Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies




1
Texas Biomedical Research Institute, Southwest National Primate Research Center, San Antonio, TX 78227, USA
2
Department of Pharmacology, Renaissance School of Medicine, Stony Brook University, Stony Brook, NY, 11794-8651, USA
3
Tulane National Primate Research Center, Covington, LA 70433, USA
4
PreCliniTria, LLC., Mandeville, LA 70471, USA
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(15), 5407; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155407
Submission received: 8 July 2020
/
Revised: 23 July 2020
/
Accepted: 24 July 2020
/
Published: 29 July 2020
(This article belongs to the Special Issue Role of Nutraceuticals in Metabolic and Gastrointestinal Disorders)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Although celiac disease (CD) is an autoimmune disease that primarily involves the intestinal tract, mounting evidence suggests that a sizeable number of patients exhibit neurological deficits. About 40% of the celiac patients with neurological manifestations have circulating antibodies against neural tissue transglutaminase-6 (tTG6). While early diagnosis and strict adherence to a gluten-free diet (GFD) have been recommended to prevent neurological dysfunction, better therapeutic strategies are needed to improve the overall quality of life. Dysregulation of the microbiota-gut-brain axis, presence of anti-tTG6 antibodies, and epigenetic mechanisms have been implicated in the pathogenesis. It is also possible that circulating or gut-derived extracellular structures and including biomolecular condensates and extracellular vesicles contribute to disease pathogenesis. There are several avenues for shaping the dysregulated gut homeostasis in individuals with CD, non-celiac gluten sensitivity (NCGS) and/or neurodegeneration. In addition to GFD and probiotics, nutraceuticals, such as phyto and synthetic cannabinoids, represent a new approach that could shape the host microbiome towards better prognostic outcomes. Finally, we provide a data-driven rationale for potential future pre-clinical research involving non-human primates (NHPs) to investigate the effect of nutraceuticals, such as phyto and synthetic cannabinoids, either alone or in combination with GFD to prevent/mitigate dietary gluten-induced neurodegeneration.
1. Introduction
As early as in 1908, the first reports of neurological abnormalities and complications in patients with gastrointestinal “sprue”, such as peripheral neuritis, ataxia, and partial degeneration of spinal cord, started to emerge [1]. The first systematically corroborated evidence of dietary gluten-associated (celiac) neuropathy dates back to 1966, when Cooke and colleagues [2] described neurodegenerative lesions in muscle biopsies from adult patients with celiac disease (CD) using a combination of electron microscopy and vital (H&E) staining. Thereafter, in 1962, the first fatal case resulting from neurological disease was reported in a 57-year old patient with CD [3]. The postmortem findings in this patient were compared with nine other celiac cases that also exhibited progressive central nervous system (CNS) disorders. It was concluded that progressive neurodegeneration in the cerebellum, deep gray matter, brain stem, and spinal cord were the common histopathological characteristics in these patients [3]. Another report, involving a 47-year old CD patient with spinocerebellar degeneration and normal vitamin E levels, revealed that gluten-free diet (GFD) administered to celiac patients can have beneficial effects in stabilizing not only gastrointestinal, but also neurological symptoms [4]. Epidemiological survey of people living on the remote Micronesian islands consuming traditional grain-free versus western-type (grain-rich) diet showed that the latter group suffered with significantly higher incidence of schizophrenia [5], a neurodevelopmental but also a progressively neurodegenerative disorder [6,7]. Nonetheless, many of the initial reports pointing to an association between neurological disorders and consumption of gluten-containing diet lacked direct evidence and in-depth analysis.
The landscape has started to change in recent decades after a large body of evidence regarding the role of tissue transglutaminases (tTGs), namely the intestinal tissue transglutaminase-2 (tTG2), emerged with respect to CD pathogenesis [8]. It was established that in addition to post-translational modification of gluten/gliadin through the process of deamidation/transamidation, tTG2 functions as an autoantigen, thereby inducing the formation of tTG2 autoantibodies. Presence of tTG2 autoantibodies occurs in almost 100% of celiac patients but only in a small fraction of non-celiac gluten sensitivity (NCGS) patients [9]. Involvement of the neural isoform of tTG, i.e., tTG6 was suggested in the pathogenesis of several neurodegenerative disorders including Alzheimer’s Disease (AD) and Huntington’s Disease (HD), where tTG6 was reported to contribute to the resolution of abnormally accumulated amyloid and microtubule-associated proteins [10]. In addition, tTG6 was shown to be implicated in the pathogenesis of movement disorders, such as gluten ataxia and multiple sclerosis (MS) [11,12,13,14]. Increased levels of circulating anti-tTG6 antibodies were also detected in adult patients with schizophrenia [15]. Finally, recent human leukocyte antigen (HLA)-typing of CD and NCGS patients diagnosed with Autism Spectrum Disorders (ASD) and/or Down’s Syndrome (DS), demonstrated that there is a definitive, and significant association between specific HLA haplotypes and dysregulation of the gut-brain axis [16,17,18].
In people with psychosis and multi-episode schizophrenia, it was shown that significant proportion of these individuals have elevated anti-gliadin antibodies but not necessarily antibodies to deamidated gliadin and tTG (autoantibodies), confirming that neuro-immune responses to gliadin differ between CD and NCGS [19]. On the other hand, due to relatively low CD predictive value of anti-gliadin antibodies (~70–80%), their presence might be even less/not predictive in patients with neurodegeneration. Furthermore, antibodies to glutamic acid decarboxylase (GAD), a protein important in the synthesis of inhibitory neurotransmitter gamma-aminobutyric acid, were found to be present in gluten-sensitive people with neurological disorders, but not in CD patients [20]. In addition to autoantibodies recognizing the neural tTG6 in celiac patients with neurological disorders, autoantibodies reactive to synapsin I (neuronal synaptic vesicles), neuroglial, and Purkinje cells were identified [21,22,23].
Taken together, it became evident that dietary gluten-induced neurodegeneration is a serious and understudied phenomenon, the pathogenesis of which differs between CD and NCGS. TG6 has been proposed to play an important role in the neurological (both central and peripheral nervous system) manifestations of CD, as high TG6 antibody levels have been detected in CD patients with gluten ataxia [12] and peripheral neuropathy [24]. Due to the absence of tTG2/6 involvement in NCGS patients, it was proposed that a leaky intestinal epithelial barrier that allows partially digested immunotoxic gluten peptides to enter the systemic circulation and then cross the blood brain barrier, where these peptides along with HLA-DQ2/DQ8 restricted CD4 T cells infiltrating the brain from the intestine may produce proinflammatory cytokines driving CNS disease in NCGS patients [25,26,27,28]. Alternatively, circulating or gut derived extracellular structures, including biomolecular condensates and extracellular vesicles from CD and NCGS patients may use a leaky intestinal epithelial barrier to deliver their proinflammatory cargo to the brain.
2. Results
2.1. Preclinical Evidence
Currently, there is limited preclinical evidence to demonstrate the link between gluten and neurodegeneration. Despite attempts to develop gluten ataxia in laboratory mice, such efforts were limited or not successful [29,30].
In studies using the experimentally-induced rhesus macaque model of CD, i.e., gluten-sensitive enteropathy (GSE), it was demonstrated that several tight junction and their associated proteins, such as zonulin and haptoglobin-2, known to be expressed also in human blood brain barrier, were dysregulated in celiac rhesus macaques [31]. In CD patients, reduced expression of intestinal tight junction proteins including zonulin and occludin were linked to increased epithelial permeability, i.e., leaky gut [32]. Further, zonulin receptor has been identified as the precursor for haptoglobin-2 [28]. Similarly, we also reported significant downregulation of intestinal tight junction proteins zona occludens-1 (ZO1) and claudin-1 in duodenum of celiac macaques [32]. In recent studies, we detected markedly reduced occludin protein expression in the duodenal epithelium of celiac macaques (Figure 1). A complete loss of occludin protein expression from the apical and basal side of the duodenal epithelium of celiac macaques was noted (Figure 1A) while its high basal expression was present in healthy control macaques (Figure 1B). Evidence from studies involving psychotic patients confirmed increased levels of serum haptoglobin-2, consistent with findings from celiac macaques [28,31]. Dysregulated and impaired expression of zonulin in celiac macaques suggests that both zonulin and haptoglobin-2 could negatively impact the functioning of the blood brain barrier [28,31]. Finally, a group of dysregulated genes associated with perturbed neurological functions were also identified in celiac macaques fed a gluten-containing diet [33]. The predisposition genes potentially linked to the neurological form of CD were identified by messenger-RNA (mRNA) profiling: CADPS2 (ASD), CAPN13 (AD), BACE2 (AD, DS), DSCR5 (DS), and PINK1 (Parkinson’s Disease (PD) [33]. Expression levels of these mRNAs were not perturbed in healthy macaques and were only minimally so in celiac macaques on GFD, suggesting that consumption of dietary gluten in susceptible primates is linked, besides other effects, to neurological dysfunction. To corroborate and to further expand these findings, more translational studies employing the rhesus macaque celiac model are needed.
