Antimicrobial Activity of Small Synthetic Peptides Based on the Marine Peptide Turgencin A: Prediction of Antimicrobial Peptide Sequences in a Natural Peptide and Strategy for Optimization of Potency

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
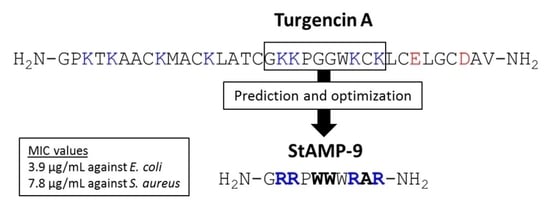
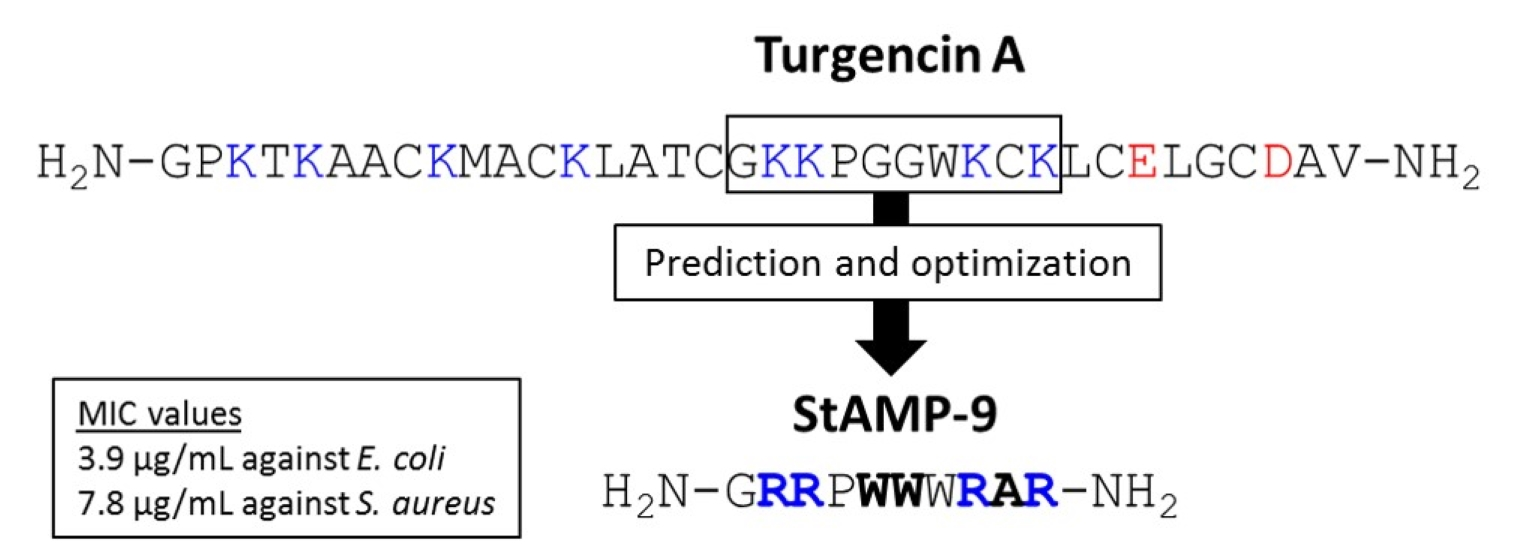
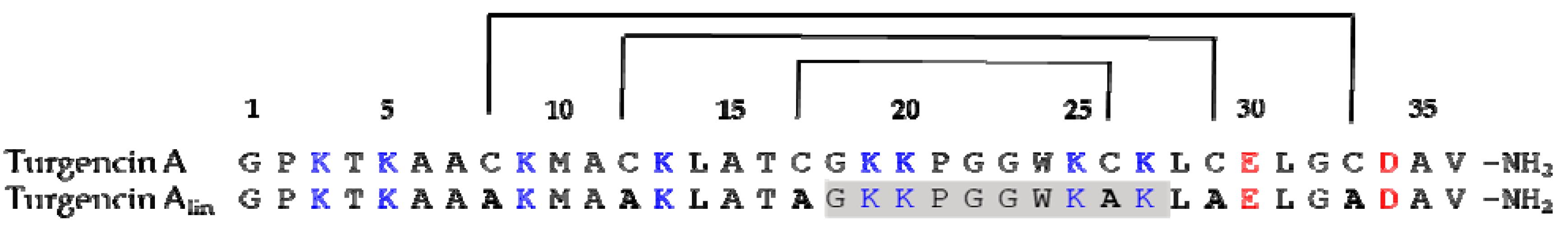
2.1. Sequence Analysis and AMP Prediction
2.2. Peptide Design and Antibacterial Screening
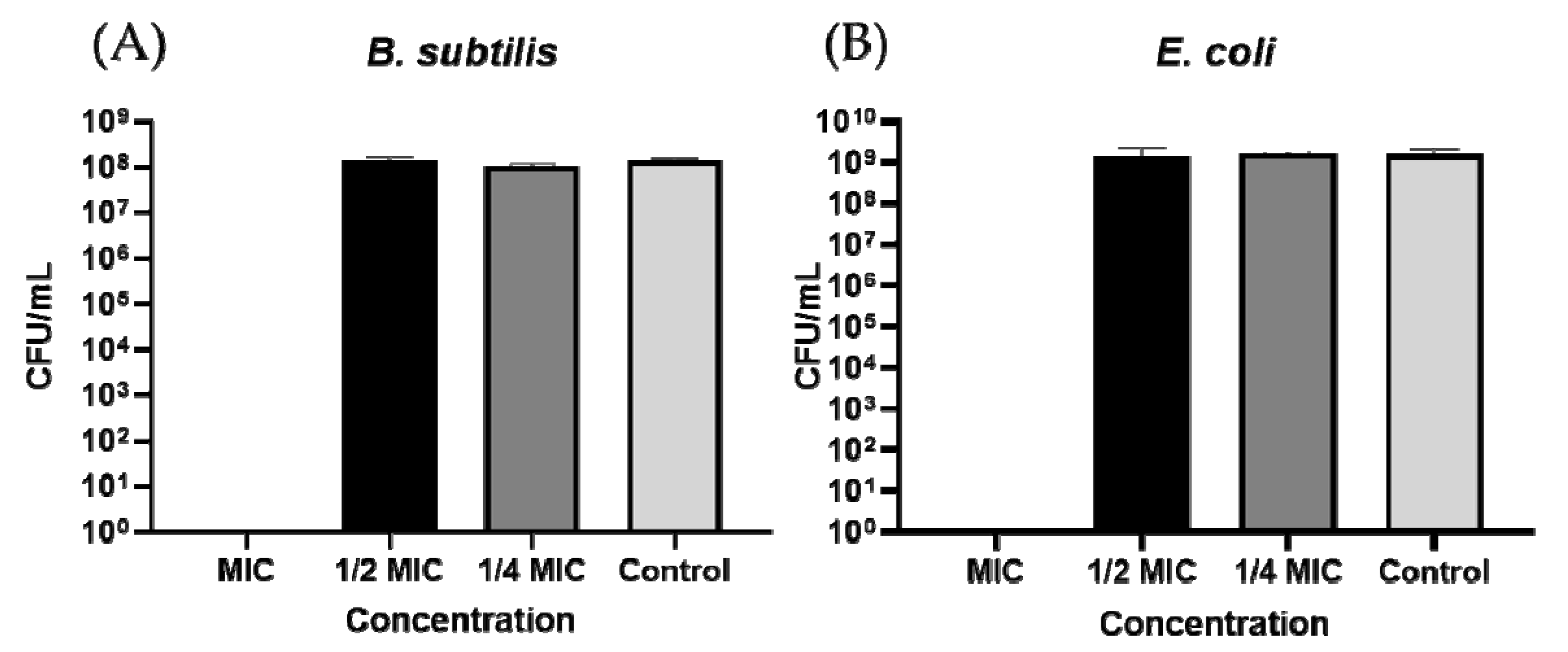
2.2.1. Bacterial Killing Experiments
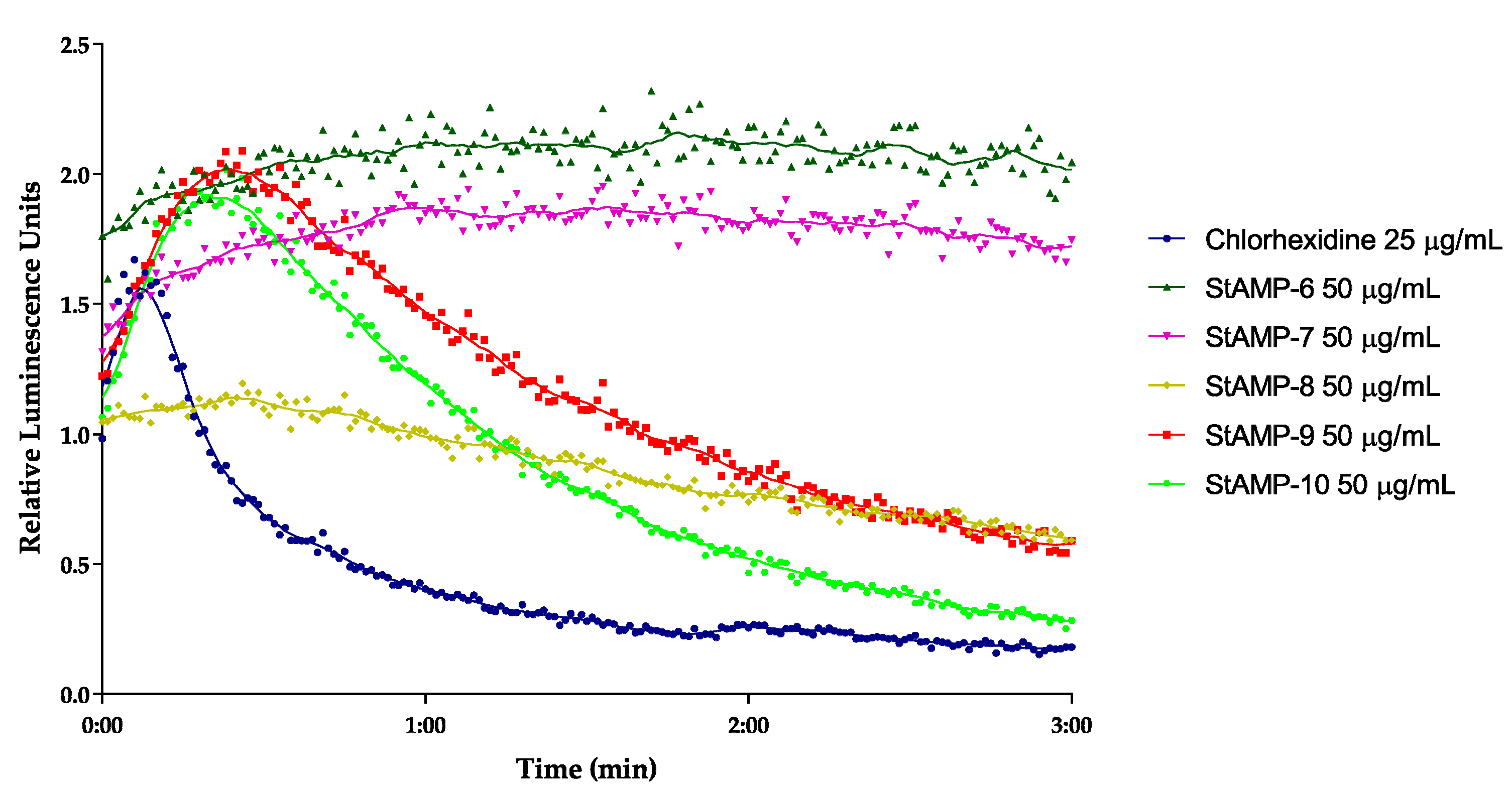
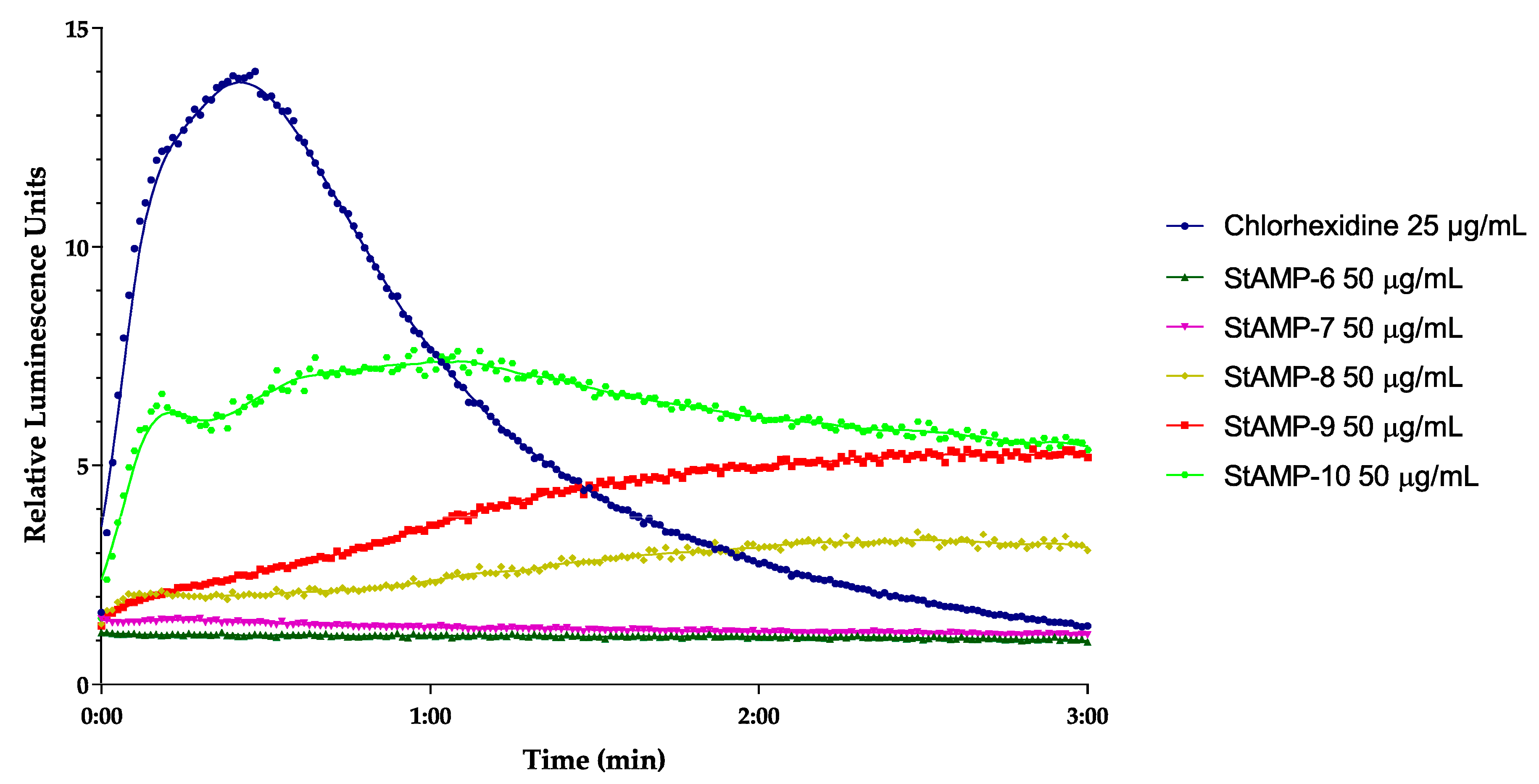
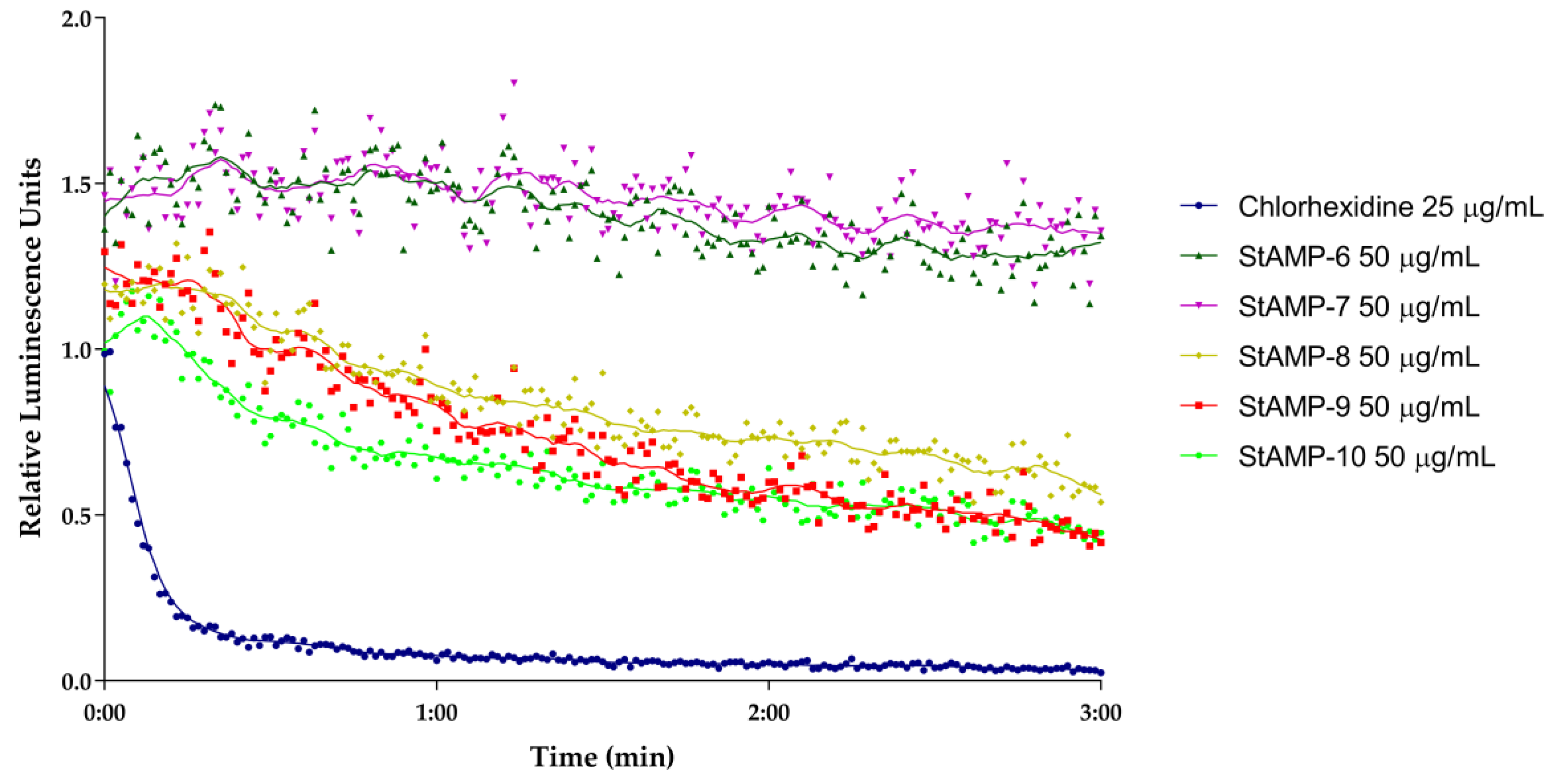
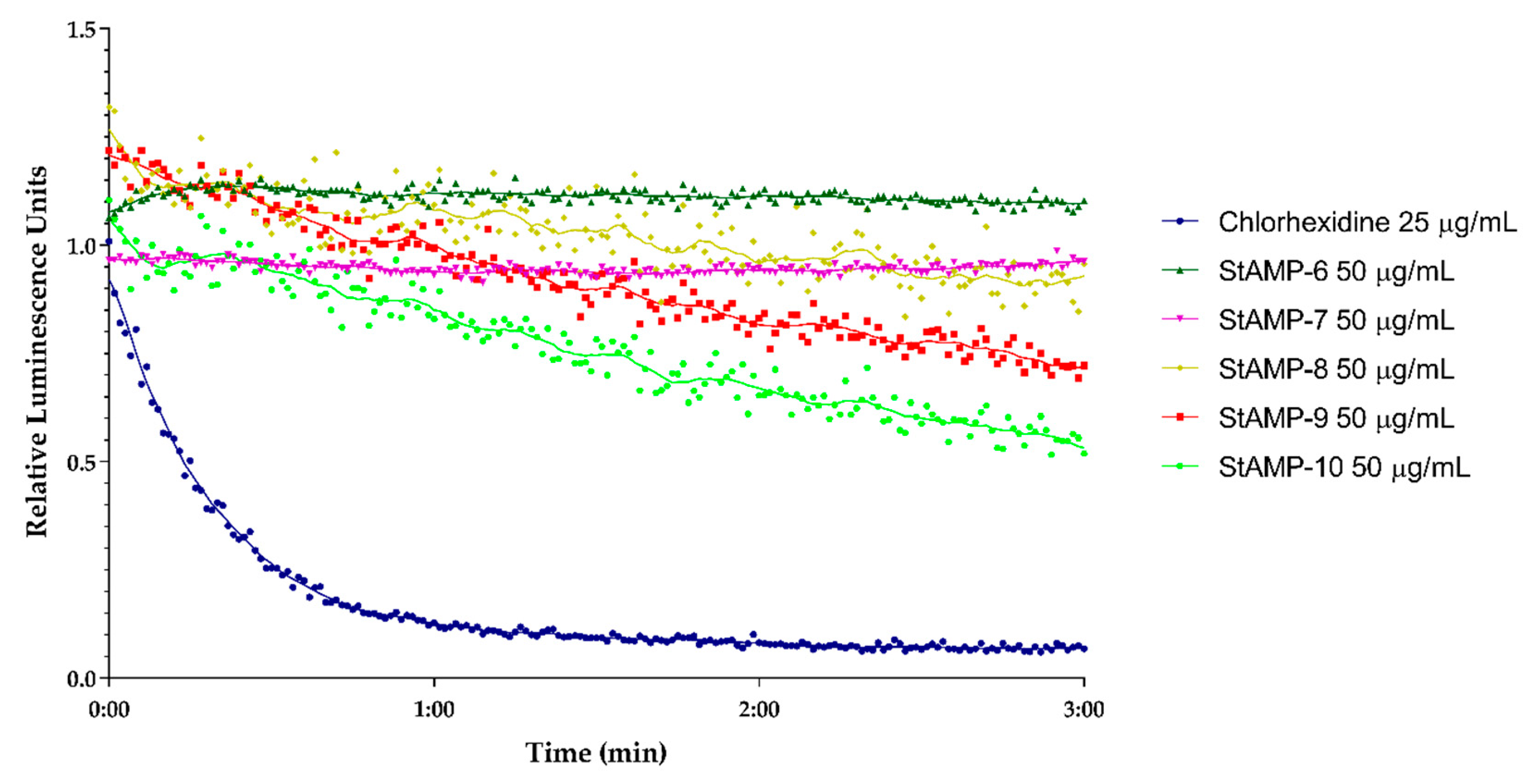
2.2.2. Membrane Integrity and Viability Investigations
2.3. Antifungal Activity
2.4. Hemolytic, Cytotoxic and Anti-Inflammatory Properties
3. Materials and Methods
3.1. Sequence Analysis and Peptide Design
3.2. Peptide Synthesis
3.3. Peptide Purification and Verification
3.4. Antibacterial Assay (Growth Inhibition)
3.5. Real-Time Assay Measuring Immediate Bacteria Membrane Disruption
3.6. Real-Time Assay Measuring Immediate Bacterial Cell Viability
3.7. Antifungal Assay
3.8. Hemolytic Activity Assay
3.9. Human Cell Viability Assay
3.10. Anti-Inflammatory Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hassan, M.; Kjos, M.; Nes, I.F.; Diep, D.B.; Lotfipour, F. Natural antimicrobial peptides from bacteria: Characteristics and potential applications to fight against antibiotic resistance. J. Appl. Microbiol. 2012, 113, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Domalaon, R.; Zhanel, G.G.; Schweizer, F. Short antimicrobial peptides and peptide scaffolds as promising antibacterial agents. Curr. Top. Med. Chem. 2016, 16, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govender, T.; Kruger, H.G.; Torre, B.G.; Albericio, F. Short AntiMicrobial Peptides (SAMPs) as a class of extraordinary promising therapeutic agents. J. Pept. Sci. 2016, 22, 438–451. [Google Scholar] [CrossRef] [PubMed]
- DeNegre, A.A.; Ndeffo Mbah, M.L.; Myers, K.; Fefferman, N.H. Emergence of antibiotic resistance in immunocompromised host populations: A case study of emerging antibiotic resistant tuberculosis in AIDS patients. PLoS ONE 2019, 14, e0212969. [Google Scholar] [CrossRef] [PubMed]
