex in solution. J. Mol. Biol. 1992, 223, 259–279. [Google Scholar] [CrossRef]
- Hou, M.H.; Lu, W.J.; Lin, H.Y.; Yuann, J.M. Studies of sequence-specific DNA binding, DNA cleavage, and topoisomerase I inhibition by the dimeric chromomycin A3 complexed with Fe(II). Biochemistry 2008, 47, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, F.M. Oligonucleotide studies of sequence-specific binding of chromomycin A3 to DNA. Biochemistry 1994, 33, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.L.; Marks, J.N.; Fox, K.R. DNA-sequence binding preference of the GC-selective ligand mithramycin. Deoxyribonuclease-I/deoxyribonuclease-II and hydroxy-radical footprinting at CCCG, CCGC, CGGC, GCCC and GGGG flanked by (AT)n and An.Tn. Eur. J. Biochem. 1993, 215, 561–566. [Google Scholar] [CrossRef]
- Hampshire, A.J.; Fox, K.R. The effects of local DNA sequence on the interaction of ligands with their preferred binding sites. Biochimie 2008, 90, 988–998. [Google Scholar] [CrossRef]
- Fletcher, M.C.; Fox, K.R. Visualising the dissociation of sequence selective ligands from individual binding sites on DNA. Febs Lett. 1996, 380, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, S.; Kawai, G.; Watanabe, K.; Miura, K.; Hirao, I. GNA trinucleotide loop sequences producing extraordinarily stable DNA minihairpins. Biochemistry 1997, 36, 4761–4767. [Google Scholar] [CrossRef]
- Crothers, D.M. Kinetics of Binding of Drugs to DNA. In Proceedings of the Research Symposium on Complexes of Biologically Active Substances with Nucleic Acids and Their Modes of Action: Held at the Walter Reed Army Institute of Research Washington, 16–19 March 1970; Hahn, F.E., Ed.; Springer: Berlin/Heidelberg, Germany, 1971; pp. 10–20. [Google Scholar] [CrossRef]
- Muller, W.; Crothers, D.M. Studies of the binding of actinomycin and related compounds to DNA. J. Mol. Biol. 1968, 35, 251–290. [Google Scholar] [CrossRef]
- Behr, W.; Honikel, K.; Hartmann, G. Interaction of the RNA polymerase inhibitor chromomycin with DNA. Eur. J. Biochem. 1969, 9, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.R.; Brassett, C.; Waring, M.J. Kinetics of dissociation of nogalamycin from DNA: Comparison with other anthracycline antibiotics. Biochim. Et Biophys. Acta 1985, 840, 383–392. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Vazquez-Laslop, N.; Mankin, A.S. Kinetics of drug-ribosome interactions defines the cidality of macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2017, 114, 13673–13678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, L.M.; Fried, M.G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef]