2.2. Mechanisms of Dietary Gluten-Induced Neuropathy
It was established that increased tTG2 activity leads to autoimmune reaction and GSE, i.e., CD in genetically-predisposed individuals [34,35,36]. Besides gluten digestion, tTG-mediated glutamine deamidation can, in some celiac patients, lead to the aggregation of cerebral β-amyloid, one of the hallmarks of neurodegeneration in people with PD, HD, and AD [37,38,39,40,41,42]. It is not clear however, if neuronal dysfunction occurs in all individuals with CD or if this is limited only to a subset of these patients. Due to tTG’s capability to be i) recognized as autoantigen in not only intestinal but also systemic tissues including CNS, ii) to cause cerebral β-amyloid polymerization, and iii) to facilitate inflammation and cancer, it became an attractive drug target for a multitude of diseases [36,37,39,43]. A substantial number of inhibitors, probes, and substrates were chemically engineered with the purpose to better understand the pathogenesis of CD and to use some of the tTG inhibitors as therapeutics [44,45]. Notwithstanding, in vivo use of these compounds in the treatment of tTG-associated illnesses is not straight-forward and requires thorough translational validation using a model that faithfully recapitulates human disease. Moreover, the contribution of dysbiotic microbial metabolome to post-translational modifications of CD-relevant proteins, such as tTGs, was suggested to influence functioning of the gut-brain axis [46,47].
2.3. MicroRNA Evidence
The evaluation of the role of micro-RNAs (miRNAs) is of great interest in CD as they represent an important epigenetic mechanism with immense potential to regulate the inflammatory response associated with CD pathogenesis. MiRNAs are ~20–23 nucleotide long, small RNA molecules that regulate gene expression post-transcriptionally by binding to homologous sequences on the 3’ untranslated regions (UTRs) (homologous base pairings between miRNA seed nucleotides 2 to 7 and the 3’ UTR). MiRNAs are known to regulate majority of cellular processes that include but are not limited to cell proliferation, differentiation, apoptosis, cell signaling, immune, and inflammatory responses. Over the past decade, the role of miRNAs in CD pathogenesis has been studied in immune cells isolated from intestinal biopsies and peripheral blood. Using duodenal pinch biopsies, Magni and colleagues [48] identified significant downregulation of miR-192-5p, miR-31-5p, miR-338-3p, and miR-197 in patients with celiac disease with severe histopathological lesions. Consistent with miR-192-5p downregulation, several bioinformatically predicted targets with critical roles in innate immune response, namely, chemokine C-X-C motif ligand 2 (CXCL2) and nucleotide oligomerization domain-2 (NOD2) showed marked upregulation at both the mRNA and protein level. In a separate study, Vaira and colleagues [49] further confirmed inflammation induced downregulation of miR-192-5p. Interestingly, miR-192-5p is also downregulated in patients with ulcerative colitis, which suggests that it plays a crucial role in maintaining intestinal homeostasis [50]. In addition, forkhead box P3 (FOXP3), Run-related transcription factor 1, and interleukin-18 which are predicted targets of miR-31-5p, miR-338-3p, and miR-197 also showed significantly high expression in biopsy tissues. More recently, expression of miR-192/215 together with the miR-200 families were shown to be progressively reduced and those of miR-17/92 and C19MC miRNAs to be upregulated in refractory CD and intestinal T-cell lymphomas associated with CD [51]. A link between downregulation of these miRNAs resulting in constitutive signal transducer and activator of transcription 3 (STAT3) activation and MYC proto-oncogene BHLH transcription factor (c-Myc) mediated oncogenic signaling in refractory CD and subsequent lymphomagenesis was proposed [51].
Unlike miR-192-5p, in pediatric CD, miR-449a expression was significantly increased [52]. Notch receptor 1 (NOTCH1) and kruppel like factor 4 (KLF4) that are associated with goblet cell proliferation and differentiation was validated as direct targets of miR-449a. Consistent with miR-449a mediated negative regulation of NOTCH1 and KLF4, goblet cell numbers were markedly reduced in the small intestines of children with CD. Interestingly, miR-21 and miR-31 have also been identified as circulating non-invasive biomarkers of pediatric CD [53]. Increased miR-21 and decreased miR-31 was confirmed in serum of pediatric CD patients. Additionally, miR-21 correlated with the presence of IgA-tTG2 autoantibodies.
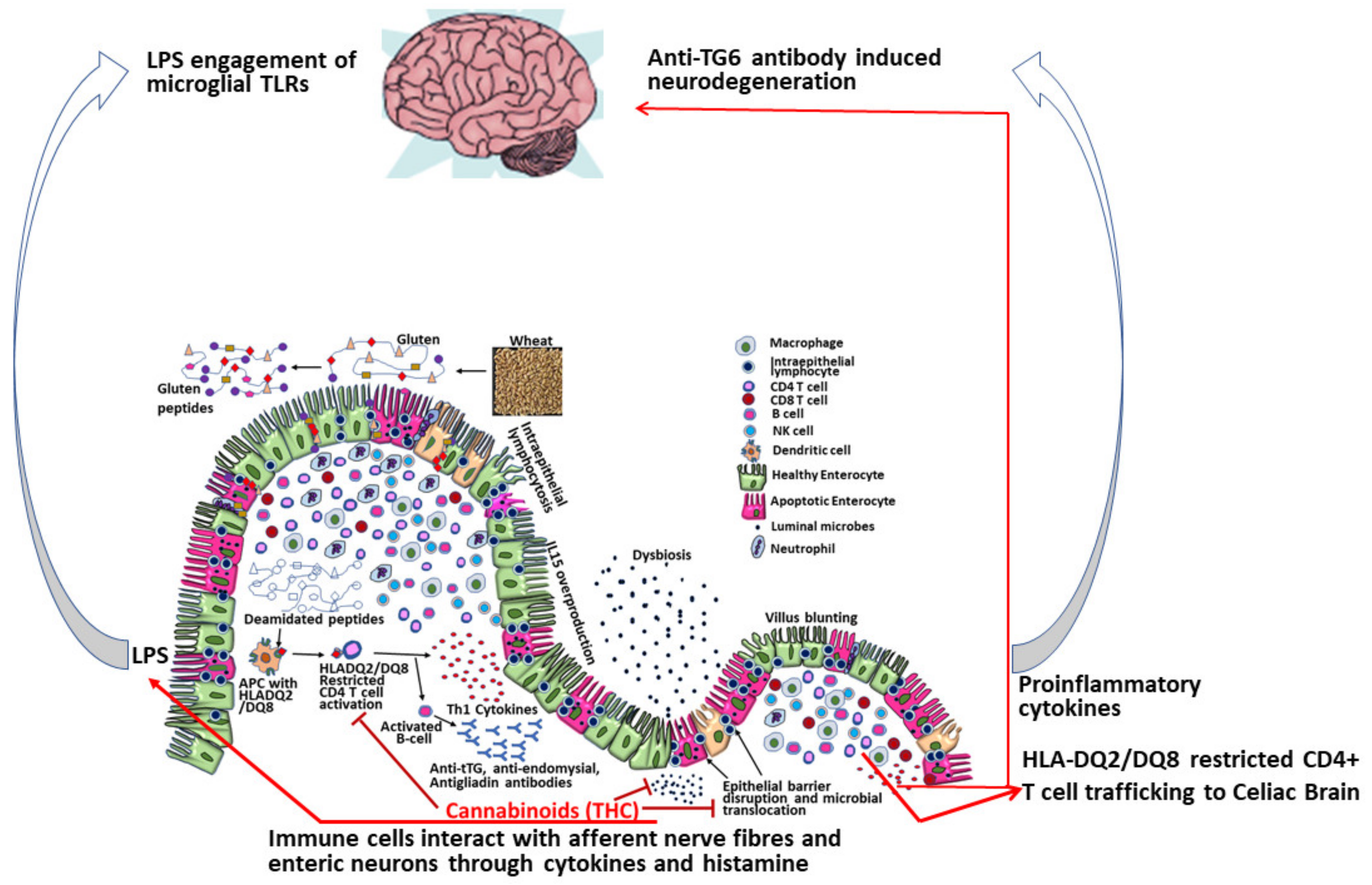
Apart from targeting innate immune response genes, our own studies using the rhesus macaque model of GSE confirmed elevated miR-204 to directly target the intestinal tight junction protein claudin-1 resulting in its significantly reduced protein expression in the duodenum of celiac macaques [32]. Further, intestinal inflammation coupled with barrier disruption was accompanied by marked gut dysbiosis [32]. Disruption of the intestinal barrier can facilitate translocation of intestinal microbes and microbial by-products into the systemic circulation. In longstanding cases of CD, prolonged microbial translocation can exceed hepatic clearance thereby increasing the potential for lipopolysaccharide (LPS) to accumulate, cross the blood brain barrier and activate microglia cells in the CNS resulting in neuroinflammation and neuronal damage (Figure 2). Overall, these findings identify a putative miRNA mediated mechanism that can facilitate intestinal inflammation and eventually trigger neurological injury in CD patients. Interestingly, our previously published studies using the simian immunodeficiency virus (SIV)-infected rhesus macaque model of acquired immune deficiency syndrome (AIDS) demonstrated the ability of phytocannabinoids to inhibit intestinal inflammation by inducing the expression of anti-inflammatory miRNAs, inhibiting T cell proliferation/activation and preserving intestinal tight junction protein expression [54,55]. These latter findings accentuate the potential of cannabinoids as a viable nutraceutical either alone or in combination with other immunobiologics to reduce dietary gluten-induced intestinal inflammation, epithelial barrier disruption, and extraintestinal complications including neurological disease in CD patients (Figure 2).