- Dumford, D.M.; Skalweit, M. Antibiotic-resistant infections and treatment challenges in the immunocompromised host. Infect. Dis. Clin. N. Am. 2016, 30, 465–489. [Google Scholar] [CrossRef] [PubMed]
- Teillant, A.; Gandra, S.; Barter, D.; Morgan, D.J.; Laxminarayan, R. Potential burden of antibiotic resistance on surgery and cancer chemotherapy antibiotic prophylaxis in the USA: A literature review and modelling study. Lancet Infect. Dis. 2015, 15, 1429–1437. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance; HM Government and the Wellcome Trust: London, UK, 2016.
- Simpkin, V.L.; Renwick, M.J.; Kelly, R.; Mossialos, E. Incentivising innovation in antibiotic drug discovery and development: Progress, challenges and next steps. J. Antibiot. 2017, 70, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [Green Version]
- Shabir, U.; Ali, S.; Magray, A.R.; Ganai, B.A.; Firdous, P.; Hassan, T.; Nazir, R. Fish antimicrobial peptides (AMP’s) as essential and promising molecular therapeutic agents: A review. Microb. Pathog. 2018, 114, 50–56. [Google Scholar] [CrossRef]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011, 40, 387–397. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Semreen, M.H.; El-Gamal, M.I.; Abdin, S.; Alkhazraji, H.; Kamal, L.; Hammad, S.; El-Awady, F.; Waleed, D.; Kourbaj, L. Recent updates of marine antimicrobial peptides. Saudi Pharm. J. 2018, 26, 396–409. [Google Scholar] [CrossRef]
- Arias, M.; Piga, K.B.; Hyndman, E.M.; Vogel, H.J. Improving the activity of Trp-rich antimicrobial peptides by Arg/Lys substitutions and changing the length of cationic residues. Biomolecules 2018, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Tincu, J.A.; Taylor, S.W. Antimicrobial peptides from marine invertebrates. Antimicrob. Agents Chemother. 2004, 48, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M. Antimicrobial peptides under clinical trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef]
- Mikut, R.; Ruden, S.; Reischl, M.; Breitling, F.; Volkmer, R.; Hilpert, K. Improving short antimicrobial peptides despite elusive rules for activity. BBA Biomembr. 2016, 1858, 1024–1033. [Google Scholar] [CrossRef]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E.W. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef]