- Fried, M.; Crothers, D.M. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 1981, 9, 6505–6525. [Google Scholar] [CrossRef] [Green Version]
- Beniaminov, A.D.; Dezhenkova, L.G.; Mamaeva, O.K.; Shchyolkina, A.K.; Tevyashova, A.N.; Kaluzhny, D.N.; Shtil, A.A. Divalent cations are dispensable for binding to DNA of a novel positively charged olivomycin A derivative. PLoS ONE 2018, 13, e0191923. [Google Scholar] [CrossRef] [Green Version]
- Van Dyke, M.W.; Dervan, P.B. Chromomycin, mithramycin, and olivomycin binding sites on heterogeneous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA)iron(II). Biochemistry 1983, 22, 2373–2377. [Google Scholar] [CrossRef]
- Barcelo, F.; Scotta, C.; Ortiz-Lombardia, M.; Mendez, C.; Salas, J.A.; Portugal, J. Entropically-driven binding of mithramycin in the minor groove of C/G-rich DNA sequences. Nucleic Acids Res. 2007, 35, 2215–2226. [Google Scholar] [CrossRef] [Green Version]
- Trantirek, L.; Stefl, R.; Vorlickova, M.; Koca, J.; Sklenar, V.; Kypr, J. An A-type double helix of DNA having B-type puckering of the deoxyribose rings. J. Mol. Biol. 2000, 297, 907–922. [Google Scholar] [CrossRef]
- Stefl, R.; Trantirek, L.; Vorlickova, M.; Koca, J.; Sklenar, V.; Kypr, J. A-like guanine-guanine stacking in the aqueous DNA duplex of d(GGGGCCCC). J. Mol. Biol. 2001, 307, 513–524. [Google Scholar] [CrossRef]
- Protozanova, E.; Yakovchuk, P.; Frank-Kamenetskii, M.D. Stacked-unstacked equilibrium at the nick site of DNA. J. Mol. Biol. 2004, 342, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Vologodskii, A.; Frank-Kamenetskii, M.D. DNA melting and energetics of the double helix. Phys. Life Rev. 2018, 25, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cons, B.M.; Fox, K.R. The GC-selective ligand mithramycin alters the structure of (AT)n sequences flanking its binding sites. FEBS Lett. 1990, 264, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Gause, G.F. Chromomycin, Olivomycin, Mithramycin. In Antineoplastic and Immunosuppressive Agents: Part II; Sartorelli, A.C., Johns, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 1975; pp. 615–622. [Google Scholar] [CrossRef]
- Chen, F.M. Methods for the studies of drug dissociation from DNA. Methods Mol. Biol. 1997, 90, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.R.; Crothers, D.M. Kinetics and sequence specificity of drug-DNA interactions: An in vitro transcription assay. Biochemistry 1986, 25, 7355–7362. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