2.4. Gut Dysbiosis-Neurodegeneration Link
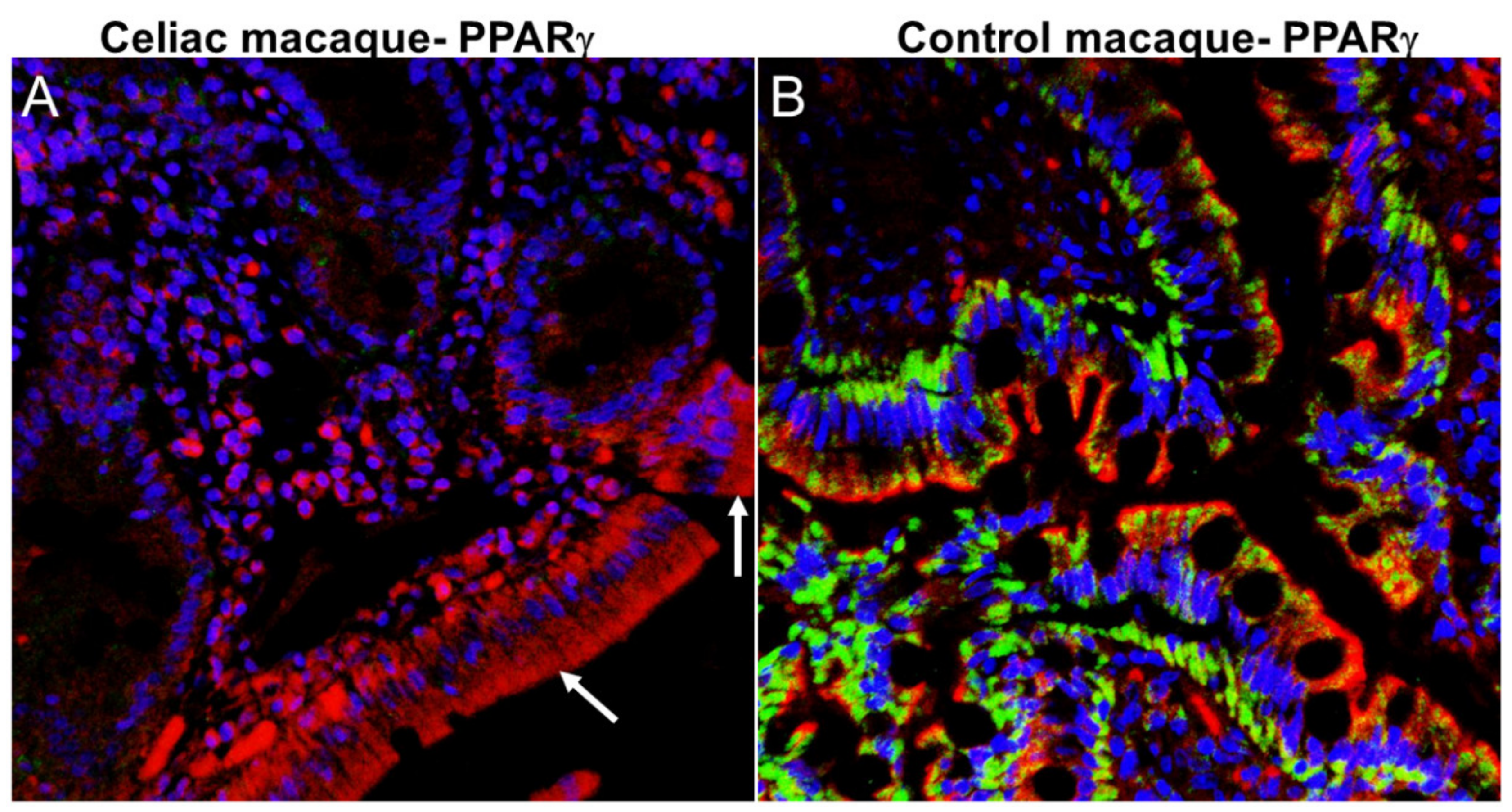
It was postulated that homeostasis of the gut-brain axis can be disrupted in association with progressed age, obesity, diet, and drug use [26]. Chronic intestinal inflammation that is triggered in predisposed (CD/NCGS) individuals by consumption of gluten-containing diet is intimately linked with intestinal dysbiosis and leaky gut [32,54]. We and others have demonstrated that dietary gluten-induced inflammation and dysbiosis are linked with perturbations of genetic regulatory factors of neuroinflammation, cognition, and neurodegeneration [33,56,57]. From a molecular mechanistic perspective, the expression of peroxisome proliferator activated receptor gamma (PPARγ), a key gene with anti-inflammatory (peripheral, intestinal and neuroinflammation) [58] and anti-dysbiotic effects [59] is considerably reduced in ulcerative colitis [60] and celiac disease [61,62,63] patients. Byndloss et al. [59] demonstrated that PPARγ downregulation was associated with dysbiotic expansion of bacteria belonging to the family Enterobacteriaceae (phylum Proteobacteria) and reduction in the relative abundance of obligate anerobic bacteria. Like celiac patients, we detected markedly reduced PPARγ expression in duodenal epithelium of celiac macaques (Figure 3A) relative to healthy control macaques (Figure 3B). Similar to occludin protein expression (Figure 1A), considerable loss of PPARγ protein expression from the duodenal epithelium was detected in celiac macaques. Accordingly, PPARγ downregulation may promote intestinal inflammation and subsequent dysbiosis in celiac macaques and by extension in patients with CD.
It would be an oversimplification to suggest that gut dysbiosis-associated neuropathology can be resolved by a strict adherence to GFD. Nevertheless, research regarding the inflammatory pathways and microbial taxa in affected individuals is already paving the way for the formulation of new prevention and treatment strategies [32]. Accordingly, it was suggested that remission of dysbiosis in individuals with autism and other neurodevelopmental disorders could be the first step in the treatment of these illnesses [64]. Although gluten-free diets (GFD) have shown success in restoration of PPARγ expression in celiac patients [62], the risks of gluten contamination coupled with the persistence of intestinal inflammation in patients with CD on GFD underscores the need to develop better therapeutic solutions. Accordingly, reducing chronic inflammation, promoting intestinal mucosal healing and repair, and restoring epithelial barrier integrity and the microbiome are the topmost priorities of the celiac research community. Because PPARγ was described to be the key functional receptor transducing the effects of commonly prescribed anti-inflammatory amino salicylates in inflammatory bowel disease (IBD) patients [65], augmenting PPARγ expression represents a promising approach for the clinical management of gluten induced intestinal inflammation [66]. In this context, mesalazine also known as 5-amino salicylic acid (5-ASA) was shown to reduce oxidative burst and induce PPARγ expression in ex vivo cultures of duodenal biopsies obtained from newly diagnosed celiac patients [66]. In addition, mesalazine treatment also reduced protein levels of nuclear factor kappa B (NFκB) and nitric oxide synthase 2 (NOS2), an enzyme that produces nitric oxide [66]. Obligate anerobic bacteria have been shown to activate PPARγ signaling to limit the production of nitrate and oxygen electron acceptors by host epithelial cells, thereby preventing the expansion of facultative anerobic bacteria belonging to the family Enterobacteriaceae (phylum Proteobacteria) [59]. Therefore, reduced PPARγ signaling in celiac patients and macaques may promote the activation of NOS2, resulting in increased production of nitric oxide, which can then be broken down to non-toxic nitrates in the intestinal lumen and made abundantly available as a respiratory electron acceptor to members of the Enterobacteriaceae family that encode nitrate reductases. Although 5-ASA has been effective, it also has produced adverse effects [67], which makes it critical to develop newer approaches to restore and activate PPARγ signaling to reduce intestinal dysbiosis and prevent microbial products from reaching the brain and activate neuroinflammatory signaling. Moreover, in this context, we recently demonstrated the ability of cannabinoids to directly trans activate PPARγ expression in in vitro cultured cell lines and in ex vivo colon explant cultures [68]. Most notably, using the SIV-infected rhesus macaque model of AIDS, we found that long-term controlled low dose administration of naturally available phytocannabinoids like delta-9-tetrahydrocannabinol (THC) not only inhibited intestinal inflammation [55] but also inflammation of the minor salivary glands in the oral cavity [69]. Specifically, THC treated chronically SIV-infected rhesus macaques had better preservation of oral commensal bacteria species that are critical to the maintenance of oral microbial homeostasis (Mohan personal communication). In addition, chronic THC significantly reduced the relative abundance of pathogenic bacteria associated with periodontitis (Mohan personal communication). These findings are exciting and promising as it represents a safe yet pharmacologically feasible new approach to inhibit intestinal and neuroinflammation, restore epithelial barrier integrity, and prevent dysbiosis in not only celiac, but also other chronic inflammatory diseases of the intestine.