- Knappe, D.; Henklein, P.; Hoffmann, R.; Hilpert, K. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob. Agents Chemother. 2010, 54, 4003–4005. [Google Scholar] [CrossRef] [Green Version]
- Strøm, M.B.; Rekdal, Ø.; Svendsen, J.S. The effects of charge and lipophilicity on the antibacterial activity of undecapeptides derived from bovine lactoferricin. J. Pept. Sci. 2002, 8, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, V.S.; Blencke, H.-M.; Benincasa, M.; Haug, T.; Eksteen, J.J.; Styrvold, O.B.; Scocchi, M.; Stensvåg, K. Structure-activity relationships of the antimicrobial peptide arasin 1-and mode of action studies of the N-terminal, proline-rich region. PLoS ONE 2013, 8, e53326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, I.K.Ø.; Isaksson, J.; Poth, A.G.; Hansen, K.Ø.; Andersen, A.J.C.; Richard, C.S.M.; Blencke, H.-M.; Stensvåg, K.; Craik, D.J.; Haug, T. Isolation and characterization of antimicrobial peptides with unusual disulfide connectivity from the colonial ascidian Synoicum turgens. Mar. Drugs 2020, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-T.; Lee, C.-C.; Yang, J.-R.; Lai, J.Z.C.; Chang, K.Y. A large-scale structural classification of antimicrobial peptides. Biomed Res. Int. 2015, 2015, 475062. [Google Scholar] [CrossRef] [PubMed]
- Björn, C.; Håkansson, J.; Myhrman, E.; Sjöstrand, V.; Haug, T.; Lindgren, K.; Blencke, H.-M.; Stensvåg, K.; Mahlapuu, M. Anti-infectious and anti-inflammatory effects of peptide fragments sequentially derived from the antimicrobial peptide centrocin 1 isolated from the green sea urchin, Strongylocentrotus droebachiensis. AMB Express 2012, 2, 67. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Fan, L.; Sun, J.; Lao, X.; Zheng, H. Computational resources and tools for antimicrobial peptides. J. Pept. Sci. 2017, 23, 4–12. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, S. Comparative genomics analysis of five families of antimicrobial peptide-like genes in seven ant species. Dev. Comp. Immunol. 2012, 38, 262–274. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. BBA Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [Green Version]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef]