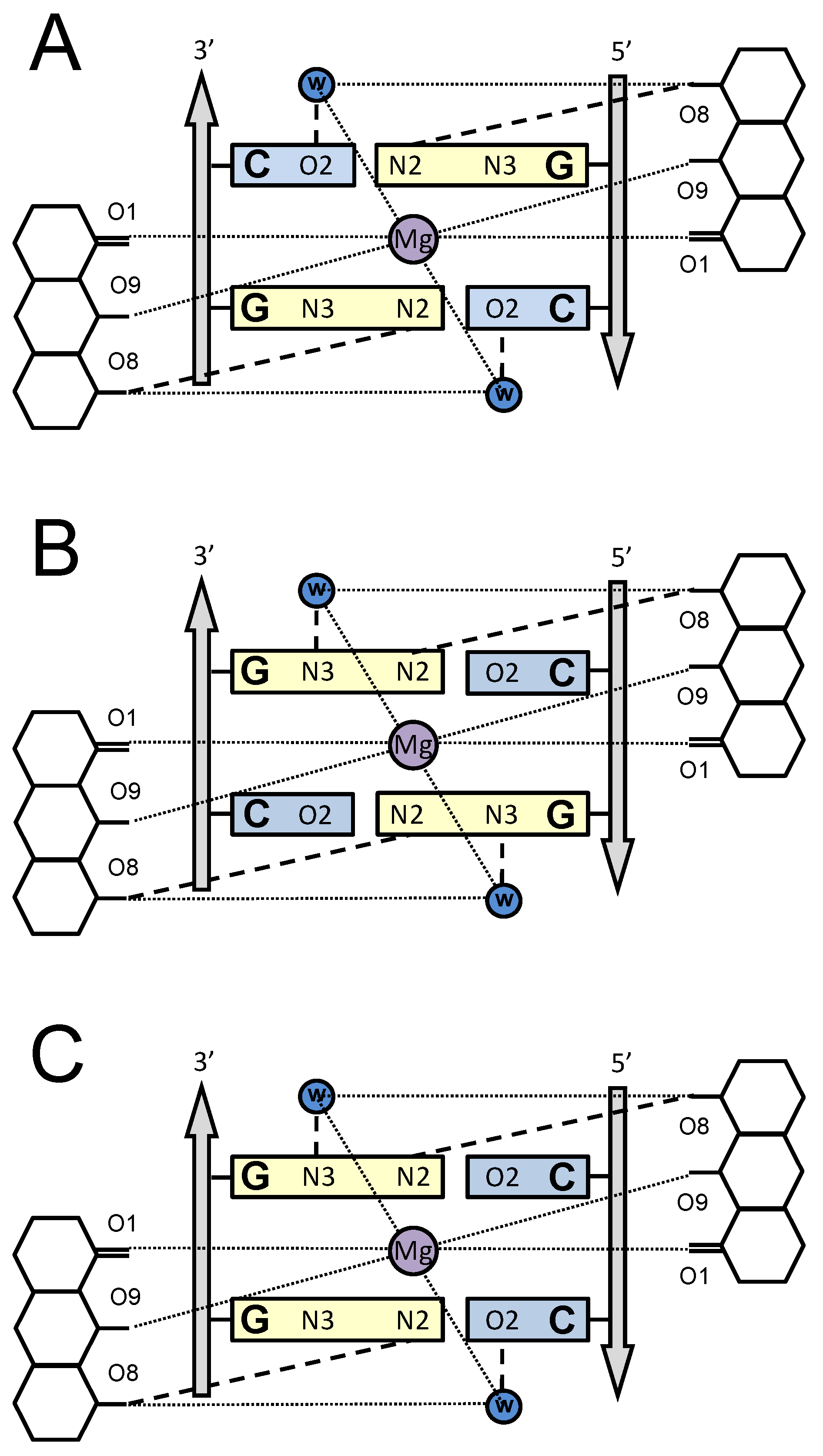{kind=link}
| Oligo Number | N (DNA/OA Dimer) | Ka, 107 (M−1) | ΔH (kcal/mol) | TΔS (kcal/mol) |
|---|---|---|---|---|
| 1 | 0.9 | 2.08 ± 0.24 | −6.97 ± 0.05 | 3.0 |
| 2 | 1.0 | 1.88 ± 0.15 | −7.60 ± 0.04 | 2.3 |
| 3 | 0.7 | 2.3 ± 0.7 | −9.68 ± 0.18 | 0.4 |
| 4 | 1.0 | 1.94 ± 0.22 | −6.50 ± 0.05 | 3.5 |
| 5 | 1.0 | 2.21 ± 0.18 | −7.39 ± 0.04 | 2.6 |
| 6 | 1.1 | 1.8 ± 0.3 | −7.18 ± 0.08 | 2.7 |
| 7 | 0.9 | 1.9 ± 0.3 | −5.11 ± 0.06 | 4.8 |
| 8 | 0.9 | 1.26 ± 0.15 | −4.42 ± 0.04 | 5.3 |
| 9 | 0.9 | 1.16 ± 0.17 | −4.76 ± 0.05 | 4.9 |
| 10 | 0.9 | 1.11 ± 0.10 | −5.39 ± 0.03 | 4.2 |
| Central Dinucleotide | Binding Site (Oligo Number) | k+, 104 M−1 s−1 | k−(SDS), 10−4 s−1 | Lifetime of OA–DNA Complexes, Min * |
|---|---|---|---|---|
| GG | GGGG (1) | 3.8 ± 0.4 | 1.3 ± 0.2 | 128 |
| GGGC (2) | 4.5 ± 0.2 | 2.5 ± 0.1 | 66 | |
| CGGG (3) | 3.9 ± 0.3 | 1.4 ± 0.4 | 122 | |
| CGGC (4) | 3.4 ± 0.2 | 3.8 ± 0.2 | 44 | |
| GC | GGCG (5) | 3.0 ± 0.2 | 1.3 ± 0.1 | 129 |
| GGCC (6) | 2.7 ± 0.3 | 1.7 ± 0.4 | 99 | |
| CGCG (7) | 1.8 ± 0.3 | 1.4 ± 0.1 | 118 | |
| CG | GCGG (8) | 3.6 ± 0.1 | 16 ± 1 | 10 |
| GCGC (9) | 3.5 ± 0.4 | 17 ± 1 | 10 | |
| CCGG (10) | 4.7 ± 0.2 | 10 ± 1 | 17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beniaminov, A.D.; Chashchina, G.V.; Livshits, M.A.; Kechko, O.I.; Mitkevich, V.A.; Mamaeva, O.K.; Tevyashova, A.N.; Shtil, A.A.; Shchyolkina, A.K.; Kaluzhny, D.N. Discrimination between G/C Binding Sites by Olivomycin A Is Determined by Kinetics of the Drug-DNA Interaction. Int. J. Mol. Sci. 2020, 21, 5299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155299
Beniaminov AD, Chashchina GV, Livshits MA, Kechko OI, Mitkevich VA, Mamaeva OK, Tevyashova AN, Shtil AA, Shchyolkina AK, Kaluzhny DN. Discrimination between G/C Binding Sites by Olivomycin A Is Determined by Kinetics of the Drug-DNA Interaction. International Journal of Molecular Sciences. 2020; 21(15):5299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155299
Chicago/Turabian StyleBeniaminov, Artemy D., Galina V. Chashchina, Mikhail A. Livshits, Olga I. Kechko, Vladimir A. Mitkevich, Olga K. Mamaeva, Anna N. Tevyashova, Alexander A. Shtil, Anna K. Shchyolkina, and Dmitry N. Kaluzhny. 2020. "Discrimination between G/C Binding Sites by Olivomycin A Is Determined by Kinetics of the Drug-DNA Interaction" International Journal of Molecular Sciences 21, no. 15: 5299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155299