2.5. Potential Role of Extracellular Structures—Biomolecular Condensates and Extracellular Vesicles in Pathogenesis of CD-Associated Neurodegeneration
It is becoming increasingly evident that all cells create discrete biochemical environments in the form of extracellular structures (ES), used to organize and orchestrate complex biochemical reactions that regulate host health and disease. For the purpose of this review, ES are defined as non-replicative organism-derived acellular structures that consist of two archetypes—namely, i) the membrane-less biomolecular condensates (BMCs), and ii) the membrane-encased extracellular vesicles (EVs). While BMCs assemble by thermodynamic-mediated liquid-liquid phase separation, the liquid BMCs are known to transform into reversible amyloid fibers or insoluble aggregates [70]. While the details of BMC biology is still evolving, it has been shown that RNA and proteins with intrinsically disordered regions are key factors that influence the intracellular composition, size, and morphology of BMCs [71,72]. Unlike BMCs, EVs contain at least one membrane. EVs are comprised of at least three different subtypes, including apoptotic bodies, released as a product of apoptotic cell disassembly; microvesicles, generated by the outward budding and fission of membrane vesicles from the cell surface; and exosomes, generated by reverse budding of multivesicular bodies [73,74]. These EV subtypes are characterized by their size, although size range overlaps exist. Apoptotic bodies are generally larger (500–4000 nm) in size, microvesicles usually range in size from 50–2000 nm, most being larger than 200 nm, and exosomes from 30 to 100 nm [75,76,77]. Despite the size range, different cells, tissues, and organisms release vesicles that vary in size. Another characteristic of EVs is their ability to encase bioactive cargo that includes subsets of diverse proteins, different RNA species (mRNAs, miRNA, tRNA, etc), dsDNA, extrachromosomal DNA, lipids, pathogen-derived products, and illicit and licit drugs [78,79,80]. Given the diverse nature of EVs and the overlap in their size and cargo composition, we will use the term EVs to encompass apoptotic bodies, microvesicles, and exosomes. Both BMCs and EVs regulate host response to disease, immunological, and environmental assaults. Hence, BMCs play important roles in various physiological contexts, such as transcription and stress response [81,82], and in pathophysiological conditions, including cancer and neurodegenerative diseases [83,84,85]. EVs mediate distal and proximal intercellular communications [86,87,88] and organism-to-cell interactions [89,90,91], and are involved in neuroinflammation [92], irritable bowel syndrome (IBS) [93], and, potentially, CD [94].
2.6. ES and Neurodegeneration
ES—BMCs and EVs display both beneficial and detrimental roles in various disease contexts. With regards to the CNS that relies on complex cross talk amongst CNS cellular and acellular components, it has been shown that BMCs and EVs display both anti- and pro- inflammatory responses in inflammation-associated neurodegenerative diseases, such as AD, frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS), HD, and PD. It is undisputable that neurodegenerative disorders involve BMCs, in the form of regional aggregation of cytosolic or nuclear proteins, which are believed to drive neurodegeneration [95]. Examples of the neuro-associated BMCs include the proteinaceous assemblies of the microtubule-associated protein Tau (MAPT), cytosolic inclusions of the RNA-binding proteins TAR DNA binding protein of 43 kDa (TDP-43), fused in Sarcoma (FUS), and poly-glutamine (polyQ) aggregates [96]. Tau aggregates are mostly found in AD, TDP-43 and FUS are found in ALS and FTD, while polyQ aggregates more in HD [96]. Interestingly, BMC aggregates are thought to spread from one brain region to another, in line with the progressive nature of these diseases [97]. Similar to BMCs, EVs have been implicated in the pathogenesis and these membrane structures are integral component of neuroimmune interaction. For example, CNS-derived EVs are present in peripheral circulation [95,96]. The CNS EVs play a role in assessing cerebral neuroimmune status. For example, it is known that the initiation, propagation, and resolution of inflammatory response to CNS injury or neurodegenerative disease relies on cytokines, miRNAs, or microbial products, all of which have been shown to be present in EVs [79,98,99,100,101,102]. Additionally, EVs mediate the transfer of peripheral inflammatory molecules to the CNS [103]. Although BMCs and EVs are different in structure and function, there seem to be convergence in their function. Both BMCs and EVs have been implicated in contributing to the pathogenesis of neurodegenerative diseases where the pathology is dictated in part by neuroimmune mechanisms, such as the presence of amyloid-β peptide (Aβ) [104,105,106] and Tau [95,107].
2.7. ES and the Gut-Brain Barrier
Although the gut and brain are anatomically distinct, cross-talk exists between these organs and we hypothesize that gut microbiota derived BMCs and/or EVs may have important role on the intestinal immunity and its regulation and interaction with the brain. Alteration in the gut microbiome either by infectious agents or use of licit and illicit substances has been linked to altered immune responses [108]. Moreover, gut dysbiosis promotes inflammatory and metabolic disorders [109], which may be imprinted in gut or blood derived BMCs and EVs, such as seen in EVs from saccharibacteria, formerly known as TM7, Akkermansia muciniphila, Bacteroides acidifaciens bacteria [110], or Bacteroides fragilis-derived EVs [111]. Thus, ES-based network may represent a link between the gut and the brain. Such an ES-mediated role may contribute to the pathogenesis of neurodegeneration, including the CD-associated neurodegeneration.
Indeed, the gut microbiota influences gut-brain cross talk as reviewed by Van Den Elsen [112]. Multiple studies have shown that both host and microbiota derived EVs enter systemic circulation and such EVs mediate cross-talk between inflammatory pathways on both sides of the blood brain barrier (BBB) [113,114]. EVs generated by peripheral cells have been shown to contribute to various neurological deficits [115,116,117,118]. On the other hand, mesenchymal stem cell derived EVs that possess anti-inflammatory properties against myocardial inflammation, arthritis, and inflammatory lung diseases [119,120,121,122] also reduce inflammation in various CNS pathologies [123,124,125,126,127].
Although how EVs breach the BBB is unknown, we and others have shown that EVs facilitate release of metalloproteases, promote extracellular matrix reorganization, epithelial barrier regulation, and inflammatory cell recruitment [128]. Moreover, EVs containing immune/inflammatory and epithelial cell specific markers are elevated in the blood and intestinal lumen of patients suffering from IBD and such high EV concentrations are correlated with disease severity [129]. In this context, a causal relationship between increased number of cargo-carrying EVs in blood/intestinal tissue and enhanced intestinal permeability has been reported in IBS [93]. It is therefore clear that communication between the brain and the periphery is a bidirectional interaction as demonstrated by the entrance of blood-derived EVs into the CNS and uptake by microglia cells [103] vis a vis the detection of CNS-derived EVs in the blood [130]. Therefore, both of these cross interactions must have occurred following EV-mediated transport through the BBB.
Although how gut- or neurodegenerative-linked BMCs or EVs arise, as well as the exact mechanisms behind their ability to regulate gut-directed neurodegeneration, is still not well understood, we propose multiple possibilities based on our studies and the literature. First, ES released by intestinal cells and/or microbiota may be decorated with degradative enzymes such as matrix metalloproteinases (MMPs) or microbiota-derived ES-mediated degradation of polysaccharides and inositol polyphosphates. Second, such degradative molecules may promote the ability of ES to access and cross the epithelial barrier, gain access to systemic circulation, and reach other organs, and the brain. Third, compromised intestinal epithelial barrier and BBB may facilitate transcellular transmigration and deposition of inflammatory cargo. Alternatively, peripheral ES may use other mechanisms to reach the CNS or vice versa via paracellular transmigration, micropinocytosis, clathrin-mediated endocytosis, or caveolin-mediated endocytosis.