- Joo, H.; Chavan, A.G.; Phan, J.; Day, R.; Tsai, J. An amino acid packing code for α-helical structure and protein design. J. Mol. Biol. 2012, 419, 234–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouellette, A.J.; Satchell, D.P.; Hsieh, M.M.; Hagen, S.J.; Selsted, M.E. Characterization of luminal paneth cell alpha-defensins in mouse small intestine. Attenuated antimicrobial activities of peptides with truncated amino termini. J. Biol. Chem. 2000, 275, 33969–33973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides-using a sequence template to guide structure-activity relationship studies. BBA Biomembr. 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabere, M.N.; Noble, W.S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic α-helical cationic antimicrobial peptides. Pept. Sci. 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef]
- Juretić, D.; Chen, H.-C.; Brown, J.H.; Morell, J.L.; Hendler, R.W.; Westerhoff, H.V. Magainin 2 amide and analogues. Antimicrobial activity, membrane depolarization and susceptibility to proteolysis. FEBS Lett. 1989, 249, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Wolfenden, R.; Lewis, C.A.; Yuan, Y.; Carter, C.W. Temperature dependence of amino acid hydrophobicities. Proc. Natl. Acad. Sci. USA 2015, 112, 7484–7488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virta, M.; Åkerman, K.E.O.; Saviranta, P.; Oker-Blom, C.; Karp, M.T. Real-time measurement of cell permeabilization with low-molecular-weight membranolytic agents. J. Antimicrob. Chemother. 1995, 36, 303–315. [Google Scholar] [CrossRef]
- Igumnova, E.M.; Mishchenko, E.; Haug, T.; Blencke, H.-M.; Sollid, J.U.E.; Fredheim, E.G.A.; Lauksund, S.; Stensvåg, K.; Strøm, M.B. Synthesis and antimicrobial activity of small cationic amphipathic aminobenzamide marine natural product mimics and evaluation of relevance against clinical isolates including ESBL–CARBA producing multi-resistant bacteria. Bioorgan. Med. Chem. 2016, 24, 5884–5894. [Google Scholar] [CrossRef]