3. Conclusions
In summary, there are several avenues for shaping the dysregulated gut homeostasis in individuals with CD, NCGS and/or neurodegeneration. In addition to GFD [62], probiotics [131], and nutraceuticals, including potentially synthetic cannabinoids, such as selective cannabinoid receptor agonists, there are other evolving [132] strategies that could shape the host microbiome towards better prognostic outcomes of associated neuropathological disorders. To narrow down these approaches to the most promising ones, preclinical trials with the rhesus macaque model of CD can help to achieve these goals.
Author Contributions
All three authors contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this publication was funded by the National Institutes of Health Award Numbers R01DA042524 and R56DE026930 to M.M., R01DA042348 to C.M.O., R01DA050169 to M.M. and C.M.O. and P51OD011133. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Acknowledgments
The authors would like to thank Kyle Shannon, Eunhee Lee, Maurice Duplantis, Faith R. Schiro, Cecily C. Midkiff, and Xavier Alvarez for their technical assistance in the study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| CD | Celiac Disease |
| GFD | Gluten-Free Diet |
| tTG2 | Intestinal Tissue Transglutaminase |
| tTG6 | Neural Tissue Transglutaminase |
| NCGS | Non-Celiac Gluten Sensitivity |
| CNS | Central Nervous System |
| BBB | Blood-Brain Barrier |
| HLA | Human leukocyte antigen |
| mRNA | messenger RNA |
| miRNA | micro RNA |
| tRNA | transfer RNA |
| dsDNA | double-stranded DNA |
| SIV | Simian Immunodeficiency Virus |
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| HD | Huntington’s Disease |
| ASD | Autism Spectrum Disorder |
| DS | Down’s Syndrome |
| IBD | Inflammatory Bowel Disease |
| IBS | Irritable Bowel Syndrome |
| ZO1 | Zona Occludens Protein 1 |
| PPARγ | Peroxisome proliferator activated receptor gamma |
| STAT3 | Signal transducer and activator of transcription 3 |
| c-MYC | MYC proto-oncogene BHLH transcription factor |
| NOTCH1 | Notch receptor 1 |
| KLF4 | Kruppel like factor 4 |
| NFκB | Nuclear factor kappa B |
| NOS2 | Nitric oxide synthase 2 |
| THC | Delta-9-Tetra-Hydrocannabinol |
| ES | Extracellular Structure |
| BMS | Biomolecular Condensate |
| EV | Extracellular Vesicle |
| FTD | Frontotemporal Dementia |
| ALS | Amyotrophic Lateral Sclerosis |
| MAPT | Microtubule-Associated Protein Tau |
References
- Cooke, W.T.; Smith, W.T. Neurological disorders associated with adult coeliac disease. Brain 1966, 89, 683–722. [Google Scholar] [CrossRef] [PubMed]
- Cooke, W.T.; Johnson, A.G.; Woolf, A.L. Vital staining and electron microscopy of the intramuscular nerve endings in the neuropathy of adult coeliac disease. Brain 1966, 89, 663–682. [Google Scholar] [CrossRef] [PubMed]
- Kinney, H.C.; Burger, P.C.; Hurwitz, B.J.; Hijmans, J.C.; Grant, J.P. Degeneration of the central nervous system associated with celiac disease. J. Neurol. Sci. 1982, 53, 9–22. [Google Scholar] [CrossRef]
- Ward, M.E.; Murphy, J.T.; Greenberg, G.R. Celiac disease and spinocerebellar degeneration with normal vitamin E status. Neurology 1985, 35, 1199. [Google Scholar] [CrossRef] [PubMed]
- Dohan, F.C.; Harper, E.H.; Clark, M.H.; Rodrigue, R.B.; Zigas, V. Is schizophrenia rare if grain is rare? Boil. Psychiatry 1984, 19, 385–399. [Google Scholar]
- Ashe, P.C.; Berry, M.; Boulton, A. Schizophrenia, a neurodegenerative disorder with neurodevelopmental antecedents. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2001, 25, 691–707. [Google Scholar] [CrossRef]
- Rao, J.; Chiappelli, J.; Kochunov, P.; Regenold, W.T.; Rapoport, S.I.; Hong, L.E. Is schizophrenia a neurodegenerative disease? Evidence from age-related decline of brain-derived neurotrophic factor in the brains of schizophrenia patients and matched nonpsychiatric controls. Neurodegener. Dis. 2014, 15, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, D.; Currò, M.; Ientile, R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 989–1003. [Google Scholar] [CrossRef]
- Sestak, K.; Fortgang, I. Celiac and Non-Celiac Forms of Gluten Sensitivity: Shifting Paradigms of an Old Disease. Br. Microbiol. Res. J. 2013, 3, 585–589. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [Green Version]
- Baizabal-Carvallo, J.F.; Jankovic, J. Movement disorders in autoimmune diseases. Mov. Disord. 2012, 27, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Aeschlimann, P.; Sanders, D.S.; Mäki, M.; Kaukinen, K.; Grünewald, R.; Bandmann, O.; Woodroofe, N.; Haddock, G.; Aeschlimann, D. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013, 80, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Vörös, P.; Sziksz, E.; Himer, L.; Ónody, A.; Pap, D.; Frivolt, K.; Szebeni, B.; Lippai, R.; Győrffy, H.; Fekete, A.; et al. Expression of PARK7 is increased in celiac disease. Virchows Archiv 2013, 463, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Gratch, D.; Pagano, B.; McDermott, K.; Huang, J.; Jian, J.; Bates, D.; A Sadiq, S. Transglutaminase-6 is an autoantigen in progressive multiple sclerosis and is upregulated in reactive astrocytes. Mult. Scler. J. 2016, 23, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Cascella, N.; Kryszak, D.; Gregory, P.; Fasano, D.L.K.A.; Eaton, W.W. Increased Prevalence of Transglutaminase 6 Antibodies in Sera From Schizophrenia Patients. Schizophr. Res. 2012, 136, S12. [Google Scholar] [CrossRef] [Green Version]
- Bavykina, I.A.; Zvyagin, A.A.; Petrova, I.V.; Nastausheva, T.L. Markers of gluten intolerance in children with autism spectrum disorders and Down’syndrome. Zhurnal Nevrol. Psikhiatrii Im. SS Korsakova 2018, 118, 64–68. [Google Scholar] [CrossRef]
- Bennabi, M.; Gaman, A.; Delorme, R.; Boukouaci, W.; Manier, C.; Scheid, I.; Mohammed, N.S.; Bengoufa, D.; Charron, D.; Krishnamoorthy, R.; et al. HLA-class II haplotypes and Autism Spectrum Disorders. Sci. Rep. 2018, 8, 7639. [Google Scholar] [CrossRef] [Green Version]
- Rahmoune, H.; Boutrid, N. Autism & Gluten: The Proof By Regression. Pediatr. Neurol. Briefs 2018, 32, 9. [Google Scholar] [CrossRef]
- Dickerson, F.B.; Stallings, C.; Origoni, A.; Vaughan, C.; Khushalani, S.; Alaedini, A.; Yolken, R. Markers of gluten sensitivity and celiac disease in bipolar disorder. Bipolar Disord. 2011, 13, 52–58. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, D.; A Grünewald, R.; Sanders, D.S.; Sharrack, B.; Woodroofe, M.N. GAD antibody-associated neurological illness and its relationship to gluten sensitivity. Acta Neurol. Scand. 2011, 123, 175–180. [Google Scholar] [CrossRef]
- Alaedini, A.; Okamoto, H.; Briani, C.; Wollenberg, K.; Shill, H.A.; Bushara, K.O.; Sander, H.W.; Green, P.H.R.; Hallett, M.; Latov, N. Immune cross-reactivity in celiac disease: anti-gliadin antibodies bind to neuronal synapsin I. J. Immunol. 2007, 178, 6590–6595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjivassiliou, M.; Boscolo, S.; Davies–Jones, G.A.; Grünewald, R.A.; Not, T.; Sanders, D.S.; Simpson, J.E.; Tongiorgi, E.; Williamson, C.A.; Woodroofe, N. The humoral response in the pathogenesis of gluten ataxia. Neurology 2002, 58, 1221–1226. [Google Scholar] [CrossRef]
- Hu, W.T.; Murray, J.A.; Greenaway, M.C.; Parisi, J.E.; Josephs, K.A. Cognitive Impairment and Celiac Disease. Arch. Neurol. 2006, 63, 1440–1446. [Google Scholar] [CrossRef] [Green Version]
- Zis, P.; Rao, D.G.; Sarrigiannis, P.G.; Aeschlimann, P.; Aeschlimann, D.; Rigby, R.; Grünewald, R.; Hadjivassiliou, M. Transglutaminase 6 antibodies in gluten neuropathy. Dig. Liver Dis. 2017, 49, 1196–1200. [Google Scholar] [CrossRef]
- Aziz, I.; Hadjivassiliou, M.; Sanders, D.S. The spectrum of noncoeliac gluten sensitivity. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Non-celiac gluten sensitivity triggers gut dysbiosis, neuroinflammation, gut-brain axis dysfunction, and vulnerability for dementia. CNS Neurol. Disord. Drug Targets 2015, 14, 110–131. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Sanders, D.S.; A Grünewald, R.; Woodroofe, N.; Boscolo, S.; Aeschlimann, D. Gluten sensitivity: from gut to brain. Lancet Neurol. 2010, 9, 318–330. [Google Scholar] [CrossRef]
- Lionetti, E.; Leonardi, S.; Franzonello, C.; Mancardi, M.; Ruggieri, M.; Catassi, C. Gluten Psychosis: Confirmation of a New Clinical Entity. Nutrients 2015, 7, 5532–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscolo, S.; Sarich, A.; Lorenzon, A.; Passoni, M.; Rui, V.; Stebel, M.; Sblattero, D.; Marzari, R.; Hadjivassiliou, M.; Tongiorgi, E. Gluten Ataxia: Passive Transfer in a Mouse Model. Ann. N.Y. Acad. Sci. 2007, 1107, 319–328. [Google Scholar] [CrossRef]
- Tarlac, V.; Kelly, L.; Nag, N.; Allen-Graham, J.; Anderson, R.P.; Storey, E. HLA-DR3-DQ2 Mice Do Not Develop Ataxia in the Presence of High Titre Anti-gliadin Antibodies. Cerebellum 2012, 12, 370–376. [Google Scholar] [CrossRef]
- Mazumdar, K.; Álvarez, X.; Borda, J.T.; Dufour, J.; Martin, E.; Bethune, M.T.; Khosla, C.; Sestak, K. Visualization of Transepithelial Passage of the Immunogenic 33-Residue Peptide from α-2 Gliadin in Gluten-Sensitive Macaques. PLoS ONE 2010, 5, e10228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, M.; Chow, C.-E.T.; Ryan, C.N.; Chan, L.S.; Dufour, J.; Aye, P.P.; Blanchard, J.; Moehs, C.P.; Sestak, K. Dietary Gluten-Induced Gut Dysbiosis Is Accompanied by Selective Upregulation of microRNAs with Intestinal Tight Junction and Bacteria-Binding Motifs in Rhesus Macaque Model of Celiac Disease. Nutrients 2016, 8, 684. [Google Scholar] [CrossRef] [PubMed]
- Sestak, K.; Conroy, L.; Aye, P.P.; Mehra, S.; Doxiadis, G.G.; Kaushal, D. Improved Xenobiotic Metabolism and Reduced Susceptibility to Cancer in Gluten-Sensitive Macaques upon Introduction of a Gluten-Free Diet. PLoS ONE 2011, 6, e18648. [Google Scholar] [CrossRef] [PubMed]
- Du Pré, M.F.; Blazevski, J.; Dewan, A.E.; Stamnaes, J.; Kanduri, C.; Sandve, G.K.; Johannesen, M.K.; Lindstad, C.B.; Hnida, K.; Fugger, L.; et al. B cell tolerance and antibody production to the celiac disease autoantigen transglutaminase 2. J. Exp. Med. 2019, 217. [Google Scholar] [CrossRef]
- Klöck, C.; DiRaimondo, T.R.; Khosla, C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 2012, 34, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Sulic, A.-M.; Kurppa, K.; Rauhavirta, T.; Kaukinen, K.; Lindfors, K. Transglutaminase as a therapeutic target for celiac disease. Expert Opin. Ther. Targets 2014, 19, 335–348. [Google Scholar] [CrossRef]
- Gentile, V.; Cooper, A.J.L. Transglutaminases—Possible drug targets in human diseases. Curr. Drug Target -CNS Neurol. Disord. 2004, 3, 99–104. [Google Scholar] [CrossRef]
- Schmid, A.W.; Condemi, E.; Tuchscherer, G.; Chiappe, D.; Mutter, M.; Vogel, H.; Moniatte, M.; Tsybin, Y.O. Tissue Transglutaminase-mediated Glutamine Deamidation of β-Amyloid Peptide Increases Peptide Solubility, Whereas Enzymatic Cross-linking and Peptide Fragmentation May Serve as Molecular Triggers for Rapid Peptide Aggregation. J. Boil. Chem. 2011, 286, 12172–12188. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; de Jager, M.; Bakker, E.N.; Drukarch, B. Tissue transglutaminase in Alzheimer’s disease: involvement in pathogenesis and its potential as a therapeutic target. J. Alzheimers Dis. 2014, 42, S289–S303. [Google Scholar] [CrossRef]
- Junn, E.; Ronchetti, R.D.; Quezado, M.M.; Kim, S.Y.; Mouradian, M.M. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; Verhaar, R.; Andringa, G.; Bol, J.G.; Cras, P.; Shan, L.; Hoozemans, J.J.; Drukarch, B. Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson’s disease brain. Brain Pathol. 2011, 21, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Chung, K.C. New insight into transglutaminase 2 and link to neurodegenerative diseases. BMB Rep. 2018, 51, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, N.; Mehta, K. Transglutaminase-2: evolution from pedestrian protein to a promising therapeutic target. Amino Acids 2016, 49, 425–439. [Google Scholar] [CrossRef] [PubMed]
- DiRaimondo, T.R.; Klöck, C.; Khosla, C. Interferon-γ activates transglutaminase 2 via a phosphatidylinositol-3-kinase-dependent pathway: implications for celiac sprue therapy. J. Pharmacol. Exp. Ther. 2012, 341, 104–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, R.; Khosla, C. Substrates, inhibitors, and probes of mammalian transglutaminase 2. Anal. Biochem. 2020, 591, 113560. [Google Scholar] [CrossRef]
- Aaron, L.; Torsten, M.; Patricia, W. Autoimmunity in celiac disease: Extra-intestinal manifestations. Autoimmun. Rev. 2019, 18, 241–246. [Google Scholar] [CrossRef]
- Lerner, A.; Neidhöfer, S.; Matthias, T. The Gut Microbiome Feelings of the Brain: A Perspective for Non-Microbiologists. Microorganisms 2017, 5, 66. [Google Scholar] [CrossRef]
- Magni, S.; Comani, G.B.; Elli, L.; Vanessi, S.; Ballarini, E.; Nicolini, G.; Rusconi, M.; Castoldi, M.; Meneveri, R.; Muckenthaler, M.U.; et al. miRNAs Affect the Expression of Innate and Adaptive Immunity Proteins in Celiac Disease. Am. J. Gastroenterol. 2014, 109, 1662–1674. [Google Scholar] [CrossRef]
- Vaira, V.; Roncoroni, L.; Barisani, D.; Gaudioso, G.; Bosari, S.; Bulfamante, G.; Doneda, L.; Conte, D.; Tomba, C.; Bardella, M.T.; et al. microRNA profiles in coeliac patients distinguish different clinical phenotypes and are modulated by gliadin peptides in primary duodenal fibroblasts. Clin. Sci. 2013, 126, 417–423. [Google Scholar] [CrossRef]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs Are Differentially Expressed in Ulcerative Colitis and Alter Expression of Macrophage Inflammatory Peptide-2α. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef]
- Vaira, V.; Gaudioso, G.; Laginestra, M.A.; Terrasi, A.; Agostinelli, C.; Bosari, S.; Di Sabatino, A.; Vanoli, A.; Paulli, M.; Ferrero, S.; et al. Deregulation of miRNAs-cMYC circuits is a key event in refractory celiac disease type-2 lymphomagenesis. Clin. Sci. 2020, 134, 1151–1166. [Google Scholar] [CrossRef] [PubMed]
- Capuano, M.; Iaffaldano, L.; Tinto, N.; Montanaro, D.; Capobianco, V.; Izzo, V.; Tucci, F.; Troncone, G.; Greco, L.; Sacchetti, L. MicroRNA-449a Overexpression, Reduced NOTCH1 Signals and Scarce Goblet Cells Characterize the Small Intestine of Celiac Patients. PLoS ONE 2011, 6, e29094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amr, K.; Bayoumi, F.; Eissa, E.; Abu-Zekry, M. Circulating microRNAs as potential non-invasive biomarkers in pediatric patients with celiac disease. Eur. Ann. Allergy Clin. Immunol. 2019, 51, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, L.C.; Kumar, V.; Torben, W.; Stouwe, C.V.; Winsauer, P.; Amedee, A.; Molina, P.E.; Mohan, M. Chronic Administration of Δ9-Tetrahydrocannabinol Induces Intestinal Anti-Inflammatory MicroRNA Expression during Acute Simian Immunodeficiency Virus Infection of Rhesus Macaques. J. Virol. 2014, 89, 1168–1181. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Torben, W.; Mansfield, J.; Alvarez, X.; Stouwe, C.V.; Li, J.; Byrareddy, S.N.; Didier, P.J.; Pahar, B.; Molina, P.E.; et al. Cannabinoid Attenuation of Intestinal Inflammation in Chronic SIV-Infected Rhesus Macaques Involves T Cell Modulation and Differential Expression of Micro-RNAs and Pro-inflammatory Genes. Front. Immunol. 2019, 10, 914. [Google Scholar] [CrossRef]
- De Palma, G.; Cinova, J.; Stepankova, R.; Tuckova, L.; Sanz, Y. Pivotal Advance: Bifidobacteria and Gram-negative bacteria differentially influence immune responses in the proinflammatory milieu of celiac disease. J. Leukoc. Boil. 2009, 87, 765–778. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2014, 30, 350–358. [Google Scholar] [CrossRef]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2017, 38, 121–132. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Vetuschi, A.; Pompili, S.; Gaudio, E.; Latella, G.; Sferra, R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8839–8848. [Google Scholar]