- Igumnova, E.M.; Mishchenko, E.; Haug, T.; Blencke, H.-M.; Sollid, J.U.E.; Fredheim, E.G.A.; Lauksund, S.; Stensvåg, K.; Strøm, M.B. Amphipathic sulfonamidobenzamides mimicking small antimicrobial marine natural products; investigation of antibacterial and anti-biofilm activity against antibiotic resistant clinical isolates. Bioorgan. Med. Chem. 2018, 26, 4930–4941. [Google Scholar] [CrossRef]
- McDonnell, G.; Russell, A.D. Antiseptics and Disinfectants: Activity, Action, and Resistance. Clin. Microbiol. Rev. 1999, 12, 147–179. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, P.; Rosa, R.D.; Destoumieux-Garzón, D. An intimate link between antimicrobial peptide sequence diversity and binding to essential components of bacterial membranes. BBA Biomembr. 2016, 1858, 958–970. [Google Scholar] [CrossRef]
- Sperstad, S.V.; Haug, T.; Vasskog, T.; Stensvåg, K. Hyastatin, a glycine-rich multi-domain antimicrobial peptide isolated from the spider crab (Hyas araneus) hemocytes. Mol. Immunol. 2009, 46, 2604–2612. [Google Scholar] [CrossRef]
- Solstad, R.G.; Johansen, C.; Stensvåg, K.; Strøm, M.B.; Haug, T. Structure-activity relationship studies of shortened analogues of the antimicrobial peptide EeCentrocin 1 from the sea urchin Echinus esculentus. J. Pept. Sci. 2020, 26, e3233. [Google Scholar] [CrossRef] [Green Version]
- Vesterlund, S.; Paltta, J.; Lauková, A.; Karp, M.; Ouwehand, A.C. Rapid screening method for the detection of antimicrobial substances. J. Microbiol. Meth. 2004, 57, 23–31. [Google Scholar] [CrossRef]
- Radeck, J.; Kraft, K.; Bartels, J.; Cikovic, T.; Dürr, F.; Emenegger, J.; Kelterborn, S.; Sauer, C.; Fritz, G.; Gebhard, S.; et al. The Bacillus BioBrick Box: Generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J. Biol. Eng. 2013, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frackman, S.; Anhalt, M.; Nealson, K.H. Cloning, organization, and expression of the bioluminescence genes of Xenorhabdus luminescens. J. Bacteriol. 1990, 172, 5767–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperstad, S.V.; Haug, T.; Paulsen, V.; Rode, T.M.; Strandskog, G.; Solem, S.T.; Styrvold, O.B.; Stensvåg, K. Characterization of crustins from the hemocytes of the spider crab, Hyas araneus, and the red king crab, Paralithodes camtschaticus. Dev. Comp. Immun. 2009, 33, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.Ø.; Andersen, J.H.; Bayer, A.; Pandey, S.K.; Lorentzen, M.; Jørgensen, K.B.; Sydnes, M.O.; Guttormsen, Y.; Baumann, M.; Koch, U.; et al. Kinase chemodiversity from the Arctic: The breitfussins. J. Med. Chem. 2019, 62, 10167–10181. [Google Scholar] [CrossRef] [PubMed]
- Michael, P.; Hansen, E.; Isaksson, J.; Andersen, J.H.; Hansen, K.Ø. Dendrobeaniamine A, a new alkaloid from the Arctic marine bryozoan Dendrobeania murrayana. Nat. Prod. Res. 2019, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Anuwongcharoen, N.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C. Roles of d-amino acids on the bioactivity of host defense peptides. Int. J. Mol. Sci. 2016, 17, 1023. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Region | Sequence | Net Charge | Hydro-Phobic Ratio (%) | Boman Index (kcal/mol) | CAMPR3 1 | ADAM 2 | |||
|---|---|---|---|---|---|---|---|---|---|
| SVM | RF | ANN | DA | SVM | |||||
| 1–10 | GPKTKAAAKM | +3 | 40 | 1.04 | 1.000 | 0.479 | AMP | 0.681 | 1.49 |
| 2–11 | PKTKAAAKMA | +3 | 50 | 0.96 | 0.548 | 0.439 | NAMP | 0.343 | 1.95 |
| 3–12 | KTKAAAKMAA | +3 | 60 | 0.78 | 0.131 | 0.443 | AMP | 0.325 | 2.53 |
| 4–13 | TKAAAKMAAK | +3 | 60 | 0.78 | 0.972 | 0.439 | AMP | 0.170 | 2.53 |
| 5–14 | KAAAKMAAKL | +3 | 70 | 0.03 | 0.478 | 0.428 | AMP | 0.797 | 2.59 |
| 6–15 | AAAKMAAKLA | +2 | 80 | −0.70 | 0.980 | 0.363 | AMP | 0.785 | 2.64 |
| 7–16 | AAKMAAKLAT | +2 | 70 | −0.26 | 0.989 | 0.358 | AMP | 0.483 | 2.41 |
| 8–17 | AKMAAKLATA | +2 | 70 | −0.26 | 0.947 | 0.325 | AMP | 0.312 | 2.41 |
| 9–18 | KMAAKLATAG | +2 | 60 | −0.17 | 0.330 | 0.281 | AMP | 0.254 | 2.13 |
| 10–19 | MAAKLATAGK | +2 | 60 | −0.17 | 0.651 | 0.270 | AMP | 0.135 | 2.13 |
| 11–20 | AAKLATAGKK | +3 | 50 | 0.61 | 0.615 | 0.425 | AMP | 0.786 | 2.07 |
| 12–21 | AKLATAGKKP | +3 | 40 | 0.79 | 0.751 | 0.376 | AMP | 0.535 | 1.29 |
| 13–22 | KLATAGKKPG | +3 | 30 | 0.87 | 0.244 | 0.377 | AMP | 0.647 | 1.58 |
| 14–23 | LATAGKKPGG | +2 | 30 | 0.23 | 0.736 | 0.379 | AMP | 0.649 | 2.16 |
| 15–24 | ATAGKKPGGW | +2 | 30 | 0.49 | 0.075 | 0.282 | AMP | 0.781 | 2.52 |
| 16–25 | TAGKKPGGWK | +3 | 20 | 1.22 | 0.880 | 0.398 | AMP | 0.591 | 2.53 |
| 17–26 | AGKKPGGWKA | +3 | 30 | 0.78 | 0.490 | 0.427 | AMP | 0.930 | 2.85 |
| 18–27 | GKKPGGWKAK | +4 | 20 | 1.52 | 0.968 | 0.559 | AMP | 0.884 | 2.85 |