- Luciani, A.; Villella, V.R.; Vasaturo, A.; Giardino, I.; Pettoello-Mantovani, M.; Guido, S.; Cexus, O.N.; Peake, N.; Londei, M.; Quaratino, S.; et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 2010, 59, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.L.P.; Matoso, R.D.O.; Teixeira, L.G.; Menezes, Z.; Pereira, S.S.; Alves, A.C.; Batista, N.V.; Faria, A.C.; Cara, D.C.; Ferreira, A.V.M.; et al. Gluten-free diet reduces adiposity, inflammation and insulin resistance associated with the induction of PPAR-alpha and PPAR-gamma expression. J. Nutr. Biochem. 2013, 24, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Sziksz, E.; Molnár, K.; Lippai, R.; Pap, D.; Ónody, A.; Veres-Székely, A.; Vörös, P.; Szabo, D.; Győrffy, H.; Veres, G.; et al. Peroxisome proliferator-activated receptor-γ and thymic stromal lymphopoietin are involved in the pathophysiology of childhood coeliac disease. Virchows Archiv 2014, 465, 385–393. [Google Scholar] [CrossRef]
- Kang, D.-W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced Incidence of Prevotella and Other Fermenters in Intestinal Microflora of Autistic Children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Lou, X.; Zhang, F.; Xu, G. Nuclear Receptors in the Pathogenesis and Management of Inflammatory Bowel Disease. Mediat. Inflamm. 2019, 2019, 2624941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, E.; Viscido, A.; Castelli, V.; Maggiani, C.; D’Angelo, M.; Di Giacomo, E.; Antonosante, A.; Picarelli, A.; Frieri, G. Mesalazine treatment in organotypic culture of celiac patients: Comparative study with gluten free diet. J. Cell. Physiol. 2018, 233, 4383–4390. [Google Scholar] [CrossRef]
- Sehgal, P.; Colombel, J.-F.; Aboubakr, A.; Narula, N. Systematic review: safety of mesalazine in ulcerative colitis. Aliment. Pharmacol. Ther. 2018, 47, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Mansfield, J.; Fan, R.; MacLean, A.G.; Li, J.; Mohan, M. miR-130a and miR-212 Disrupt the Intestinal Epithelial Barrier through Modulation of PPARγ and Occludin Expression in Chronic Simian Immunodeficiency Virus–Infected Rhesus Macaques. J. Immunol. 2018, 200, 2677–2689. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, X.; Sestak, K.; Byrareddy, S.N.; Mohan, M. Long Term Delta-9-tetrahydrocannabinol Administration Inhibits Proinflammatory Responses in Minor Salivary Glands of Chronically Simian Immunodeficieny Virus Infected Rhesus Macaques. Viruses 2020, 12, 713. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell Boil. 2014, 206, 579–588. [Google Scholar] [CrossRef]
- Navarro, M.G.-J.; Kashida, S.; Chouaib, R.; Souquere, S.; Pierron, G.; Weil, D.; Gueroui, Z. RNA is a critical element for the sizing and the composition of phase-separated RNA-protein condensates. Nat. Commun. 2019, 10, 3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittag, T.; Parker, R. Multiple Modes of Protein-Protein Interactions Promote RNP Granule Assembly. J. Mol. Boil. 2018, 430, 4636–4649. [Google Scholar] [CrossRef] [PubMed]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 7439. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-Oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, T.; Yamamoto, T.; Sugamata, M.; Okumura, H.; Ueno, Y. The process of ultrastructural changes from nuclei to apoptotic body. Virchows. Archiv. 1998, 433, 443–447. [Google Scholar] [CrossRef]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Boil. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [Green Version]
- Welch, J.L.; Kaddour, H.; Winchester, L.; Fletcher, C.V.; Stapleton, J.T.; Okeoma, C.M. Semen Extracellular Vesicles From HIV-1–Infected Individuals Inhibit HIV-1 Replication In Vitro, and Extracellular Vesicles Carry Antiretroviral Drugs In Vivo. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 83, 90–98. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 1–9. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Altmeyer, M. Phase Separation: Linking Cellular Compartmentalization to Disease. Trends Cell Boil. 2016, 26, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Dobra, I.; Pankivskyi, S.; Samsonova, A.; Pastré, D.; Hamon, L. Relation Between Stress Granules and Cytoplasmic Protein Aggregates Linked to Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2018, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Raposo, G.; Théry, C. Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Boil. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Buzas, E.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological Functions and Biogenesis of Secreted Bacterial Outer Membrane Vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [Green Version]
- Avila-Calderón, E.D.; Villanueva, M.G.A.; Cancino-Diaz, J.C.; López-Villegas, E.O.; Sriranganathan, N.; Boyle, S.M.; Contreras-Rodríguez, A. Roles of bacterial membrane vesicles. Arch. Microbiol. 2014, 197, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nordgreen, J.; Munsterhjelm, C.; Aae, F.; Popova, A.; Boysen, P.; Ranheim, B.; Heinonen, M.; Raszplewicz, J.; Piepponen, T.P.; Lervik, A.; et al. The effect of lipopolysaccharide (LPS) on inflammatory markers in blood and brain and on behavior in individually-housed pigs. Physiol. Behav. 2018, 195, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Costinean, S.; Croce, C.M.; Brasier, A.R.; Merwat, S.; Larson, S.A.; Basra, S.; Verne, G.N. MicroRNA 29 targets nuclear factor-κB-repressing factor and Claudin 1 to increase intestinal permeability. Gastroenterology 2014, 148, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, A.; Arleevskaya, M.; Schmiedl, A.; Matthias, T. Microbes and Viruses Are Bugging the Gut in Celiac Disease. Are They Friends or Foes? Front. Microbiol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Hardy, J.; Fischbeck, K.H. Toxic Proteins in Neurodegenerative Disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Davis, D.; Yuan, H.; Liang, F.-X.; Yang, Y.-M.; Westley, J.; Petzold, C.; Dancel-Manning, K.; Deng, Y.; Sall, J.; Sehgal, P.B. Human Antiviral Protein MxA Forms Novel Metastable Membraneless Cytoplasmic Condensates Exhibiting Rapid Reversible Tonicity-Driven Phase Transitions. J. Virol. 2019, 93, 22. [Google Scholar] [CrossRef]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.; Whelan, P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. mBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozelle, D.K.; Filone, C.M.; Kedersha, N.; Connor, J.H. Activation of Stress Response Pathways Promotes Formation of Antiviral Granules and Restricts Virus Replication. Mol. Cell. Boil. 2014, 34, 2003–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-J.; Wang, B.; Kodali, M.C.; Chen, C.; Kim, E.; Patters, B.J.; Lan, L.; Kumar, S.; Wang, X.; Yue, J.; et al. In Vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflam. 2018, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2013, 21, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Halverson, K.; Fraser, P.E.; Kirschner, D.A.; Lansbury, P.T., Jr. Molecular determinants of amyloid deposition in Alzheimer’s disease: conformational studies of synthetic beta-protein fragments. Biochemistry 1990, 29, 2639–2644. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.Y.; et al. Exosome-associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J. Boil. Chem. 2011, 287, 3842–3849. [Google Scholar] [CrossRef] [Green Version]
- Willing, B.P.; Russell, S.L.; Finlay, B.B. Shifting the balance: antibiotic effects on host–microbiota mutualism. Nat. Rev. Genet. 2011, 9, 233–243. [Google Scholar] [CrossRef]