| 19–28 | KKPGGWKAKL | +4 | 30 | 1.12 | 0.165 | 0.566 | AMP | 0.815 | 2.61 |
| 20–29 | KPGGWKAKLA | +3 | 40 | 0.38 | 0.027 | 0.448 | AMP | 0.689 | 2.44 |
| 21–30 | PGGWKAKLAE | +1 | 40 | 0.51 | 0.017 | 0.190 | AMP | 0.018 | 2.00 |
| 22–31 | GGWKAKLAEL | +1 | 50 | 0.02 | 0.325 | 0.238 | AMP | 0.041 | 2.37 |
| 23–32 | GWKAKLAELG | +1 | 50 | 0.02 | 0.444 | 0.241 | AMP | 0.041 | 2.37 |
| 24–33 | WKAKLAELGA | +1 | 60 | 0.21 | 0.205 | 0.252 | NAMP | 0.024 | 2.04 |
| 25–34 | KAKLAELGAD | 0 | 50 | 0.00 | 0.004 | 0.293 | NAMP | 0.002 | 1.47 |
| 26–35 | AKLAELGADA | −1 | 60 | 0.28 | 0.281 | 0.329 | NAMP | 0.003 | 1.44 |
| 27–36 | KLAELGADAV | −1 | 60 | 0.80 | 0.799 | 0.373 | NAMP | 0.007 | 0.56 |
| Peptide | Sequence 1 | Monoisotopic Mass (Da) | Net Charge | Boman Index (kcal/mol) | Hydro-Phobic Ratio (%) | Rt 3 | |
|---|---|---|---|---|---|---|---|
| Theoretical | Measured 2 | ||||||
| StAMP-1 | GKKPGGWKAK-NH2 | 1054.64 | 1054.64 | +5 | 1.52 | 20 | 0.40 |
| StAMP-2 | GKKWGGWKAK-NH2 | 1143.67 | 1143.67 | +5 | 1.29 | 30 | 1.75 |
| StAMP-3 | GKKPWGWKAK-NH2 | 1183.70 | 1183.70 | +5 | 1.38 | 30 | 2.17 |
| StAMP-4 | GKKPGWWKAK-NH2 | 1183.70 | 1183.70 | +5 | 1.38 | 30 | 2.05 |
| StAMP-5 | GKKWWGWKAK-NH2 | 1272.72 | 1272.72 | +5 | 1.15 | 40 | 5.21 |
| StAMP-6 | GKKWGWWKAK-NH2 | 1272.72 | 1272.72 | +5 | 1.15 | 40 | 5.39 |
| StAMP-7 | GKKPWWWKAK-NH2 | 1312.76 | 1312.76 | +5 | 1.24 | 40 | 5.70 |
| StAMP-8 | GKKWWWWKAK-NH2 | 1401.78 | 1401.78 | +5 | 1.01 | 50 | 8.77 |
| StAMP-9 | GRRPWWWRAR-NH2 | 1424.78 | 1424.78 | +5 | 4.99 | 40 | 6.65 |
| StAMP-10 | GRRWWWWRAR-NH2 | 1513.81 | 1513.81 | +5 | 4.76 | 50 | 9.20 |
| StAMP-11 | GRRPLLLRAR-NH2 | 1205.79 | 1205.79 | +5 | 4.21 | 40 | 2.54 |
| Antimicrobial Activity (MIC; µg/mL) 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gram-Pos | Gram-Neg | Fungi | ||||||||
| Peptide | Bm | Bs | Cg | Ml | Sa | Ec | Pa | Ap | Ca | Rh |
| Turgencin A 2 | 0.5 | 1.5 | 1.5 | 8.0 | 23.3 | 3.0 | 5.9 | 92.6 | 46.3 | 23.2 |
| StAMP-1 | 250 | >250 | 250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 |
| StAMP-2 | 3.9 | 125 | 31.3 | 250 | >250 | >250 | >250 | 62.5 | 125 | 62.5 |
| StAMP-3 | 3.9 | >250 | 15.6 | 250 | >250 | >250 | >250 | 62.5 | 125 | 62.5 |
| StAMP-4 | 3.9 | 125 | 3.9 | 125 | >250 | >250 | >250 | 62.5 | 62.5 | 31.3 |
| StAMP-5 | 1.0 | 15.6 | 2.0 | 15.6 | >250 | 31.3 | 250 | 31.3 | 31.3 | 15.6 |
| StAMP-6 | 1.0 | 3.9 | 3.9 | 62.5 | 250 | 62.5 | >250 | 62.5 | 62.5 | 31.3 |
| StAMP-7 | 1.0 | 3.9 | 2.0 | 31.3 | 125 | 31.3 | 250 | 15.6 | 31.3 | 15.6 |
| StAMP-8 | 3.9 | 7.8 | 7.8 | 15.6 | 125 | 62.5 | 125 | 7.8 | 15.6 | 15.6 |
| StAMP-9 | 1.0 | 3.9 | 2.0 | 3.9 | 7.8 | 7.8 | 31.3 | 31.3 | 31.3 | 15.6 |
| StAMP-10 | 3.9 | 7.8 | 7.8 | 15.6 | 62.5 | 15.6 | 31.3 | 62.5 | 62.5 | 15.6 |
| StAMP-11 | 7.8 | >250 | 31.3 | 62.5 | >250 | >250 | >250 | 250 | 125 | 31.3 |
| Indolicidin | 3.1 | 6.3 | 1.6 | 12.5 | 12.5 | 25.0 | >250 | 25.0 | 100 | 25.0 |
| Oxytetracycline | 0.6 | 10.0 | 0.2 | 1.3 | 0.04 | 1.3 | 2.5 | n.t 3 | n.t | n.t. |
| Triclosan | n.t | n.t | n.t | n.t | n.t | n.t | n.t | 3.1 | 3.1 | 1.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, I.K.Ø.; Lövdahl, T.; Simonovic, D.; Hansen, K.Ø.; Andersen, A.J.C.; Devold, H.; Richard, C.S.M.; Andersen, J.H.; Strøm, M.B.; Haug, T. Antimicrobial Activity of Small Synthetic Peptides Based on the Marine Peptide Turgencin A: Prediction of Antimicrobial Peptide Sequences in a Natural Peptide and Strategy for Optimization of Potency. Int. J. Mol. Sci. 2020, 21, 5460. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155460
Hansen IKØ, Lövdahl T, Simonovic D, Hansen KØ, Andersen AJC, Devold H, Richard CSM, Andersen JH, Strøm MB, Haug T. Antimicrobial Activity of Small Synthetic Peptides Based on the Marine Peptide Turgencin A: Prediction of Antimicrobial Peptide Sequences in a Natural Peptide and Strategy for Optimization of Potency. International Journal of Molecular Sciences. 2020; 21(15):5460. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155460
Chicago/Turabian StyleHansen, Ida K. Ø., Tomas Lövdahl, Danijela Simonovic, Kine Ø. Hansen, Aaron J. C. Andersen, Hege Devold, Céline S. M. Richard, Jeanette H. Andersen, Morten B. Strøm, and Tor Haug. 2020. "Antimicrobial Activity of Small Synthetic Peptides Based on the Marine Peptide Turgencin A: Prediction of Antimicrobial Peptide Sequences in a Natural Peptide and Strategy for Optimization of Potency" International Journal of Molecular Sciences 21, no. 15: 5460. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155460