- Everard, A.; Cani, P.D. Diabetes, obesity and gut microbiota. Best Pr. Res. Clin. Gastroenterol. 2013, 27, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.-S.; Ban, M.; Choi, E.-J.; Moon, H.-G.; Jeon, J.-S.; Kim, D.-K.; Park, S.-K.; Jeon, S.G.; Roh, T.-Y.; Myung, S.-J.; et al. Extracellular Vesicles Derived from Gut Microbiota, Especially Akkermansia muciniphila, Protect the Progression of Dextran Sulfate Sodium-Induced Colitis. PLoS ONE 2013, 8, e76520. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Torchia, M.L.G.; Lawson, G.W.; Karp, C.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Elsen, L.W.J.V.D.; Poyntz, H.C.; Weyrich, L.S.; Young, W.; E Forbes-Blom, E. Embracing the gut microbiota: the new frontier for inflammatory and infectious diseases. Clin. Transl. Immunol. 2017, 6, e125. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Xu, J.; Yang, M.; Kosterin, P.; Salzberg, B.M.; Milovanova, T.N.; Bhopale, V.M.; Thom, S.R. Carbon monoxide inhalation increases microparticles causing vascular and CNS dysfunction. Toxicol. Appl. Pharmacol. 2013, 273, 410–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Milovanova, T.N.; Bogush, M.; Uzun, G.; Bhopale, V.M.; Thom, S.R. Microparticle enlargement and altered surface proteins after air decompression are associated with inflammatory vascular injuries. J. Appl. Physiol. 2011, 112, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Kosterin, P.; Salzberg, B.M.; Milovanova, T.N.; Bhopale, V.M.; Thom, S.R. Microparticles generated by decompression stress cause central nervous system injury manifested as neurohypophysial terminal action potential broadening. J. Appl. Physiol. 2013, 115, 1481–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Stoica, B.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflam. 2017, 14, 47. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Shan, A.; Wei, Z.; Xu, B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2018, 503, 2611–2618. [Google Scholar] [CrossRef]
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noël, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410. [Google Scholar] [CrossRef]
- Fujita, Y.; Kadota, T.; Araya, J.; Ochiya, T.; Kuwano, K. Clinical Application of Mesenchymal Stem Cell-Derived Extracellular Vesicle-Based Therapeutics for Inflammatory Lung Diseases. J. Clin. Med. 2018, 7, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.S.; Kim, J.O.; Ha, D.H.; Yi, Y.W. Exosomes derived from human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis. Stem Cell Res. Ther. 2018, 9, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chopp, M.; Meng, Y.; Katakowski, M.; Xin, H.; Mahmood, A.; Xiong, Y. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg. 2015, 122, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ye, Y.; Su, X.; He, J.; Bai, W.; He, X. MSCs-Derived Exosomes and Neuroinflammation, Neurogenesis and Therapy of Traumatic Brain Injury. Front. Cell. Neurosci. 2017, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic Administration of Exosomes Released from Mesenchymal Stromal Cells Promote Functional Recovery and Neurovascular Plasticity After Stroke in Rats. Br. J. Pharmacol. 2013, 33, 1711–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomi, G.; Surbek, D.; Haesler, V.; Joerger-Messerli, M.; Schoeberlein, A. Exosomes derived from umbilical cord mesenchymal stem cells reduce microglia-mediated neuroinflammation in perinatal brain injury. Stem Cell Res. Ther. 2019, 10, 105. [Google Scholar] [CrossRef]
- Long, Q.; Upadhya, D.; Hattiangady, B.; Kim, D.-K.; An, S.Y.; Shuai, B.; Prockop, D.J.; Shetty, A.K. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc. Natl. Acad. Sci. USA 2017, 114, E3536–E3545. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.M.; Mascarenhas, L.A.; Sumagin, R. Extracellular vesicles regulate immune responses and cellular function in intestinal inflammation and repair. Tissue Barriers 2018, 6, e1431038. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Feldbrügge, L.; Csizmadia, E.; Mitsuhashi, M.; Robson, S.C.; Moss, A.C. Luminal Extracellular Vesicles (EVs) in Inflammatory Bowel Disease (IBD) Exhibit Proinflammatory Effects on Epithelial Cells and Macrophages. Inflamm. Bowel Dis. 2016, 22, 1587–1595. [Google Scholar] [CrossRef]
- Kanninen, K.M.; Bister, N.; Koistinaho, J.; Malm, T. Exosomes as new diagnostic tools in CNS diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 403–410. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Krajmalnik-Brown, R.; Porazinska, R.L.; Weiss, S.J.; Knight, R. Toward effective probiotics for autism and other neurodevelopmental disorders. Cell 2013, 155, 1446–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardi, A.; Totino, V.; Cacciotti, F.; Iebba, V.; Neroni, B.; Bonfiglio, G.; Trancassini, M.; Passariello, C.; Pantanella, F.; Schippa, S. Rebuilding the Gut Microbiota Ecosystem. Int. J. Environ. Res. Public Health 2018, 15, 1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Occludin (OCLN) protein expression is significantly decreased in duodenal epithelium of celiac macaques. All panels involve triple labels with OCLN (green), cytokeratin (red) and Topro3 for nuclear staining (blue). Colocalization appears yellow. Note the marked loss of occludin protein (white arrow) staining in the duodenal epithelium (DE) of the celiac macaque (A). In contrast, occludin protein (B) staining is intense in the DE of the control macaque. Magnification for both panels is 40×. Occludin (Cat#LS-B2437) antibody that cross reacts with the rhesus macaque was purchased from Lifespan Biosciences, Seattle, WA, USA.
Figure 1.
Occludin (OCLN) protein expression is significantly decreased in duodenal epithelium of celiac macaques. All panels involve triple labels with OCLN (green), cytokeratin (red) and Topro3 for nuclear staining (blue). Colocalization appears yellow. Note the marked loss of occludin protein (white arrow) staining in the duodenal epithelium (DE) of the celiac macaque (A). In contrast, occludin protein (B) staining is intense in the DE of the control macaque. Magnification for both panels is 40×. Occludin (Cat#LS-B2437) antibody that cross reacts with the rhesus macaque was purchased from Lifespan Biosciences, Seattle, WA, USA.

Figure 2.
Immunopathology of central nervous system (CNS) disease in celiac disease patients and potential beneficial (treatment) role of phytocannabinoids as nutraceuticals to mitigate gluten induced intestinal and CNS inflammation. THC—delta-9-tetrahydrocannabinol; LPS—lipopolysaccharide; TG6—transglutaminase-6; TLR—Toll-like receptor; APC—antigen presenting cell.
Figure 2.
Immunopathology of central nervous system (CNS) disease in celiac disease patients and potential beneficial (treatment) role of phytocannabinoids as nutraceuticals to mitigate gluten induced intestinal and CNS inflammation. THC—delta-9-tetrahydrocannabinol; LPS—lipopolysaccharide; TG6—transglutaminase-6; TLR—Toll-like receptor; APC—antigen presenting cell.

Figure 3.
Peroxisome proliferator activator receptor gamma (PPARγ) protein expression is significantly decreased in DE of celiac macaques. All panels involve triple labels with PPARγ (green), cytokeratin (red) and Topro3 for nuclear staining (blue). Colocalization appears yellow. Note the marked loss PPARγ (A) (white arrow) staining in the DE of the celiac macaque. In contrast, PPARγ protein (B) staining is intense in the DE of the control macaque. Magnification for both panels is 40×. PPARγ (Cat#LS-B651-50) antibody that cross reacts with the rhesus macaque was purchased from Lifespan Biosciences, Seattle, WA, USA.
Figure 3.
Peroxisome proliferator activator receptor gamma (PPARγ) protein expression is significantly decreased in DE of celiac macaques. All panels involve triple labels with PPARγ (green), cytokeratin (red) and Topro3 for nuclear staining (blue). Colocalization appears yellow. Note the marked loss PPARγ (A) (white arrow) staining in the DE of the celiac macaque. In contrast, PPARγ protein (B) staining is intense in the DE of the control macaque. Magnification for both panels is 40×. PPARγ (Cat#LS-B651-50) antibody that cross reacts with the rhesus macaque was purchased from Lifespan Biosciences, Seattle, WA, USA.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155407
AMA Style
Mohan M, Okeoma CM, Sestak K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. International Journal of Molecular Sciences. 2020; 21(15):5407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155407
Chicago/Turabian StyleMohan, Mahesh, Chioma M. Okeoma, and Karol Sestak. 2020. "Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies" International Journal of Molecular Sciences 21, no. 15: 5407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155407
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.